organic papers
o234
G. Vasukiet al. C10H8ClNO DOI: 101107/S1600536801002574 Acta Cryst.(2001). E57, o234±o235 Acta Crystallographica Section EStructure Reports
Online
ISSN 1600-5368
3-Chloro-2-methylquinolin-4(1
H
)-one
G. Vasuki,aV. Parthasarathi,a* K. Ramamurthi,aP. Jaisankarband B. Varghesec
aDepartment of Physics, Bharathidasan
Univer-sity, Tiruchirappalli 620 024, India,b
Depart-ment of Organic Chemistry, Madras University, Chennai, India, andcRegional Sophisticated
Instrumentation Centre, IIT, Chennai, India
Correspondence e-mail: [email protected]
Key indicators Single-crystal X-ray study
T= 293 K
Mean(C±C) = 0.004 AÊ
Rfactor = 0.050
wRfactor = 0.151
Data-to-parameter ratio = 13.8
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2001 International Union of Crystallography Printed in Great Britain ± all rights reserved
The title compound, C10H8ClNO, crystallizes in the
centro-symmetric space groupC2/c. The compound exists in the keto form in the crystalline state. The heterocyclic ring is not aromatic. The N atom is sp2 hybridized. The structure is
stabilized by NÐH O intermolecular hydrogen bonds.
Comment
Quinolines are ligands which are used as complexing agents for different metals (Hensen et al., 1999). The structure determination of the title compound, (I), was undertaken to study the effect of substitutions at the 2 and 3 positions on the quinolinone ring as well as the nature of the hydrogen bonding.
The torsion angles and the least-squares plane con®rm that the quinolinone ring is planar with the largest out-of-plane displacement for C4 [0.051 (2) AÊ]. The exocyclic angles of C3ÐC2ÐC11 [124.4 (2)] and C3ÐC4ÐO4 [124.4 (2)]
deviate signi®cantly from the normal value of 120. This may
be due to the steric repulsion between the substituents at positions 2 and 3, and at 3 and 4 respectively. The C4 O4 bond length [1.255 (3) AÊ] indicates a typical double-bond character and keto form of the compound in the crystalline state. In the non-aromatic heterocyclic ring, due to conjuga-tion in N1ÐC2 C3ÐC4 O4, C3ÐC4 [1.421 (3) AÊ] shows the characteristic shortening of the bond from the normal value of 1.478 AÊ (Allen et al., 1987). In the absence of substituents at C2, the average bond distance of N1ÐC2 is 1.310 (3) AÊ in related structures (Dobson & Gerkin, 1999; Lokaj et al., 1999). In the present structure, due to the substitution of the methyl group at C2, there is a signi®cant increase in the bond length N1ÐC2 [1.342 (3) AÊ] from the average value. The N atom issp2hybridized. The structure is
stabilized by a linear intermolecular hydrogen bond N1Ð H1 O4i [symmetry code: (i) x, ÿy, z+1
2]. The
hydrogen-bond parameters are: N1ÐH1 = 0.89 AÊ, N1 O4i =
2.712 (2) AÊ, H1 O4i = 1.825 AÊ and N1ÐH1 O4i = 174.
All the other intermolecular interactions are van der Waals in nature.
Experimental
2-Methyl-4-quinolone was treated with an equimolar amount ofN -chlorosuccinimide in glacial acetic acid at 323±333 K for 30 min. The reaction mixture was poured over ice and the solid was ®ltered. It was washed with ice-cold water and dried over anhydrous calcium chloride (yield 75%). The compound was crystallized from ethanol by slow evaporation at 298±303 K.
Crystal data C10H8ClNO Mr= 193.62 Monoclinic,C2/c a= 24.493 (4) AÊ
b= 6.365 (2) AÊ
c= 12.859 (8) AÊ = 117.240 (3) V= 1782.2 (13) AÊ3 Z= 8
Dx= 1.443 Mg mÿ3 CuKradiation Cell parameters from 25
re¯ections = 20±30 = 3.42 mmÿ1 T= 293 (2) K Plate, yellow
0.350.200.15 mm Data collection
Enraf±Nonius CAD-4 diffract-ometer
!±2scans
Absorption correction: scan (Northet al., 1968)
Tmin= 0.320,Tmax= 0.603
1661 measured re¯ections 1624 independent re¯ections 1440 re¯ections withI> 2(I)
Rint= 0.050 max= 68.0 h= 0!29
k= 0!7
l=ÿ15!13 2 standard re¯ections
frequency: 120 min intensity decay: none
Re®nement Re®nement onF2 R[F2> 2(F2)] = 0.050 wR(F2) = 0.151 S= 1.08 1624 re¯ections 118 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0966P)2 + 1.8554P]
whereP= (Fo2+ 2Fc2)/3 (/)max= 0.002
max= 0.43 e AÊÿ3 min=ÿ0.36 e AÊÿ3
Table 1
Selected geometric parameters (AÊ,).
N1ÐC9 1.373 (3)
C5ÐC6 1.364 (4) C5ÐC10C9ÐC10 1.405 (3)1.400 (3)
N1ÐC2ÐC3 119.1 (2) N1ÐC2ÐC11 116.5 (2) C2ÐC3ÐCl 119.55 (18) O4ÐC4ÐC3 124.4 (2)
N1ÐC9ÐC8 120.8 (2) N1ÐC9ÐC10 119.4 (2) C9ÐC10ÐC5 119.0 (2) C9ÐC10ÐC4 120.1 (2)
C9ÐN1ÐC2ÐC3 ÿ2.8 (3) N1ÐC2ÐC3ÐC4 0.9 (3) C2ÐC3ÐC4ÐO4 ÿ178.1 (2)
ClÐC3ÐC4ÐC10 ÿ177.38 (16) C7ÐC8ÐC9ÐC10 ÿ2.2 (4) O4ÐC4ÐC10ÐC9 176.3 (2)
All H atoms, except the methyl H atoms, were located from difference Fourier maps and were included in the structure-factor calculations with isotropic displacement parameters equal to 1.1Ueq
of their respective carrier atom, but their parameters were not re®ned. The methyl H atoms were ®xed with HFIX options of the
SHELX program, using the riding model.Uiso of methyl H atoms
were taken as 1.5Ueq(C11).
Data collection: CAD-4 EXPRESS (Enraf±Nonius, 1994); cell re®nement:CAD-4EXPRESS; data reduction:MolEN(Fair, 1990); program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to re®ne structure:SHELXL97 (Sheldrick, 1997); molecular graphics: ZORTEP (Zsolnai, 1997); software used to prepare material for publication:SHELXL97.
References
Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987).J. Chem. Soc. Perkin Trans.2, pp. S1±19.
Dobson, A. J. & Gerkin, R. E. (1999).Acta Cryst.C55, 1192±1195. Enraf±Nonius (1994). CAD-4 EXPRESS. Version 5.1/1.2. Enraf±Nonius,
Delft, The Netherlands.
Fair, C. K. (1990).MolEN.Enraf±Nonius, Delft, The Netherlands. Hensen, K., Mayr-Stein, R. & Bolte, M. (1999).Acta Cryst.C55, 1565±1567. Lokaj, J., Kettmann, V., Machacek, V. & Halama, A. (1999).Acta Cryst.C55,
1101±1103.
North, A. C. T., Phillips, D. C & Mathews, F. S. (1968).Acta Cryst.A24, 351± 359.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of GoÈttingen, Germany.
Zsolnai, L. (1997).ZORTEP. University of Heidelberg, Germany.
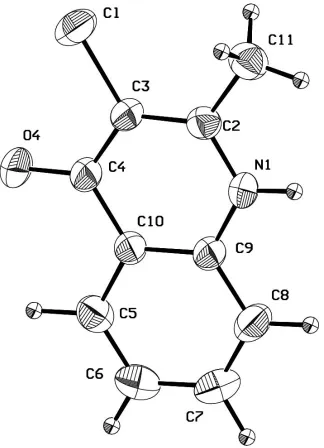
Figure 1
supporting information
sup-1
Acta Cryst. (2001). E57, o234–o235supporting information
Acta Cryst. (2001). E57, o234–o235 [doi:10.1107/S1600536801002574]
3-Chloro-2-methylquinolin-4(1
H
)-one
G. Vasuki, V. Parthasarathi, K. Ramamurthi, P. Jaisankar and B. Varghese
S1. Comment
Quinolines are ligands which are used as complexing agents for different metals (Hensen et al., 1999). The structure determination of the title compound, (I), was undertaken to study the effect of substitutions at the 2 and 3 positions on the quinolinone ring as well as the nature of hydrogen bonding. The torsion angles and the least-squares plane confirm that the quinolinone ring is planar with the largest out-of-plane displacement for C4 [0.051 (2) Å]. The exocyclic angles of C3 —C2—C11 [124.4 (2)°] and C3—C4—O4 [124.4 (2)°] deviate significantly from the normal value of 120°. This may be due to the steric repulsion between the substituents at positions 2 and 3, and at 3 and 4 respectively. The C4═O4 bond length [1.255 (3) Å] indicates a typical double-bond character and keto form of the compound in the crystalline state. In the non-aromatic heterocyclic ring, due to conjugation in N1—C2═C3—C4═O4, C3—C4 [1.421 (3) Å] shows the characteristic shortening of the bond from the normal value of 1.478 Å (Allen et al., 1987). In the absence of substituents at C2, the average bond distance of N1—C2 is 1.310 (3) Å in related structures (Dobson & Gerkin, 1999; Lokaj et al., 1999). In the present structure, due to the substitution of the methyl group at C2, there is a significant increase in the bond length N1—C2 [1.342 (3) Å] from the average value. The N atom is sp2 hybridized. The structure is stabilized by a linear
intermolecular hydrogen bond N1—H1···O4i [symmetry code: (i) x, -y, z + 0.5]. The hydrogen-bond parameters are: N1
—H1 = 0.89 Å, N1···O4i = 2.712 (2) Å, H1···O4i = 1.825 Å and N1—H1···O4i = 174°. All the other intermolecular
interactions are van der Waals in nature.
S2. Experimental
2-Methyl-4-quinolone was treated with an equimolar amount of N-chlorosuccinimide in glacial acetic acid at 323–333 K for 30 min. The reaction mixture was poured over ice and the solid was filtered. It was washed with ice-cold water and dried over anhydrous calcium chloride (yield 75%). The compound was crystallized from ethanol by slow evaporation at 298–303 K.
S3. Refinement
All H atoms, except the methyl H atoms were located from difference Fourier maps and were included in the structure-factor calculations with isotropic displacement parameters equal to 1.1Ueq of their respective carrier atom, but their
Figure 1
The molecular structure of (I) showing 50% probability displacement ellipsoids.
3-chloro-2-methylquinolin-4-one
Crystal data C10H8ClNO
Mr = 193.62
Monoclinic, C2/c a = 24.493 (4) Å b = 6.365 (2) Å c = 12.859 (8) Å β = 117.240 (3)° V = 1782.2 (13) Å3
Z = 8
F(000) = 800 Dx = 1.443 Mg m−3
Cu Kα radiation, λ = 1.54180 Å Cell parameters from 25 reflections θ = 20–30°
µ = 3.42 mm−1
T = 293 K Plate, yellow
supporting information
sup-3
Acta Cryst. (2001). E57, o234–o235Data collection Enraf-Nonius CAD-4
diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
ω–2θ scans
Absorption correction: ψ scan (North et al., 1968)
Tmin = 0.320, Tmax = 0.603
1661 measured reflections
1624 independent reflections 1440 reflections with I > 2σ(I) Rint = 0.050
θmax = 68.0°, θmin = 4.1°
h = 0→29 k = 0→7 l = −15→13
2 standard reflections every 120 min intensity decay: none
Refinement Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.050
wR(F2) = 0.151
S = 1.08 1624 reflections 118 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained w = 1/[σ2(F
o2) + (0.0966P)2 + 1.8554P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.002
Δρmax = 0.43 e Å−3
Δρmin = −0.36 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
H11C 0.0622 0.4322 1.1338 0.070* H1 0.1118 0.0496 1.3000 0.040* H5 0.1738 −0.4779 1.0530 0.045* H6 0.2196 −0.6719 1.2278 0.053* H7 0.2211 −0.5673 1.4060 0.056* H8 0.1740 −0.2445 1.4119 0.051*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Cl 0.0669 (5) 0.0536 (4) 0.0399 (4) 0.0124 (3) 0.0236 (3) 0.0171 (3) O4 0.0892 (14) 0.0518 (11) 0.0363 (10) 0.0101 (10) 0.0399 (10) 0.0030 (8) N1 0.0484 (11) 0.0381 (10) 0.0290 (9) 0.0006 (8) 0.0215 (8) −0.0015 (8) C2 0.0409 (11) 0.0315 (11) 0.0375 (12) −0.0017 (9) 0.0207 (9) 0.0012 (9) C3 0.0434 (11) 0.0345 (12) 0.0298 (11) −0.0016 (9) 0.0170 (9) 0.0040 (9) C4 0.0466 (12) 0.0350 (11) 0.0311 (11) −0.0045 (9) 0.0207 (9) 0.0001 (9) C5 0.0465 (12) 0.0363 (12) 0.0480 (13) −0.0027 (10) 0.0260 (10) −0.0031 (10) C6 0.0484 (13) 0.0360 (13) 0.0638 (16) 0.0064 (10) 0.0271 (12) 0.0053 (12) C7 0.0555 (15) 0.0495 (15) 0.0505 (16) 0.0086 (12) 0.0223 (12) 0.0196 (12) C8 0.0570 (15) 0.0521 (15) 0.0355 (13) 0.0057 (12) 0.0227 (11) 0.0099 (11) C9 0.0399 (11) 0.0342 (11) 0.0324 (11) −0.0024 (9) 0.0189 (9) 0.0011 (9) C10 0.0392 (11) 0.0314 (11) 0.0327 (12) −0.0032 (8) 0.0192 (9) −0.0005 (8) C11 0.0535 (15) 0.0390 (13) 0.0518 (15) 0.0046 (11) 0.0285 (12) 0.0000 (11)
Geometric parameters (Å, º)
Cl—C3 1.734 (2) C5—H5 1.064 O4—C4 1.255 (3) C6—C7 1.395 (4) N1—C2 1.342 (3) C6—H6 0.923 N1—C9 1.373 (3) C7—C8 1.369 (4)
N1—H1 0.890 C7—H7 0.994
C2—C3 1.371 (3) C8—C9 1.399 (3) C2—C11 1.497 (3) C8—H8 1.004 C3—C4 1.421 (3) C9—C10 1.400 (3) C4—C10 1.452 (3) C11—H11A 0.960 C5—C6 1.364 (4) C11—H11B 0.960 C5—C10 1.405 (3) C11—H11C 0.960
supporting information
sup-5
Acta Cryst. (2001). E57, o234–o235O4—C4—C10 120.5 (2) C9—C10—C4 120.1 (2) C3—C4—C10 115.10 (19) C5—C10—C4 121.0 (2) C6—C5—C10 120.4 (2) C2—C11—H11A 109.5 C6—C5—H5 121.1 C2—C11—H11B 109.5 C10—C5—H5 118.5 H11A—C11—H11B 109.5 C5—C6—C7 120.2 (2) C2—C11—H11C 109.5 C5—C6—H6 118.2 H11A—C11—H11C 109.5 C7—C6—H6 121.5 H11B—C11—H11C 109.5