IMMEDIATE ONLINE ACCEPTED (IOA)
ARTICLE
This article presented here has been peer reviewed and accepted for
publication in CCS Chemistry. The present version of this manuscript has
been posted at the request of the author prior to copyediting and
composition and will be replaced by the final published version once it is
completed. The DOI will remain unchanged.
IOA Posting Date: January 06, 2021
TITLE:
Development of Bulk Organic Chemical Processes --- History, Status
&
Opportunities for Academic Research
AUTHORS:
Carolin Schneider, Thomas Leischner, Pavel Ryabchuk, Ralf
Jackstell, Kathrin Junge, and Matthias Beller
Development of Bulk Organic Chemical Processes – History, Status &
Opportunities for Academic Research
Carolin Schneider, Thomas Leischner, Pavel Ryabchuk
†, Ralf Jackstell, Kathrin Junge, and Matthias
Beller*
Leibniz-Institut für Katalyse e.V., Albert-Einstein-Straße 29a, 18059 Rostock *Corresponding author: [email protected]
ABSTRACT: The production of bulk organic chemicals has a strong impact on our daily life. In this review, an overview of the important industrial processes using homogeneous catalysts is given. Using specific carbonylation and hydrogenation case studies, we want to show how basic research can contribute to the development of such processes and what challenges exist in this area for academic research. Catalysis - bulk chemicals – chemical processes – sustainability – methanol – carbonylation - hydrogenation.
Introduction
Without doubt industrial organic chemicals continue to have an ambiguous reputation in our society and are considered to be involved in many environmental and health problems. However, if properly produced and applied, they provide efficient solutions to many of our long-term problems. In fact, over the past 150 years the development of the chemical industry, its products and underlying processes resulted in key contributions not only improving the quality of all aspects of human living standards, but also to the significant increase of our life expectancy.1 Nowadays, chemicals are used for a
plethora of consumer goods as well as other industrial sectors, including agriculture manufacturing, constructions, rubber and plastic products, textiles, petroleum refining, pulp and paper, and primary metals.
From its beginning on, the production of synthetic organic chemical products relied to a large extent on the valorization of available and inexpensive feedstocks, which were subsequently transformed into more valuable and complex products.2
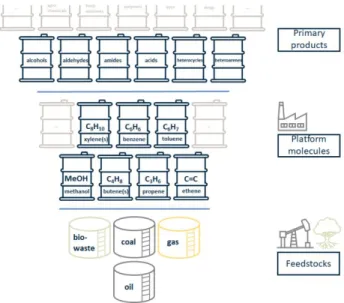
Originally, side-products of the coal industry such as coal tar were used and technically applicable processes for synthetic dyes, pharmaceuticals and polymers were developed. Following this general concept, until today the production of most organic chemicals depends on fossil-based feedstocks – coal, oil, and gas (Figure 1). However, due to current efforts towards a more sustainable world and a circular economy, an increasing number of chemical products are going to be produced from renewables in the next years.
Typically, the products of the chemical industry are subdivided into basic chemicals, fine and specialty chemicals, and consumer products. Bulk or so-called commodity chemicals are manufactured on a very large scale to cover global demand, while fine chemicals are produced in small and limited quantities (<1000 tons/year).3 Consequently, the valorization of
the applied raw materials has been improved over decades and the corresponding processes are highly optimized.
Figure 1: Valorization of basic feedstocks.
Due to the large amounts, most bulk chemicals are produced in dedicated continuous process plants. In contrast, fine chemicals are typically manufactured in batch reactors and in multipurpose plants. In bulk processes cost-efficiency is a focal point and the revenue per unit is relatively low; however, as a result of the produced amount the profit is satisfactory. Fine chemicals on the other hand are more expensive and their price is correlated with their performance. Usually, they are highly useful building blocks, influencing our everyday life through the combination with other chemicals or substrates to form for example specific materials, drugs, fragrances, or food additives. In this review, we are going to provide the reader with an overview of important bulk chemical processes featuring homogeneous catalysts. Moreover, we are going to demonstrate based on two specific case studies, how academic research can contribute to the development of related processes and what challenges exist in this area.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58
The Importance of Bulk Chemicals
In general, in this review the term bulk chemicals is used for products which are manufactured on a large scale (>100.000 ton/a) from primary feedstocks (oil, gas, coal, bio-waste) within only few steps. Other characteristics are their less complex chemical structures and a relatively low price (<1-2€/kg). In Scheme 1, a typical selection of bulk chemicals is shown.3,4
For such products, the manufacturing processes are generally known and often commercially available from engineering companies. Although the necessary technologies are accessible, an important entrance barrier for making bulk chemicals is the
high investment into any new plant. Thus, the implementation of new technologies is not simply a question of innovation or the efficiency of the respective process.
Most bulk chemical production sites operate in a continuous fashion utilizing heterogeneous catalysts. However, in several specific cases, e.g. carbonylation’s and oxidations, also homogeneous catalysts are applied. An important parameter to evaluate the efficacy of such a process is the space time yield of the catalyst system. Although the direct costs for the catalyst system are typically extremely low (<1 cent/kg product), it has a strong influence on the overall process costs, due to its control of substrate conversion and product selectivity.
O OH acetic acid 0.500.10
O
acetone 0.910.08
O OH acrylic acid 1.800.26
N
acrylonitrile 1.770.30
O HO
O OH adipic acid 1.840.32
benzene 0.880.14
butadiene 1.540.58
HO
butanol 1.100.07
O H N
caprolactam 2.100.14
cyclohexane 1.030.14
OH
ethanol 0.820.09
ethylene 1.030.08
O
ethylene oxide 1.330.14
OH
methanol 0.310.06
OH
phenol 1.250.18
O
O O
phthalic anhydride 1.230.18
propylene 0.980.08
O
propylene oxide 1.540.09
styrene 1.080.14
O OH O
HO
terephthalic acid 0.910.10
toluene 0.830.14
N C O N
C O
toluene diisocyanate 2.090.14
xylenes (mixed) 0.850.11
NH3
ammonia 0.410.07
-xylene 1.000.15
p-xylene 1.020.13
O O
n poly (methyl methacrylate) (PMM)
2.670.43
n
polyethylene 1.260.10
O
O O
O (CH2)2
n polyethylene terephthalate
1.290.14
Scheme 1: Selected examples of bulk chemicals and their average prices from January 2010 to February 2014 with standard deviations for northwestern Europe (price in €/kg).4
Industrial Processes with Homogenous Catalysts: From Past to Present
The term catalysis has been coined by Berzelius as early as 1836, though biocatalysts (enzymes) have already been used
since ancient times to produce alcoholic beverages (on large scale). The first examples of “modern” industrial catalytic processes e.g. the Deacon process (oxidation of HCl into chlorine) and the production of sulfuric acid were still 1
2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58
developed without in-depth understanding of the underlying chemical reactions.
Scheme 2: Selected Milestones of Industrial Chemistry. (Company names are given when a process was established, while concepts with later launched processes are given without company names)
Since the beginning of the 20th century, catalysis had a
tremendous impact on the chemical industry and the development of its processes. A timeline of selected milestones of industrial discoveries and realizations is shown in Scheme 2. An industrial landmark innovation was the realization of the Haber-Bosch process in 1913, allowing producing ammonia from dinitrogen and H2. This process laid the foundation for
conducting many high-pressure reactions on an industrial scale. Today’s most important example of bulk chemicals being
produced in the presence of a homogeneous catalyst was discovered by Otto Roelen at Ruhrchemie, Germany, already in 1938. Specifically, he observed the formation of aldehydes from olefins and CO in the presence of a cobalt catalyst. The underlying principals and chemical reactions were investigated in-depth in the decades after and resulted in significantly more efficient Rh-based catalyst systems. In the 1950s and 60s, processes including the Wacker oxidation of ethylene and the carbonylation of methanol to acetic acid (Monsanto process), were invented and marked the begin of the prevailing role of noble metals in industrial catalysis.
Xylene Oxidation (AMOCO process)
Based on the low price and availability of feedstocks with only C-C and C-H bonds (alkanes, alkenes, and arenes), oxidation reactions to introduce oxygen atoms or hydroxyl groups are of fundamental industrial importance for a fossil-based chemical industry. In this respect, one of the most prominent examples is the oxidation of para-xylene to give terephthalic acid (TPA). Being one of the main components in polyester industry nowadays, it is mainly used to produce polyester terephthalate (PET) and polyester fibers.
COOH
COOH Co/Mn/Br
HOAc, 175-225 °C 1.5-3.0 MPa of O2
COOH
COOH
HO O
OH COOH
O COOH
COOH
Scheme 3: Xylene oxidation under the conditions of the AMOCO process and its reaction pathway.
Initial experiments to prepare TPA can be traced back to photochemical investigations performed by Ciamician and Silber in 1912.5 They investigated the effect of light on the
oxidation of certain benzene derivatives. After a continuous reaction of one-year (!) they obtained terephthalic acid and p-toluic acid.6 Later on, further efforts in this direction were
conducted by Stephens;7 however, these studies were similarly
not applicable on an industrial scale, based on the long reaction time.
Today, 70% of terephthalic acid for PET production is obtained from the oxidation of para-xylene.8,9 Notably, for TPA
applications a highly selective process is necessary (impurities <25 ppm), which avoids costly purification steps.10,11
To design an easier and better purification process esterification with methanol was introduced, resulting in the formation of 1
2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58
dimethyl terephthalate (DMT). Initially, the oxidation reaction to produce p-toluic acid was performed with no solvent at a temperature of 180°C and a pressure of 0.8 MPa of air in the presence of a cobalt catalyst. DMT was eventually formed after esterification, second oxidation step, and another esterification reaction.12 Obviously, this process is complicated and resulted
in increased costs. However, in 1955 a cobalt-catalyzed air oxidation in the presence of bromide ions provided a solution to this problem. This so-called AMOCO process was subsequently commercialized in the late 1970s.13,14
Since then, several attempts were made to further improve this process by means of e.g. reaction in sub- or supercritical water,15,16 heterogenization of the catalyst,17-20 using carbon
dioxide as co-oxidant, or alternative promoters.21-23 However,
all efforts did not replace the original AMOCO process. Under optimal conditions, more than 98% of the para-xylene react to the desired product with selectivities around 95% within 8–24h. Although the high yield and selectivity are remarkable, there is interest towards an improved process due to environmental problems. This is especially true for the replacement of bromide which forms the highly toxic side products.24 The largest
commercial production plants are run by British Petroleum (BP), BP Zhuhai Chemical Company (Ltd.), and JBF Petrochemicals Ltd. (JBF) and produce more than 10Mt TPA per annum. In addition, the license for production is also owned by DuPont, Dow Chemical, Mitsubishi Chemical, Eastman Chemical, Hitachi, Mitsui Chemicals, Interquisa, and Grupo Petromex.25
Oxo-Synthesis
A serendipitous discovery led to the largest application of homogeneous catalysis in industry. Initially, working on Fischer-Tropsch reactions with heterogenous cobalt catalysts Otto Roelen observed the formation of aldehydes and ketones in the reaction of alkenes and synthesis gas (“syngas”).26-28
R CO/Hcat. 2
R CHO
+
R CHO
n- or linear aldehyde
iso- or branched aldehyde Scheme 4: General reaction scheme for a hydroformylation reaction.
Typically, in this reaction a mixture of linear and branched aldehydes (n-aldehydes and iso-aldehydes, respectively) are formed as major products (Scheme 4).
Remarkably, the first industrial unit was built only four years later after the discovery of the reaction at IG Farben
Leuna/Merseburg in Germany. In 2008, nearly 10.4 million metric tons of oxo chemicals had been produced,29 while each
plant is producing several hundred thousand metric tons per year. Due to the versatility of the formed aldehydes, it is easily possible to produce aliphatic alcohols, esters, as well as amines by follow-up reactions.
Analyzing the literature demonstrates, that there has been a steady increase of scientific publications on various aspects of hydroformylations, while the number of patent applications is stagnating.30 Over the years, several companies performed oxo
reactions and applied this methodology on an industrial scale. For instance, BASF, Exxon, Sasol, and Shell used cobalt catalyst systems with less syngas at temperatures from 120–190 °C, and pressure of 4–30 MPa. Furthermore, especially Shell applied cobalt-derived systems for the preparation of high boiling aldehydes or alcohols.31
For nearly 30 years cobalt-based catalysts prevailed in hydroformylation reactions. However, in the 1970s phosphine-modified Rh-complexes were introduced. Despite the much higher price of the metal, these systems proved to be superior for short chain olefins, due to the possibility to work at low pressures (1.8–6.0 MPa) and medium temperatures (85–130 °C).32 Especially, “low-pressure oxo processes” (LPOs) of
ethylene, propene, and butenes are now commonly applied in industry and cover 70% of the hydroformylation capacity (e.g. Dow Chemical, BASF, and Mitsubishi).30
With respect to reactivity of [HM(CO)xLy]-type metal
complexes in hydroformylations, the following order of activity is observed:33
Rh >> Co > Ir, Ru > Os > Pt > Pd >> Fe > Ni
Consequently, it is no surprise that today only rhodium- and cobalt-based catalysts are applied in industry. Due to the necessity of harsh reaction conditions using cobalt, rhodium is mainly used in modern developments. Interestingly, in 1980 only 10% of the industrial hydroformylation capacities were rhodium based, while only 15 years later roughly 80% featured rhodium as catalyst metal.34 In few cases, also bimetallic
systems are applied, thereby Co-salts were added in low concentrations to prevent catalyst deactivation by impurities (e.g. sulfur compounds or butadiene).35
Eventually, in hydroformylation processes the active catalyst determines the overall economics to a substantial extent.30 Thus,
there is a high demand for tailor-made ligands in these reactions. Their price and long-term stability are also crucial points for the overall cost of the industrial process.As a result, numerous purpose-made ligands were developed in industry for diverse applications (Scheme 5).30
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58
R
O P
tBu
3
R = H, Me,tBu
tBu
tBu
R1
R1
OR2 O
P O
R1=tBu, MeO; R2= Me, H
P NaO3S
NaO3S SO3Na
PAr2nPhn
PAr2nPhn
NaO3S
NaO3S Ar =m-C6H4-SO3Na
P R2
O O
H2
C
R1
R1 R1
R1
R1= Me, Ph(CMe2)
R2= alkyloxy, aryloxy
O O
MeO OMe
P P
tBu tBu
O O
O
tBu
tBu
O
R R
e.g. R,R =
Scheme 5: Selection of industrial relevant ligands.
Shell Higher Olefin Process
The Shell higher olefin process (SHOP) allows producing linear alpha olefins from simple ethylene (Schemes 6 and 7). Key step of the process is a homogeneous nickel-catalyzed oligomerization followed by isomerization and metathesis reactions.36 In the first step, a mixture of linear olefins ranging
from C4 to C30+ is produced. Via distillation it is possible to
separate a C6–C18 mixture, which is further fractionated to yield
starting materials for synthetic lubricants, plasticizer alcohols, detergent alcohols, or synthetic fatty acids. Lighter (<C6) and
heavier (>C18) alkenes undergo double-bond isomerization in
the presence of a potassium metal catalyst. In the final third step, metathesis of this isomerized mixture with ethylene is performed using an alumina-supported molybdate catalyst. The main products of interest are higher olefins, which are further used in industry to give so-called fatty alcohols, which are precursors for plasticizers and detergents.37 The annual global
production of alpha olefines through this method is over one million tons per annuum.38 Shell is selling their linear alpha and
internal olefins under the Neodene label.39,40
Originally, the process was developed by chemists at Shell Development in Emeryville (USA) in a collaboration with the Royal Shell Laboratories at Amsterdam in the Netherlands.41-49
Interestingly, this process constitutes the first industrial realization of a two-phase liquid/liquid technology, whereby in the ethene oligomerization step a nickel-phosphine catalyst (80–120 °C, 7–14 MPa) is dissolved in 1,4-butanediol, while the olefinic products form a second phase, allowing an easy product/catalyst separation.50
Evidently, the development of the active nickel catalyst was inspired to a large extent by the basic work of Ziegler and Wilke at MPI Mülheim, Germany.51,52 Based on that studies, Keim at
Shell introduced P-O chelate ligands for this process (Scheme 6)53-61 and performed detailed mechanistic studies.62-69 Although
this process was very innovative from the viewpoint of basic science, too (the first applied technology for biphasic catalysis), it did not inspire many scientists, simply because it was not known at that time to academic research groups.
(n + 2) O
NiH P O
Ph Ph
n n = 0-36
Scheme 6: Reaction scheme for the Shell Higher Olefin Process. Conditions are depending on the reaction step.
Hydrocyanation
Another example of the application of molecular catalysts for bulk chemical production in industry is the hydrocyanation reaction of 1,3-butadiene. Until today, this transformation is the basis for the production of Nylon (polyamid 66), one of the most important synthetic fibers. Already during the 1930s, researchers at DuPont, the development company of Nylon 66, looked for economic ways of a direct addition of hydrogen cyanide to 1,3-butadiene. However, initially an indirect hydrocyanation was used by DuPont (Scheme 7).70 In this
stoichiometric process, 1,3-butadiene was treated with chlorine to give 1,4-dichlorobut-2-ene, which was reacted with sodium cyanide to give adiponitrile. Obvious disadvantages of this route are the use of chlorine,71 and the number of steps. Hence,
a more efficient process was highly desired. 1
2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58
2 NaCl + 2 H2O Cl2 + 2 NaOH + H2 2 NaOH + 2 HCN 2 NaCN + 2 H2O
electrolysis
+ Cl2
+ 2 NaCl + 2 NaCN
+ H2
+ 2 HCN NC CN
CN NC
Cl Cl
Cl Cl
CN NC
CN NC
Scheme 7: Comparison of electrolysis pathway and direct hydrocyanation to give adiponitrile.
Actually, the direct addition of hydrogen cyanide to unsaturated carbon-carbon bonds was known on a small scale, but none of these routes proved to be industrially feasible.72-73 Even though
first experiments with homogeneous catalysts were not delivering adiponitrile,74 the work of Drinkard, who introduced
tetrakis(triethyl-phosphite)nickel(0) for hydrocyanation of 1-hexene,75 and further studies,75-77 finally resulted in a
commercial process at DuPont. The present adiponitrile synthesis can be divided into two steps. Firstly, 1,3-butadiene and HCN react in the presence of a tailored Ni[(ArO)3P]4
catalyst system to yield linear 3-pentenenitrile and branched 2-methyl-3-butenenitrile (approximate ratio 2:1). The branched product can be isomerized to the linear isomer in the presence of a Lewis acid promoter. Finally, kinetically controlled isomerization of the double bond with a hydridonickelcomplex and selective HCN addition leads to the desired adiponitrile with >90% yield.71,78 The first plant to produce adiponitrile
using this technology started to operate in 1971. It should be mentioned that aside from this process, adiponitrile is also manufactured by electrosynthesis using acrylonitrile as raw material. This technology is used by BASF and Monsanto.79
CN + +
HCN
CN
CN CN
CN CN
CN +
HCN NC CN
Scheme 8: Adiponitrile from 1,3-butadiene
Development of New Processes
Without doubt our civilization faces severe challenges and problems in the coming decades, which seem not to be easily solved based on today´s technologies. Indeed, we have to establish a renewable energy system, produce enough food and clean water for a growing population and at the same time use less resources and find improved ways to reduce the carbon footprint of our daily-life products, and so on. All these constraints will also put emphasis on the chemical industry to develop new and improved processes. At the same time, the
impetus of new governmental regulations should not be underestimated. As an example, climate change is one of the most vividly discussed topics in politics and society. In this context, the use of fossil-based raw materials also in the chemical industry is currently critically debated. Further there is the discussion about (real) carbon dioxide taxes to be installed in the coming years, which will provide a strong driving force towards more a sustainable and energy-efficient production of chemicals as well as a shift towards renewable feedstocks. Obviously, changing technologies for a typical plant scale of >100.000 tons/a is not an easy task, due to the large investment of the required infrastructure. Hence, “green” adaptations of existing technologies are much more likely than revolutionary process changes. For example, it is more probable that carbonylation reactions in future will be run with carbon monoxide generated by carbon dioxide electrolysis, which is powered from renewable energy, rather than performing completely new reactions. At this point, it has to be remembered that most organic bulk chemicals are in strong global competition; thus, the costs of the products cannot be higher even if the new process is more benign.
What will be the role of academia in this field? To be clear, without industrial partners for scientists at universities it is very difficult to identify the real existing challenges of a new process and often “elegant solutions for non-existing problems” are developed and published in highly reputed scientific journals. Hence, we believe cooperation (among equals) is a pre-requisite for successful joint projects. Undoubtedly, basic science can initiate game changing processes, but such developments need time. The typical cooperation project between industry and academic research groups with a duration of <1-3 years is simply too short.
We are convinced that both industry and academia strongly benefit from partnerships. In the following chapter two case studies will be presented, which show exemplarily our experiences in this area.
Case Studies
The following two case studies will give some insights reflecting our experience on the synergies, which can be realized when academic and industrial researchers work together hand in hand on the development of new processes. The first case is based on a long term (>20 years) cooperation with an international leading company in the field of carbonylation reactions, while in the second example a cooperation with a small/medium enterprise (SME) initiated our recent work on methanol synthesis. In both cases the importance of improved catalyst systems will be highlighted.
Improved Functional Catalysts for Carbonylation Reactions
Transition metal-catalyzed carbonylation reactions represent an important toolbox to produce aldehydes and a variety of carboxylic acid derivatives. Hence, using starting materials such as olefins,80-83 alcohols,84 alkyl or aryl halides85 the
corresponding esters, acids, aldehydes, amides, and C1
-extended alcohols86 are straightforwardly available depending
on the type of carbonylation reaction. Industrially most relevant 1
2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58
are carbonylation’s of olefins, which have been pioneered both by industrial chemists Otto Roelen (hydroformylation)87,88 and
Walter Reppe (hydroxyl- and alkoxycarbonylation)89. Shortly
after the original discovery vide supra the first oxo plant started – without knowing the underlying catalysis!30 In addition to
olefins, carbonylation of methanol to produce acetic acid is second most important homogeneous carbonylation process. Here, methanol reacts in situ with HI to methyl iodide, which then is activated by a rhodium carbonyl (Monsanto process90,91)
or iridium carbonyl species (Cativa process92,93).
Both for olefin carbonylation processes as well as for methanol carbonylation, the evolution of the respective catalyst system over the years allowed for major economic and ecologic improvements as well as the invention of new processes.94 As
an example, Drent et al. discovered a highly efficient alkoxycarbonylation reaction using propyne to give methyl methacrylate (MMA) in the presence of a specific palladium complex (Scheme 9).95
L =
20-60 bar CO, 45-115°C MeOH
O O Pd(OAc)2/L /MSA
P N
Scheme 9: Reaction pathway using acetylene to produce MMA by Drent et al..
Unfortunately, problems with the feedstock 1-propyne precluded a final commercial realization. Due to the interest in methyl methacrylate, the carbonylation of ethylene was further investigated by Tooze et al..96 After successful optimization,
this reaction became the basis of the so-called Lucite Alpha process (Scheme 11), which was first commercialized in 2008 and is producing currently >100.000 tons of MMA per annum.97-101 Crucial for its implementation was the use of the
tailored 1,2-bis(di-tert-butylphophino)xylene (dtbpx) ligand in the presence of an acidic co-catalyst. To be clear, without this ligand development no process could have been realized. According to reported data using less than 1 kg catalyst could produce 200 tons of the intermediate methyl propionate with a selectivity > 99.9% under mild conditions (10 bar, 100 °C). From the economical side, there is a capital cost advantage of approximate 30-40% compared to cyanohydrin or other C4 pathways.102, 103
Looking at the Lucite Alpha process, it is understandable why researcher’s all-over the world continue to be active in the exploration of ligands for advancing molecular catalysts in carbonylation and related processes. In this respect, our group has a tradition of nearly 25 years to prepare new phosphine ligands and evaluate the corresponding metal complexes in various catalytic reactions. More specifically, we became interested in the Lucite Alpha process first within the framework of a long-term industrial cooperation with Evonik Performance Materials. In several meetings with industrial
colleagues the question was discussed if this reaction can be used for other less reactive olefins than ethylene. Especially, we were eager to know if it would be possible to address also carbonylation’s of internal, branched, and/or sterically hindered olefins. Such olefins represent a major part of “di-butene”, which is an important industrial feedstock.
CO/MeOH
HCHO Cs/SiO2
[Pd]/ L /MSA O
O O
O
P P
L = 80 °C,
10 atm ethene + CO
Scheme 10: Reaction scheme for the Lucite Alpha process for MMA.
Hence, we initiated a catalyst search program for the alkoxycarbonylation of 2,3-dimethyl-2-butene as a model system for highly unreactive tetra-substituted olefins. After testing a selection of commercially available phosphines including privileged ligand structures and state-of-the-art catalysts, unfortunately we did not identify any suitable ligand. Obviously, to realize the alkoxycarbonylation of for demanding ‘non-reactive’ olefins, the development of a new catalyst system was imperative.104 In order to do so, we focused on the
improvement of key elementary reactions steps of the known catalytic cycles that needed to be tackled for improving the reactivity. According to the hydride mechanism, the formation of a metal hydride complex is mandatory for a catalytic alkoxycarbonylation cycle.105-109 A major problems for
multi-substituted olefins are the required isomerization to a more reactive intermediate, as well as the irreversible alcoholysis of the final Pd-acyl species.101 Based on mechanistic studies, we
realized that the alcoholysis of the Pd-acyl species had to be improved, which can be achieved by a base-promoted system.80,95,110-112 On the other hand, a strong acid is needed to
form the active palladium-hydride complex. Obviously, this constitutes a contradiction that needed to be solved. We thought, the use of an integrated base within the ligand structure could help to unravel this problem. Hence, inspired by the work of Drent a pyridyl-derivative of the dtbpx ligand was prepared (Scheme 11). Indeed, with this designed ligand in hand it became possible to carbonylate a wide variety of highly substituted olefins in good to excellent yields. With respect to reactivity, this catalyst surpasses known carbonylation catalysts by far. To be clearer, in the presence of the designed ligand olefins such as ethylene and propylene are converted in quantitative yields at 80 °C within 10 min and even at room temperature reaction was completed in 30 min. Notably, the dtbpx ligand showed no conversion in the latter case. In addition to industrially important bulk alkenes, more advanced synthetic building blocks can be alkoxycarbonylated with excellent activity.
The key feature of our active catalyst is the combination of amphoteric and sterically hindered moieties on the P atoms. On one hand, the integrated base acts as a proton shuttle for the 1
2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58
formation of the active palladium hydride and the rate-determining N-assisted alcoholysis. On the other hand, the nitrogen atom can improve the durability of the catalyst via hemilabile coordination to the palladium center in the catalytic cycle. Both experimental and DFT computational studies support the metal–ligand cooperativity in these alkene carbonylation reactions. Hence, we believe this work should stimulate the more rational design of new carbonylation catalysts, too.
P P N
N
Scheme 11: Ligand- and crystal structure for the LIKAT ligand
Following our concept, we introduced this structural motif also in other bidentate ligands, including ferrocenyl phosphines (Scheme 12).113 Some of the obtained ligands made it possible
to perform the methoxycarbonylation of ethylene under very mild conditions, especially lower temperature (rt) and without the use of an additional acid. Interestingly, catalysts with such ligands also tolerate diene impurities in the olefin carbonylation process. Indeed, the system showed higher stability for the alkoxycarbonylation of C4 mixtures.114
Fe P
P N N
Fe P
P X X Fe
P
P N N Fe
P
P NN
N
N Fe
P
P N N
N N Fe
P
P N N
Fe P
P N N N
N Fe
P
P N
N Fe
P
P NMe2
NMe2
Fe P
P N N N N
N N
N N X = O, S
Scheme 12: Selected ferrocenyl-based ligand structures with amphoteric and sterically hindered moieties on the P atoms.113
Using carbonylation reactions, not only the production of esters is important, similarly carboxylic acids find many applications (e.g. polymers, agrochemicals, pharmaceuticals).114-117
However, known catalyst systems for the direct introduction of an acid function by hydroxycarbonylation do not fulfill industrial requirements.111,118-124 Existing protocols often
involve special substrates and/or multi-step sequences to succeed in the production of the desired product.125-127 For
example, Oxea, BASF, and KH Neochem use a hydroformylation/oxidation sequence to form 3,5,5-trimethylhexanoic acid (TMHAc) from di-isobutylene (DIB) in more than 85.000 tons per annum (2017).128,129 Obviously, a
direct hydroxycarbonylation process has advantages in such cases. However, to be industrially viable major improvements are required, especially regarding catalyst stability and recycling. To our delight, the “built-in-base” LIKAT-ligand allowed for a very efficient conversion of a mixture of internal and terminal DIB olefins in the presence of a palladium-metal-precursor (Scheme 13). With 26 recycling runs this system showed an excellent recycling behavior and provides a basis for a cost-competitive process for manufacturing carboxylic acids.129 Consequently, as a next step towards realization a
continuous mini plant is planned.
tBu
+
tBu
81% 19%
tBu
OH O
[Pd]/ L /acid (0.25 / 1.00 / 3.75 mol%) solvent (2.0 mL), CO (40 bar)
100°C, 20 h
Scheme 13: Pd-catalyzed hydroxycarbonylation of di-iso-butene.
A long-term dream reaction in the field of carbonylation reactions is the direct synthesis of adipic acid starting from 1,3-butadiene. Adipic acid is used most importantly, with respect to scale, to produce nylon. In addition, adipates are applied as plasticizers, perfumes, lubricants, solvents, and for several pharmaceutical ingredients. In general, adipic acid and adipates are produced in a complex multi-stage, energy- and cost-intensive synthesis. The current process involves oxidation of a mixture of cyclohexanol and cyclohexanone, the so-called ‘KA oil’ (ketone-alcohol oil), by an excess of nitric acid. Notably, special equipment (stainless steel, type 304L or better, or even titanium)130 owing to the acid’s corrosiveness is required and
stoichiometric amounts of nitrous oxide (N2O) are produced,
which is a main greenhouse gas and has nearly 300 times the atmospheric heat-trapping capacity of CO2.131
OH O
+
CuII, NH 4VO3, HNO3
O
O OH HO
70-90 °C
Scheme 14: Main process for adipic acid.
Another disadvantage of this process is the formation of undesired by-products, e.g. glutaric and succinic acid, and to a lesser extent pentanoic and hexanoic acid. Due to all these drawbacks, a significant interest in more environmentally benign and cost-efficient alternative processes for adipic acid 1
2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58
and its derivatives exist and for several decades research in academia and industry has been performed in such direction. For example, the carbonylation of 1,3-butadiene has been investigated since the early 1980s. Nevertheless, previously existing catalyst systems did not allow for an industrially viable process, although several companies including BASF, Shell, and DSM tried to achieve this.132-137 Interestingly, again it was
the development of new ligands, which inspired us to explore this area. To study their potential, a small library of diphosphine ligands with sterically hindered and basic substituents was synthesized. Although such preparations always look straightforward afterwards, it took us nearly a year finishing the program. In the end this patience paid off and finally an efficient, direct dicarbonylation of 1,3-butadiene could be realized. The optimal catalyst system makes use of the specific tailored ligand HeMaRaPhos, which showed excellent results for both activity (≥ 95%) and selectivity (≥ 97%).138
nBuO
O
OnBu
O Pd(TFA)2/ L / PTSA
CO, nBuOH, 120 °C, 60 h
P P
N
HeMaRaphos L =
Scheme 15: Direct synthesis of adipates by carbonylation of 1,3-butadiene.
Under optimized conditions, the selectivity of the overall transformation is very high and no special materials resisting high corrosivity are needed. Currently, a continuous lab scale unit is installed to evaluate the long-term catalyst stability and to improve the activity further on. If these studies will be successful, the next step would be the transfer to pilot plant scale.
Apart from the industrial relevance, this work also served for us as an inspiration for basic methodological developments. Having such more active ligands in hand, it was possible to develop previously unknown carbonylation reactions. As an example, the domino double carbonylation of allylic alcohols is realized for the first time in the presence of HeMaRaPhos.139
New defined Catalysts for Hydrogenation Reactions
In the past decade many exciting developments of new molecularly-defined hydrogenation catalysts took place. This opens also new avenues for the industrial synthesis of fine and bulk chemicals. In this respect, methanol is an especially interesting example. It is among the top 5 most important basic chemicals produced in nearly 100 million tons per year in 2019.140 Today, methanol is used as raw material for the
production of a number of valuable basic chemicals such as formaldehyde, formic and acetic acid, dimethyl ether, and
others.141 Moreover, aliphatic and aromatic hydrocarbons are
obtained from methanol using methanol-to-olefin and methanol-to-gasoline processes on a million-ton scale. Besides serving as a C1 precursor in organic synthesis, methanol is also
used as a fuel additive and is a promising liquid hydrogen carrier with 12.6 wt% hydrogen content.142 Due to its high
energy density and physical properties, methanol serves as a synthetic combustion fuel for applications where battery technology cannot compete.
Methanol has been produced on an industrial scale since the 1920s from syngas via the BASF process, using a heterogeneous zinc chromite (Cr2O3−ZnO) catalyst. This
process occurs under harsh reaction conditions (300−400 °C, pressures > 300 atm).143 Later, in 1960s ICI introduced a more
efficient Cu/ZnO/Al2O3 catalyst, which operates at milder
conditions, but still elevated pressures (50−100 atm) and temperatures (200−300 °C) are needed.144 Until today, this
state-of-the-art catalytic system is still used for the syngas to methanol conversion, although there is a strong need for novel more efficient methods. Due to the process conditions and raw materials, methanol is among top three chemicals for energy consumption and greenhouse gas emissions.145
In general, instead of applying a heterogeneous material, a more active, soluble molecularly defined catalyst could be a solution to this problem since processes of this kind are typically more selective and operate at milder conditions than their heterogeneous counterparts. Yet, the development of homogeneous catalysts for CO hydrogenation to methanol proved to be extremely difficult. Initial attempts to design such a catalytic process date back to 1950s, when DuPont filed patents in which cobalt carbonyl species have been reported to convert syngas to methanol at prohibitively harsh conditions (1500−5000 atm).146,147 Furthermore, a lot of efforts have been
made in 1970–80s when a series of catalytic systems based on noble metals have been reported; however, they also operated at extreme pressures (> 1000 atm).148-150 The key challenges in
the hydrogenation of CO with defined organometallic species is the high binding affinity of CO to the metal centers and high bond strength of CO, which is in fact the strongest bond found in a neutral molecule (BDECO = 1076 kJ mol−1).151 Additionally,
for direct carbon monoxide reduction, migratory insertion of CO into an M−H bond, which is the key elementary step, is highly endothermic.152
In 2018, we had discussions with Marek Checinski and colleagues from Creative Quantum, a startup company specialized in predictive molecular modelling, on the conversion of carbon dioxide to methanol. During a meeting, the idea came up that the mechanistic difficulties can be overcome by using a conceptually different route of indirect CO hydrogenation. In this strategy methanol would be obtained in a stepwise fashion: At first CO is converted into a more reactive intermediate, generated in situ under reaction conditions, which is subsequently hydrogenated to give methanol. Interestingly, this strategy has been recognized by the Danish chemist Jens A. Christiansen as early as 1919.153 He proposed methyl formate
as such an intermediate, which is easily obtained by base-catalyzed carbonylation of methanol and might be further hydrogenated to two equivalents of methanol under milder conditions. Nonetheless, the first catalytic examples of such reactions appeared only in the 1980s. More specifically, two catalytic systems based on nickel154,155 and copper156 have been
suggested, but these systems suffered from excessive use of 1
2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58
moisture sensitive base. Moreover, the use of the nickel catalyst in industrial carbonylations is highly unlikely due to the volatility and high toxicity of the Ni(CO)4 formed in situ during
the reaction. As for the copper-based catalytic system, its main disadvantage is the extremely low activity resulting in a slow process and catalyst deactivation (TONs <10).
Hence, so far alkyl formates are not suitable intermediates for the indirect CO hydrogenation. Alternatively, we thought that formamides are better candidates for such stepwise process, due to their convenient handling and fast formation from CO.157
Although, the direct carbonylation of amines to the corresponding formamides is known, the hydrogenolysis of (form)amides to amines and alcohols remained elusive for decades. Indeed, amides are the most stable carboxylic acid derivatives.158 Their inert nature is a consequence of the
difference in the electronic character of nitrogen and oxygen, leading to the resonance stabilization. The earliest example of amide hydrogenolysis dates to 2003, when Crabtree filed a patent employing a Ru/Triphos for the hydrogenation of propionamide.159 However, also this catalytic system suffered
from low conversions and poor chemoselectivity. The breakthrough occurred later in 2010, when the Milstein group developed ruthenium-based catalyst for highly selective hydrogenolysis of (form)amides to corresponding amines and alcohols.160 In the past 10 years several other catalytic systems
based on noble (Ru,161,162 Ag163) and non-noble (Fe,164,165 Mo,166
Mn167) metals have been reported.
CO + 2 H2 CH3OH
5 µmol Mn-1
CO + 2 H2 CH3OH
50 µmol Ru-MACHO-BH A. Prakash, 2019
B. Beller, 2019
N H
NH2
H2N
145°C, 80 bar (CO/H2)
toluene/EtOH, 168 h
N H
150°C, 50 bar (CO/H2)
C6H12,18 h
N H or
Ru CO HBH3
H
P(Ph)2
(Ph)2P
N H
Ru-MACHO-BH
Mn CO CO Br
P(i-Pr)2
(i-Pr)2P
N H
TON - 539 TOF - 2.7 h-1
TON - 3170 (2550) TOF - 176 h-1(142 h-1)
Mn-1
Scheme 16: Comparison of recently disclosed approaches for a homogeneously catalyzed methanol synthesis.
The first low-temperature CO hydrogenation to methanol via formamide hydrogenolysis was reported by Prakash and co-workers in August 2019.168 In this work, formamides were
generated in situ via base catalyzed carbonylation of aliphatic amines. Subsequently, they were converted to methanol in the presence of a Ru-MACHO catalyst under 80 bar CO/H2 (10:70)
at 145°C during an extended period (148 h), giving an overall maximum TON of 539. A detailed mechanistic investigation by the Prakash group revealed a dramatic drop in activity for Ru-PNP complexes even in the presence of trace amounts of CO.169
The catalyst deactivation pathway was identified, which involves the formation of ruthenium biscarbonyl monohydride cationic complexes [RuHPNPR(CO)2]+. For comparison, this
catalyst in the absence of CO has been previously reported to be much more active for the hydrogenation of CO2 to methanol
via formamide hydrogenolysis (TON = 9900 at 145 °C, 75 bar CO2/H2), indicating that Ru-based catalysts are much less suited
for CO hydrogenation compared to CO2.
Independently from the Prakash group, our group together with Creative Quantum studied the same reaction in the past two years. According to modelling results, we predicted ruthenium-based catalysts to be less active. Interestingly, related iron- or 1
2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58
manganese-based complexes should have improved activity as the theoretical calculations indicated that manganese/iron pincer complexes are more robust and tolerant to CO than Ru. Interestingly, in case of manganese these theoretical predictions could be verified by experimental results, which showed that the key reactive manganese dihydride species are formed even in the presence of CO. Thus, a more efficient system for the indirect CO hydrogenation to methanol using manganese pincer complexes and azole additives under milder reaction conditions (TON up to 3170, at 150 °C, 50 bar CO/H2).170,171 Another
crucial aspect for the achievement of low-temperature CO hydrogenation was the use of amine-based promoters. The latter capture CO to give formamides, which then undergo manganese-catalyzed hydrogenolysis, giving methanol and regenerating the promoter. Among various N-based reagents studied, different azoles, in particular pyrrole and 3-methylindole, were found to be most optimal for methanol generation. The advantage of using azoles compared to more nucleophilic amines is the formation of weaker amide bond prone to hydrogenolysis.
After demonstrating the feasibility of this stepwise approach for carbon monoxide hydrogenation to methanol, currently we are engaged in the development of a continuous process and the construction of a lab demonstration unit will start in November 2020. Clearly, the current development is still very far away from real process, but to prove the opportunities, cooperation with colleagues from industry is a pre-requisite. What has to be achieved next? Although Prakash and co-workers as well as our group demonstrated that the reaction temperature and pressures for syngas to methanol conversion can be significantly reduced, the stability of the catalyst system and the N-promotor must be studied in more detail and has to be improved. We are aiming here for total turnover numbers of >106. In addition, the activity
must be increased, too.
Apart from hydrogenation of carbon monoxide, the catalytic reduction of carbon dioxide offers interesting potential as a future route for sustainable production of methanol. Currently, many efforts are devoted improving the existing heterogeneous catalysts for this transformation, but also new approaches are investigated applying homogenous catalysts, enzymes, or electro-catalytic routes. Because this area has been reviewed extensively in the past years, we do not cover this area here. 172-175
Outlook & Perspectives
Due to the inherent effectiveness of their products and their role as enabler for many other industries, the production of bulk chemicals will continue to be a crucial part of our global economy. Indeed, a sustainable society with more than 8 billion people will be impossible without synthetic chemicals and materials, which enable efficient energy generation, food production, safe mobility, communication, transport, and so on. Since its beginning in last century, the production of bulk organic chemicals has steadily increased and is expected to grow further on after the current pandemic crisis (Scheme 17). In the last two decades, especially China’s substantial economic growth contributed to this development.176 In the next years,
this trend will continue especially in Asia, but midterm also in other areas of the world, e.g. South America, Africa, and Middle East where market demands will grow and/or feedstock and energy costs are low. Because of the world-wide competition
within this industry and the price of their products as the main factor for selecting a certain manufacturer, bulk chemical processes must be state-of-the-art and as cost efficient as possible. To be clear, under these circumstances it is not an easy task to develop new processes or even more difficult to come up with completely new products. Nevertheless, in the context of a circular economy, the use of renewables or recycled materials is of growing interest and will stimulate many future efforts also for bulk chemicals. An illustrative example is the current case to replace (partly) fossil-based terephthalic acid (PTA) in polyesters by furan-2,5-dicarboxylic acid (FDCA), which can be produced from carbohydrates.177-181 Moreover,
governmental legislation, e.g. a global CO2 tax, will be an
important driver for innovations in this area! For example, such tax will provide not only opportunities for a new methanol (see above) or formic acid process, but also make sustainable Fischer Tropsch reactions attractive.
Scheme 17: Growth in world chemical sales.176
Is it meaningful for academic research to be involved in such endeavors? For us, there is no doubt that the answer should be yes, because it offers many synergies. We believe there is a clear win-win situation for both industry and academia in these important projects. To provide a state-of-the-art process for a certain chemical product, it needs a complete molecular understanding, which is impossible without the tools of advanced basic research. It is particularly at the early stages, from the drawing board to the realization and optimization in a laboratory, where academic research can provide significant expertise and opportunities. In this way, basic research can directly contribute to the benefits of our society. In addition, the knowledge transfer from industry to academia should not be underestimated.
Obviously, there are also many challenges for cooperation in this field. Due to the potentially large financial impact of the respective project, it needs trust (!) and patience (!) among the (scientific) partners. The present focus of many companies on their daily business is an obvious obstacle. If you want to develop a new bulk chemical process within in one year, you better not start working and safe the money. In this field longer term cooperation is a pre-requisite to be successful. Also, our understanding of science in general at the universities and 1
2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58
research institutes is impeding to some extent cooperation’s in this area. Very often, academic scientists consider a “novel” chemical transformation or a sophisticated material or molecular complex to be “real” science, while the improvement of a certain step of a >50 years old process by 2% seems to be at best boring. This is by no means true as it requires very hard work and detailed investigations. Moreover, such developments can result in significant ecological and economic advantages and thus in increased market shares of the respective product. Consequently, efforts to advance existing manufacturing processes deserve appropriate attention from industrial and research chemists, respectively, but also from society in general.
In conclusion, humanity is going to face an increasing necessity to develop all kinds of more environmentally benign processes. This is also true for the production of all kinds of chemicals chemicals. In this respect, bulk chemical processes have an enormous impact and should be considered. Using the complementary expertise of research, institutes, universities, and companies we will be faster and more innovative to make things happen: Together we stand, divided we might fall.
AUTHOR INFORMATION Present Addresses
†Galapagos NV, Generaal De Wittelaan L11 A3, 2800 Mechelen ORCID
Pavel Ryabchuk: 0000-0003-2859-3867 Matthias Beller: 0000-0001-5709-0965
Conflict of Interest
There are no conflicts to declare.
Funding Sources
European Research Council (ERC), NoNaCat grant number 670986.
ACKNOWLEDGMENT
We gratefully acknowledge the support of the European Research Council (ERC NoNaCat - 670986), the Federal Ministry of Education and Research (BMBF), the State of Mecklenburg-Vorpommern.
REFERENCES
(1) ourworldindata.org/life-expectancy
(2) Arpe, H.-J. Industrial Organic Chemistry, Fifth, Completely Revised Edition, Wiley-VCH Weinheim: Weinheim, Germany, 2010.
(3) https://www.syntor.co.uk/bulk-chemicals/
(4) Straathof, A.J.J.; Bampouli, A. Potental of commodity chemicals to become bio-based according to miximum yields and petrochemical prices. Biofuels, Bioprod. Bioref., 2017, 11, 798-810.
(5) Ciamician, G.; Silber, P. Chemische Lichteinwirkungen. Ber. Dtsch Chem. Ges. 1912, 45, 38.
(6) Xiao, Y. Zhang, X.Y.; Wang, Q.B.; Tan, Z.; Guo, C.C.; Deng, W.; Liu, Z. G.; Zhang, H.F. The preparation of terephthalic acid by solvent-free oxidation of p-xylene with air over T(p -Cl)PPMnCl and Co(OAc)2. Chin Chem Lett, 2011, 22,
135-138.
(7) Stephens, H.N. Oxidations in the benzene series by gaseous oxygen II. Alkyl benzenes with two or more carbon atoms in the side chain. J. Am. Chem. Soc., 1926, 48, 2920-2922. (8) Partenheimer, W. Methodology and scope of metal/bromide
autoxidation of hydrocarbons. Catal Today, 1995, 23, 69-158. (9) Li, M.; Niu, F.; Zuo, X.; Metelski, P.D.; Subramaniam, B. A
spray reactor concept for catalytic oxidation of p-xylene to produce high-purity terephthalic acid. Chem Eng Sci, 2013,
104, 93-102.
(10) Raghavendrachar, P.; Ramachandran S. Liquid-phase catalytic oxidation of p-xylene. Ind. Eng. Chem. Res., 1992, 31, 2, 453-462.
(11) Scheirs, J.; Long, T.E. Modern Polyesters: Chemistry and Technology of Polyesters and Copolyesters. Chichester, Wiley, 2003.
(12) Saffer, A.; Barker, R.S. Process for the production of aromatic polycarboxylic acids. GB Patent 807091A, 1959.
(13) Saffer, A.; Barker, R.S. Preparation of aromatic polycarboxylic acids. US Patent 2833816, 1958.
(14) Saffer, A.; Barker, R.S. Oxidation chemical process. US Patent 3089906A, 1963.
(15) Gupta, M.; Paul, S.; Gupta, R.; Loupy, A. ZnO: a versatile agent for benzylic oxidations. Tetrahedron lett, 2005, 46, 4957-4960.
(16) Shaabani, A.; Rahmati, A.; Aerobic oxidation of alkyl arenes using a combination of N-hydroxy phthalimide and recyclable cobalt(II) tetrasulfophthalocyanine supported on silica. Catal Commun, 2008, 9, 8, 1692-1697.
(17) Sabater, M.J.; Corma, A.; Domenech, A.; Fornés, V.; García, H. Chiral salen manganese complex encapsulated within zeolite Y: a heterogeneous enantioselective catalyst for the epoxidation of alkenes. Chem. Commun., 1997, 14, 1285-1286.
(18) Hutchings, G.J. New approaches to rate enhancement in heterogeneous catalysis. Chem. Commun., 1999, 4, 301-306. (19) Chavan, S.A.; Srinivas, D.; Ratnasamy, P. Selective Oxidation
of para-Xylene to Terephthalic Acid by µ3-Oxo-Bridges Co/Mn Cluster Complexes Encapsulated in Zeolite-Y. J. Catal., 2001, 204, 409-419.
(20) Ghiaci, M.; Mostajeran, M.; Gil. A. Synthesis and Characterization of Co-Mn Nanoparticles Immobilized on a Modified Bentonite and Its Application for Oxidation of p-Xylene to Terepthalic Acid. Ind. Eng. Chem. Res., 2012, 51,
49, 15821-15831.
(21) Brill, W. F. Terepthalic Acid by Single-Stage Oxidation. Ind. Eng. Chem., 1960, 52, 837-840.
(22) Yoo, J.S.; Jhung, S.H.; Lee, K.H.; Park, Y.S. An advanced MC-type oxidation process – the role of carbon dioxide. Appl. Catal. A, 2002, 223, 239-251.
(23) Saha, B.; Koshino, N.; Espenson, J.H. N-Hydroxyphthalimides and Metal Cocatalysts for the Autoxidation of p-Xylene to Terephthalic Acid. J. Phys. Chem. A, 2004, 108, 3, 425-431. (24) Horst, A; Holmstrand, H; Andersson, P; Thornton, B F;
Wish-kerman. A; Keppler, F; Gustafsson, O.; Geochim Cosmochim Ac-ta, 2014, 125, 186.
(25) Fadzil, N.A.M.; Rahim, M.H.A.; Maniam, G.P.; Chinese Journal of Catalysis, 2014, 35, 1641-1652.
(26) Roelen, O. A process for the production of oxygen-containing compounds. DE Patent 849548, 1938.
(27) Roelen, O. Production of oxygenated carbon compounds. US Patent 2327066, 1943.
(28) Adkins, H.; Krsek, G.; Hydroformylation of Unsaturated Compounds with a Cobalt Carbonyl Catalyst. J. Am. Chem. Soc., 1949, 71, 9, 3051-3055.
(29) Naqvi, S. Oxo Alcohols. Process Economics Program Report No. 21E; SRI Consulting: Menlo Park, CA, 2010.
(30) Franke, R.; Selent, D.; Börner, A. Applied Hydroformylation.
Chem. Rev., 2012, 112, 5675-5732.
(31) Drent, E.; Suykerbuyk. J.C.L.J. Hydroformylation process for the conversion of an ethylenically unsaturated compound to an alcohol. WO Patent 2004054946A1, 2004.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58
(32) Bohnen, H.-W.; Cornils, B. Hydroformylation of alkenes: A industrial view of the status and importance. Adv. Catal., 2002,
47, 1-64.
(33) Pruchnik, F. P. Organometallic Chcemistry of the Transition Elements; Plenum Press: New York, 1990; p.691.
(34) Beller, M.; Cornils, B.; Frohning, C.D.; Kohlpaintner, C.W. Progress in hydroformylation and carbonylation. J. Mol. Catal. A: Cem., 1995, 104, 17-85.
(35) Kirk, F.A.; Whitfield, G.H.; Miles, D.H. Process for production of oxygenated organic compounds. WO Patent 2002072520, 2004.
(36) Turner, A.H. Purity Aspects of Higher Alpha Olefins. J. Am. Oil Chem. Soc., 1983, 60, 623-627.
(37) Mol, J.C. Industrial Application of Olefin Methathesis. In Imamoglu Y., Bencze L. (eds) Novel Metathesis Chemistry: Well-Defined Initiator Systems for Specialty Chemical Synthesis, Tailored Polymers and Advanced Material Application. NATO Science Series (Series II: Mathematics, Physics and Chemistry), vol 122. Springer, Dordrecht, 2003. (38) Keim, W. Oligomerisierung von Ethen zu α-Olefinen:
Erfindung und Entwicklung des Shell-Higher-Olefin Prozesses (SHOP). Angew. Chem.2013, 125, 12722-12726.
(39)
www.shell.com/business-customers/chemicals/our- products/higher-olefins-and-derivatives/neodene-linear-alpha-and-internal-olefins.html.
(40) Shell Chemicals Information Handbook, Shell Chemicals Ltd., 2002.
(41) Mason, R.F. (by Shell Oil), alpha-olefin production. US Patent 3.676.523, 1972.
(42) Mason, R.F. (by Shell Oil), alpha-olefin production. US Patent 3.686.351, 1972.
(43) Berger, A.J. Olefin production. US Patent 3.726.938, 1973. (44) Mason, R.F. (by Shell Oil), alpha-olefin production. US Patent
3.737.475, 1973.
(45) Mason, R.F.; Wicker, G.R. (by Shell Oil), Catalyst, process for its manufacture and its use. DE Patent 2.264.088, 1973. (46) Lutz, E.F. (by Shell Oil), Ethylene Oligomerization Process.
US Patent 3.825.615, 1974.
(47) Keim, W., in Fundamental Research in Homogeneous Catalysis, Plenum, New York, 1984, Vol. 4. p.131.
(48) Lutz, E.F. (by Shell Oil), Ethylene Oligomerization process carried out in a monohydric/dihydric alcohol solvent mixture. US Patent 4.528.416, 1985.
(49) Lutz, E.F. Shell higher olefins process. J. Chem. Educ., 1986,
63, 3, 202-203.
(50) Steinborn, D. in Grundlagen der Metallorganischen Komplexkatalyse (p.148), Vieweg+Teubner, Wiesbaden 2010. (51) Wilke, G., in Fundamental Research in Homogeneous
Catalysis, Plenum, New York, 1979, Vol.3.
(52) Wilke, G. Contributions to Organo-Nickel Chemistry, Angew. Chem. Int. Ed. Engl., 1988, 27, 185-206.
(53) Bauer, R.S.; Chung, H.; Glockner, P.W.; Keim, W.; van Zwet, H. (by Shell Oil), Ethylene Polymerization. US Patent 3.635.937, 1972.
(54) Bauer, R.S.; Chung, H.; Cannell, L.G.; Keim, W.; van Zwet, H. (by Shell Oil), Ethylene polymerization in the presence of complex nickel catalysts containing benzoic acid derivative ligands. US Patent 3.637.636, 1972.
(55) Bauer, R.S.; Chung, H.; Glockner, P.W.; Keim, W.; van Zwet, H. (by Shell Oil), Ethylene Oligomerization. US Patent 3.644.563, 1972.
(56) van Zwet, H.; Bauer, R.S.; Keim, W. (by Shell Oil), ethylene Oligomerization in the presence of complex nickel-fluorinecontaining catalysts. US Patent 3.644.564, 1972. (57) Glockner, P.W.; Keim, W.; Mason, R.F. (by Shell Oil),
ethylene oligomerization. US Patent 3.647.914, 1972. (58) Bauer, R.S.; Glockner, P.W.; Keim, W.; Mason, R.F. (by Shell
Oil), Ethylene Oligomerization. US Patent 3.647.915, 1972. (59) Bauer, R.; Chung, H.; Keim, W.; van Zwet, H. (by Shell Oil),
ethylene polymerization in the presence of complex nickel
catalysts containing a glycolic acid, thioglycolic, or thiolactic acid ligand. US Patent 3.661.803, 1972.
(60) Bauer, R.; Chung, H.; Barnett, K.W.; Glockner, P.W.; Keim, W. (by Shell Oil), ethylene polimerization. US Patent 3.686.159, 1972.
(61) Spitzer, E.L.T.M. The Shop olefin process. Seifen, Oele, Fette, Wachse, 1981, 107, 141-143.
(62) Keim, W.; Kowaldt, F.H.; Goddard, R.; Krüger, C. Novel Coordination of (benzoylmethylene)triphenylphosphorane in a Nickel Oligomerization Catalyst. Angew. Chem. Int. Ed. Engl., 1978, 17, 466-467.
(63) Keim, W.; Behr, A.; Limbäcker, B.; Krüger, C. Novel Nickel-Oligomerization Catalysts with Arsenic-Oxygen Chelate Ligands. Angew. Chem. Int. Ed. Engl., 1983, 22, 503. (64) Keim, W. Nickel Hydrides: Catalysis in Oligomerization and
Polymerization Reactions of Olefins. Ann. N. Y. Acad. Sci., 1983, 415, 191-200.
(65) Keim, W.; Behr, A.; Gruber, B.; Hoffmann, B.; Kowaldt, F.H.; Kürschner, U.; Limbäcker, B.; Sistig, F.P. Reaction of Chelate Ylides with Nickel(0) Complexes. Organometallics, 1986, 5, 2356-2359.
(66) Keim, W. Organometallic complexes as catalyst precursors: advantages and disadvantages. J. Mol. Catal., 1989, 52, 19-25. (67) Müller, U.; Keim, W.; Krüger, C.; Betz, P.
[{Ph2PCH2C(CF3)2O}NiH(PCy3)]: Support for a Nickel
hydride Mechanism in Ethene Oligomerization. Angew. Chem. Int. Ed. Engl., 1989, 28, 1011-1013.
(68) Keim, W. Nickel: An Element with Wide Application in industrial Homogeneous Catalysis. Angew. Chem. Int. Ed. Engl., 1990, 29, 235-244.
(69) Keim, W.; Schulz, P. Chelate control in the nickel-complex catalysed homogeneous oligomerization of ethylene. J. Mol. Catal., 1994, 92, 21-33.
(70) Parshall, G.W., in Homogeneous Catalysis. (p. 219) Wiley, New York, 1980.
(71) Tolman, C.A.; McKinney, R.J.; Seidel, W.C.; Druliner, J.D.; Stevens, W.R. Homogeneous Nickel-Catalyzed Olefin Hydrocyanation. Advances in Catalysis,1985, 33, 1-46. (72) Schulze, W.A.; Mahan, J.E. (by Phillips Petroleum Company),
Production od Nitriles of Unsaturated Acids. US Patent 2.422.859, 1947.
(73) Harris, C.R; DeAtley, W.W. (by DuPont), Production of Nitriles from certain olefins and HCN. US Patent 2.455.995, 1948.
(74) ArthurJr., P.; England, D.C.; Pratt, B.C.; Whitman, G.M. Addition of Hydrogen Cyanide to Unsaturated Compounds. J. Am. Chem. Soc., 1954, 76, 5364-5367.
(75) Drinkard, W.C.; Lindsey Jr., R.V (by DuPont), Hydrocyanation of Olefins using selected Nickel Phosphite Catalysts. US Patent 3.496.215, 1970.
(76) Drinkard Jr., W.C.; Kassal, R.J. (by DuPont), Hydrocyanation of Olefins. US Patent 3.496.217, 1970.
(77) Drinkard Jr., W.C. (by DuPont), Hydrocyanation of Olefins. US Patent 3.496.218, 1970.
(78) Steinborn, D. Grundlagen der metallorganischen Komplexkatalyse p. 262
(79) Cardoso, D.S.; Slijukic, B., Santos, D.M.F.; Sequeira, C.A.C. Organic Electrosynthesis: From Laboratorial Practice to Industrial Applications. Org. Process Res. Dev.,2017, 21, 9, 1213-1226.
(80) Margo, A.A.N.; Robb, L-M.; Pogorzelec, P.J.; Slawin, A.M.Z.; Eastham, G.R.; Cole-Hamilton, D.J. Highly selective formation of unsaturated esters or cascade reactions to α,ω -diesters by the methoxycarbonylation of alkynes catalysed by
palladium complexes of
1,2-bis(ditertbutylphosphinomethyl)benzene. Chem. Sci., 2010, 1, 723-730.
(81) Suleiman, R.; Tijani, J.; ElAli, B. Palladium(II)-catalyzed catalytic aminocarbonylation and alkoxycarbonyaltion of terminal alkynes: regioselectivity controlled by the nucleophiles. Appl. Organometal. Chem.2010, 24, 38-46.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58