Copyright © 2002, American Society for Microbiology. All Rights Reserved.
The Mold-Specific
MS8
Gene Is Required for Normal Hypha
Formation in the Dimorphic Pathogenic Fungus
Histoplasma capsulatum
Xianbin Tian and Glenmore Shearer, Jr.*
Department of Biological Sciences, Center for Molecular and Cellular Biosciences, University of Southern Mississippi, Hattiesburg, Mississippi 39406-5018
Received 2 August 2001/Accepted 22 January 2002
The dimorphic fungusHistoplasma capsulatum is the etiologic agent of one of the most common systemic
mycoses of humans, histoplasmosis. In the environment,H. capsulatumgrows in a differentiated mold form and
shifts to an undifferentiated yeast form after mold fragments or spores are inhaled. This mold-to-yeast shift is
required for disease. Little is known about the molecular biology of dimorphism inHistoplasma, and most
studies have been directed toward yeast-specific genes. While it is important to examine the role of genes upregulated in the yeast morphotype, genes which are silenced in the yeast (i.e., mold-specific genes) may also play a critical role in dimorphism. To begin to examine this hypothesis, we report here the first misexpression
and knockout analysis of a mold-specific gene inHistoplasma. The strongly expressedMS8gene encodes a
predicted 21-kDa protein extremely rich in glycine and glutamine. Forced expression ofMS8driven by the
TEF1promoter in yeast did not alter the yeast morphology at 37°C or mold formation at 25°C. Yeast expressing
MS8did exhibit clumping in liquid medium and formed “sticky” colonies on agar plates. Allelic replacement
ofMS8was accomplished by a positive-negative selection procedure.ms8knockout mutants formed apparently
normal yeast at 37°C but gave rise to aberrant mycelia at 25°C. The mold colonies of the knockouts were less than half as large as normal, had a granular surface, produced a dark-red pigment, and formed short hyphae which were 40% wider with a distinctive twisted “zig-zag” shape.
The dimorphic fungus Histoplasma capsulatumis the
etio-logic agent of one of the most common systemic mycoses of humans, histoplasmosis. Integral to pathogenesis is the mold-to-yeast (M-Y) dimorphic shift. The organism grows in soil as a differentiated multicellular mold. When mold fragments or spores are inhaled, the fungus shifts to an undifferentiated single-cell yeast growth form in the lungs of the host. If this M-Y shift is blocked, the disease cannot progress (11). In the laboratory, this reversible dimorphism can be observed by changing the incubation temperature to favor the desired form: 25°C for mold or 37°C for yeast.
The molecular basis of the shift from the differentiated mul-ticellular form to the undifferentiated unicellular form is un-known. Investigators have searched for genes upregulated in the pathogenic yeast form to identify genes required for di-morphism and virulence, and several interesting genes have been identified in these studies, as summarized in the recent review by Retallack and Woods (13). Keath and Abidi (4), for
example, reported that theyps-3 gene is specific to the yeast
form and is expressed at higher levels in the most virulent strains. Sebghati et al. (16) demonstrated that the calcium
binding protein geneCBP1is required for survival within host
phagocytic cells. Neither gene is required for dimorphism,
however, since strains that do not express yps-3 and cbp1
knockout mutants still exhibit apparently normal dimorphism. Although the search for yeast-specific genes as regulators of
dimorphism is quite logical, it is possible that genes critical for the dimorphic shift are being overlooked due to the assump-tion that yeast-specific or yeast-upregulated genes are the most important regulators. It may be necessary, for example, to downregulate certain mold-specific genes in order to form the in vivo yeast morphotype. One can envision a scenario in which mold-specific (MS) genes would exert an overriding effect, i.e., expression of a mold-specific gene(s) would shift growth to the mold form and a shutdown of this gene(s) would be required to allow the shift to the undifferentiated yeast form. We have begun to examine this hypothesis by isolating a number of genes which are silent in the yeast morphotype (18). Here, we report the first misexpression and knockout analysis of a
mold-specific gene,MS8, inH. capsulatumand show thatms8
knock-out mutants form aberrant mycelia.
MATERIALS AND METHODS
Strains and growth conditions. (i) Yeast strains.TheH. capsulatumstrains G186AS and G184AS (7) were used in these studies. Both strains represent restriction fragment length polymorphism class 3 isolates (5) and exhibit the same expression profile ofMS8. A uracil auxotroph of G184AS, G184ASura5
-11, the kind gift of Jon Woods (University of Wisconsin, Madison), was used for knockout experiments. Routine cultures were grown in GYE medium (2% [wt/ vol] glucose, 1% [wt/vol] yeast extract) at 37°C with 150-rpm gyratory shaking for yeast or at 25°C with 100-rpm gyratory shaking for mold. Cultures for transfor-mation or knockout analysis were grown in HMM, a rich synthetic medium based on F-12 tissue culture medium supplemented with cysteine and glutamic acid and buffered to pH 7.5 with HEPES (22). HMM plates were made by adding 1% (wt/vol) agarose (A-5093; Sigma). To reduce the chance of bacterial contamina-tion during the prolonged incubacontamina-tions, 50g of ampicillin/ml and 100g of streptomycin/ml were added to the HMM. The following additions to HMM were made when necessary: HMM/u, 50g of uracil/ml; HMM/h, 200g of hygromycin/ml; and HMM/f, 1 mg of 5-fluoroorotic acid/ml.
Yeast cultures for DNA or RNA extraction were grown to mid-log phase and * Corresponding author. Mailing address: Department of Biological
Sciences, Center for Molecular & Cellular Biosciences, University of Southern Mississippi, Hattiesburg, MS 39406-5018. Phone: (601) 266-4722. Fax: (208) 379-0426. E-mail: [email protected].
249
on September 8, 2020 by guest
http://ec.asm.org/
used for routine plasmid preparations. Telomere plasmids prepared in other strains occasionally exhibit sequence rearrangements, presumably because of the telomeric repeats.
Preparation of DNA.Plasmids fromE. coliwere isolated by alkaline lysis and Celite chromatography with the Wizard kit (Promega, Madison, Wis.) according to the manufacturer’s directions. Purification of DNA from agarose gels or after enzyme reactions was done by silica adsorption with the Zymoclean kit (Zymo Research, Orange, Calif.) according to the manufacturer’s directions.
Large-scale genomic DNA preparations fromHistoplasmawere done as fol-lows. Approximately 35 ml of mid-log-phase yeast was harvested by centrifuga-tion at 2,000⫻gfor 5 min, resuspended in 35 ml of ice-cold water, and collected by centrifugation for 5 min. The cell pellet was resuspended in 0.6 ml of DNA extraction buffer (100 mM Tris [pH 8.0], 100 mM EDTA, 250 mM NaCl). Phenol-chloroform (5:1; pH 7.9) (0.6 ml) and 0.8 ml of 0.45-mm-diameter glass beads were added, and the tubes were vortexed on a Vortex Genie 2 (Fisher Scientific) at maximum speed for 1 min. The tubes were cooled on ice for 1 min, followed by vortexing for 1 min. The cycles of vortexing and cooling were repeated for a total vortex time of 5 min. The cell debris and beads were pelleted by centrifugation at 11,000⫻gfor 10 min. The supernatant was removed to a new tube, and 30l of a 5-mg/ml RNase A solution was added. The tubes were incubated at 37°C for 60 min to digest the RNA. The mixture was extracted with an equal volume of phenol-chloroform (5:1; pH 7.9), and the aqueous phase was removed to a new tube. DNA was precipitated from the aqueous phase by the addition of 2 volumes of ethanol followed by centrifugation at 11,000⫻gfor 10 min. The DNA pellet was dissolved in 10 mM Tris (pH 7.8)–1 mM EDTA, and the concentration was determined by absorbance at 260 nm. The DNA typically migrated as a single large band slightly larger than a 23-kb lambdaHindIII marker when analyzed by agarose gel electrophoresis.
DNA miniprep for PCR testing of putative knockout colonies was as follows. A single colony was scraped from an HMM plate with a sterile toothpick and resuspended in 50l of H2O in a 500-l microcentrifuge tube. DNA extraction buffer (100l), 0.45-mm-diameter glass beads (50l), and 100l of phenol-chloroform (5:1; pH 8.0) were added, and the tube was vortexed for 3 min. The tube was centrifuged at 10,000⫻gfor 5 min, and the aqueous phase was recovered. RNase A was added to the aqueous phase to a final concentration of 100g/ml, and the tube was incubated at room temperature for 10 min. The DNA was then ethanol precipitated as described above and dissolved in sterile water.
Preparation of RNA.RNA was isolated by extraction with acidic phenol, which partitions RNA in the aqueous phase and DNA and protein in the lower phases (8). Mid-log-phase yeast cells were collected by centrifugation at 400⫻gfor 5 min. Actively growing mold was vacuum filtered onto 0.45-m-pore-size nitro-cellulose membranes and washed with ice-cold sterile water. The mycelium was scraped from the filter with a plastic spatula. Yeast or mold cells were resus-pended in 0.4 ml of ice-cold RNA extraction buffer (0.1 M sodium acetate [pH 5.0], 0.2 M NaCl, 0.2% [wt/vol] sodium dodecyl sulfate [SDS]) in a 1.5-ml screw cap microcentrifuge tube. An equal volume of acidic phenol-chloroform (5:1; equilibrated with RNA extraction buffer) and 300l of 0.45-mm-diameter glass beads were added. The tubes were vortexed at maximum speed for 10 min at room temperature and centrifuged for 3 min at 10,000⫻g, and the aqueous phase was recovered to a new tube. RNA was precipitated from the aqueous phase by the addition of 2 volumes of ethanol followed by centrifugation at 10,000⫻gfor 10 min. The RNA pellet was washed once with 75% ethanol and dissolved in 10 mM Tris (pH 7.5)–1 mM EDTA–0.2% (wt/vol) SDS by heating the solution at 65°C for 10 min. Any insoluble material was removed by centrif-ugation at 10,000⫻gfor 10 min, and the RNA in the supernatant was quanti-tated by absorbance at 260 nm.
Southern and Northern blotting.For Southern blotting, approximately 1g of
H. capsulatumgenomic DNA was digested with restriction enzymes (New En-gland Biolabs) and electrophoresed on a 0.7% (wt/vol) agarose gel. The DNA was transferred onto charged nylon membranes (Hybond N⫹; Amersham Corp.) by downward blotting with 0.4 N NaOH using a Turboblotter (Schleicher and Schuell, Keene, N.H.). The membrane was neutralized with several washes of 2⫻
SSC (1⫻SSC is 0.15 M NaCl–0.015 M sodium citrate, pH 7.0) and allowed to air dry. The membranes were prehybridized in 0.5 M sodium phosphate (pH
standard methods (9). RNA was transferred onto charged nylon membranes (Hybond N⫹) with 20⫻SSC by using a Turboblotter. After the transfer, the membrane was soaked in 50 mM NaOH for 4 min and neutralized with 2⫻SSC. RNA was cross-linked to the membrane by exposure to 254-nm UV light for a dosage of 120 mJ/cm2. Probe labeling, prehybridization, hybridization, and washes were done as described for Southern blots.
MS8genomic walking and sequencing.TheMS8gene and flanking sequence were isolated by genomic-walking PCR (3). Forward and reverse primers specific for the previously clonedMS8cDNA (GenBank accession no. AF292398) were used to isolate approximately 2 kb of upstream and 1.6 kb of downstream sequence. Primers were then designed to isolate the entire genomic DNA for
MS8via PCR. DNA was sequenced by the dideoxy chain termination method. To reduce sequence errors from PCR, a proofreading polymerase (Advantage 2; Clontech) was used and three independent PCR products were sequenced.
Plasmid construction.AnH. capsulatumtelomere vector was made based on the successful plasmid constructs of Woods and Goldman (20). A DNA construct containing (in 5⬘-3⬘ order) a NotI site, 12H. capsulatum telomeric repeats (GGGTTA), aPmeI site, a 1.3-kbHindIII-StyI tetracycline resistance marker from pBR322, aPmeI site, 12H. capsulatumtelomeric repeats (TAACCC), and aNotI site was cloned into theNotI site of pGem11 Zf⫹(Promega) to generate pG11TelTet. A 1.7-kb EcoRI fragment containing the Pa Ura5 gene from pWU55 (21) was cloned into theEcoRI site of pG11TelTet to generate pXTU1. A 1.0-kb fragment ofH. capsulatumgenomic DNA (representing approximately 950 nucleotides [nt] of 5⬘untranscribed flanking sequence and 50 nt of the 5⬘
untranslated region of theH. capsulatumtranslation elongation factor 1 [TEF1] gene) containing the strong constitutiveH. capsulatum TEF1 promoter was cloned into theXhoI site of pXTU1 to generate pXTU2. ApaI linkers were ligated to theMS8cDNA (GenBank accession no. AF292398), which was then inserted into theApaI site of pXTU2 immediately downstream of theTEF1
promoter to generate pXTU3. Several clones were isolated and analyzed by restriction cutting to select the sense orientation relative theTEF1promoter.
AnMS8knockout vector was constructed as follows. A 4.3-kbH. capsulatum
genomic DNA PCR product containing theMS8gene and 2 kb of 5⬘flanking sequence plus 1.5 kb of 3⬘flanking sequence was cloned into pGemTeasy (Pro-mega) to generate pMS8g. A 760-ntBclI/BstBI fragment in pMS8g representing most of the MS8coding sequence was replaced with a 2.9-kb BamHI/ClaI hygromycin resistance marker from pMAD93 (21) to generate pMS8g::hph. pXTU3 was cut withSmaI andApaI to remove theTEF1promoter andMS8
cDNA. The remaining vector was blunted with Klenow and ligated toNotI adapters. A 6.4-kbNotI fragment containing the knockout construct was isolated from pMS8g::hph and ligated to the resulting vector to form the knockout vector pXTU4⌬. The vector pXTU3g was constructed forms8knockout complemen-tation experiments as follows. pXTU3 was cut withSmaI andApaI to remove the
TEF1promoter andMS8cDNA. The remaining vector was blunted with Klenow and ligated toNotI adaptors. The 4.3-kbMS8genomic fragment was cut from pMS8g withNotI and ligated to the remaining pXTU3 vector to generate pXTU3g.
Histoplasmatransformation.Plasmids were digested withPmeI to remove the Tetrmarker and expose the telomeric repeats. The resulting linear vector was purified by agarose gel electrophoresis and recovered from the gel using a silica gel column (Zymoclean).H. capsulatumyeast was transformed by electropora-tion by a modificaelectropora-tion of the method of Woods et al. (21). Briefly, 5 ml of mid-log-phase yeast cells from an HMM broth culture was centrifuged at room temperature at 300⫻gfor 5 min to harvest the cells. The cells were washed in 5 ml of 10% (wt/vol) mannitol and collected by centrifugation. The cell pellet was resuspended in 350l of 10% (wt/vol) mannitol, and 0.5g of linearized vector (contained in less than 10l of sterile water) was added and mixed. The cell-DNA mixture was transferred to a 0.2-cm electroporation cuvette (Bio-Rad). Electroporation was done in an ElectroPorator (Invitrogen) at the following settings: 750V; 71F; 150⍀(which yields a pulse of 3,750 V/cm for 10.6 ms). For uracil selection, the cells were immediately plated on HMM plates. For hygro-mycin selection, the cells were mixed with 0.8 ml of HMM and incubated on a roller drum at 37°C for 12 h to allow expression of thehphmarker, followed by spreading the mixture on HMM/h plates. The plates were incubated in a humid-ified chamber at 37°C. Small colonies were typically visible in about 7 days.
on September 8, 2020 by guest
http://ec.asm.org/
Construction of knockout mutants.Genomic replacement of the nativeMS8
locus with thehph-disruptedMS8construct was accomplished by a modification of the positive-negative selection method of Sebghati et al. (16). G184ASura5-11
yeast was transformed with linearized pXTU4⌬. The cells were spread on HMM and incubated for 7 to 10 days at 37°C. Ten colonies were picked and inoculated into 1 ml of HMM broth in 10 disposable 15-ml plastic screw cap centrifuge tubes. A small hole was made in the cap of each tube with a 26-gauge needle, and the tubes were incubated on a roller drum at 37°C for 1 week to allow time for the double-crossover event to occur. Positive-negative selection was imposed by spreading 200l from each tube onto HMM/uhf plates to select for the hygro-mycin marker and against theUra5marker. The plates were incubated at 37°C for 7 to 10 days, and 10 colonies were selected for miniprep DNA extraction and analysis by PCR to identify knockout mutants.
RESULTS
Sequence analysis of MS8. The complete nucleotide
se-quence of theMS8gene is shown in Fig. 1. The gene is
inter-rupted by a single intron of 95 nt with canonical GT/AG splice junction sequences and a putative splice box (GCTAAC), which matches the filamentous-fungus consensus sequence
RCTRAC (2, 19), starting 27 nt upstream of the 3⬘terminus of
the intron. The predicted protein is 203 residues long with a mass of 21,332 Da and an estimated pI of 6.76. Sequencing of several cDNA clones identified two poly(A) sites: one site at nt 1360, 20 nt downstream of a putative poly(A) signal
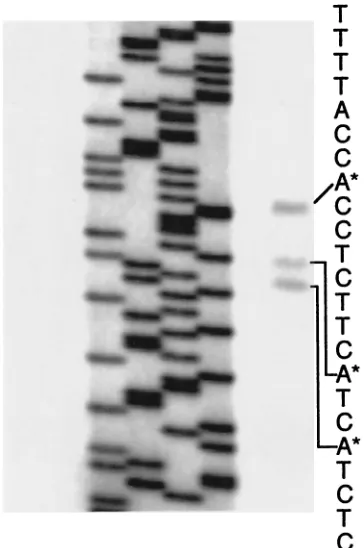
(AATAAA), and a second poly(A) site 42 nt upstream of the first site. Primer extension experiments (Fig 2) identified three transcriptional start points, each starting at the A in a CAY
motif, located at⫹1,⫹9, and⫹12. A TATA box (TATATAA)
was found starting 44 nt upstream of the first transcriptional start site.
Expression ofMS8during the Y-M shift.TheMS8transcript
is very abundant in the mold morphotype but is not detectable
on Northern blots with RNA from the yeast growth form ofH.
capsulatum(18). To determine the time course of induction, yeast cells growing at 37°C were downshifted to 25°C, which results in a switch to the mold growth form. As shown in Fig. 3, weak expression was first detected 11 h after the tempera-ture shift, corresponding to the first emergence of germ tubes. Expression then increased dramatically, particularly at day 3, corresponding to branch formation in the growing germ tubes.
MS8transcript levels remained high in stable mold cultures. In
several lanes, some signal was present in a slightly higher molecular weight band, which we believe represents a longer transcript from the secondary poly(A) site.
MS8 expression in yeast results in clumping and altered
colony texture.To test the hypothesis that expression ofMS8in
yeast might shift the morphology to become more mold-like at
FIG. 1. Sequence of theMS8gene (GenBank accession no. AY049031). Nucleotide numbers are shown on the right, and amino acid numbers are shown in parentheses. The first transcriptional start site is designated⫹1 and underlined. Two additional start sites were found at⫹9 and⫹12. A putative TATA box (underlined) was found 44 nt upstream of the⫹1 site. The single intron is shown in lowercase letters and includes a putative splice box (underlined). Two poly(A) sites were found at nt 1318 (single underline) and 1360 (double underline). A putative poly(A) signal was found starting at nt 1335 (double underline). Asterisk, stop codon.
on September 8, 2020 by guest
http://ec.asm.org/
37°C, we forced yeast cells to express the MS8 transcript at
levels similar to that seen in mold cultures. TheH. capsulatum
telomere vector pXTU3 (Fig. 4) was constructed with theMS8
cDNA (GenBank accession no. AF292398), including a 30-nt
poly(A) tail, fused to the strong constitutive H. capsulatum
TEF1promoter in the sense orientation. G184ASura5-11cells
were transformed with the linearized plasmid, and transfor-mants were identified by uracil selection on HMM plates. Six transformants were selected at random, and the expression
level ofMS8was determined by Northern blotting, as shown in
Fig. 5. Transformants S1, S4, and S6 had levels similar to that seen in mold cells, while S2, S3, and S5 showed less expression than in mold. All six transformants, maintained by uracil se-lection, exhibited normal yeast morphology as determined by
FIG. 2. Primer extension analysis of theMS8gene. The four lanes on the left show DNA sequencing reactions used as size markers. The rightmost lane shows a primer extension reaction, with the correspond-ing sequence of theMS8gene.
FIG. 3. Expression analysis ofMS8during the Y-M shift. Total RNA was extracted from cells shifted from 37 to 25°C from 11 h (hr) to 11 days (d) as indicated and electrophoresed on a denaturing agarose gel (20g/lane). The blot was probed withMS8cDNA (top), stripped, and probed with aHistoplasma18S ribosomal DNA probe (bottom) as a control. The rightmost lane (M) shows RNA extracted from fully developed mold. FIG. 4. Plasmid for forced expression ofMS8in yeast.MS8cDNA was fused to the strong constitutiveHistoplasma TEF1promoter. The plasmid was cut withPmeI (to remove the Tet marker and expose the telomeric repeats), and cells were transformed with the linear vector. A,ApaI; E,EcoRI; P,PmeI; X,XhoI; Tel, telomeric repeats.
on September 8, 2020 by guest
http://ec.asm.org/
microscopic examination. In HMM broth culture, however,
yeast expressingMS8, but not yeast transformed with the
vec-tor lackingMS8 sequences, exhibited pronounced clumping.
Yeast colonies expressingMS8on HMM plates had no obvious
alteration in size or color but were quite sticky, i.e., when touched with an inoculating loop, the cells tended to stick to the loop like very thick honey. These transformants formed apparently normal mold after the growth temperature was shifted to 25°C. No detectable change in the Y-M transforma-tion or in the microscopic morphology of the mold was noted.
Positive-negative selection of knockout mutants.To test the
effect ofMS8loss of function on H. capsulatum growth and
dimorphism, knockout mutants were constructed by a modifi-cation of the positive-negative selection method of Sebghati et
al. (16).H. capsulatumG184ASura5-11yeast (which shows the
sameMS8 expression pattern as G186AS [data not shown])
was transformed with a construct containing theMS8gene with
a large section of the coding sequence replaced by a
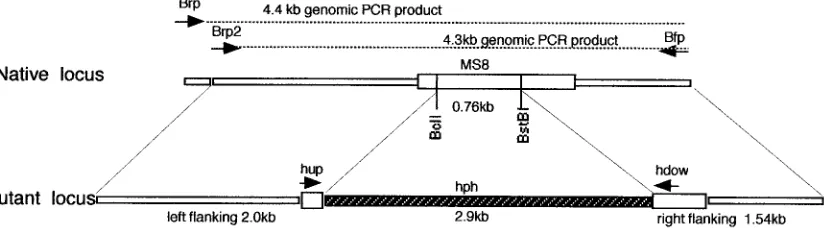
hygromy-cin marker (hph), as shown in Fig. 6. The knockout construct
contained 2 kb of 5⬘flanking sequence and 1.5 kb of 3⬘flanking
sequence relative to the open reading frame. Approximately
0.8 kb of theMS8coding region was replaced with the 2.9-kb
hph marker. After 1 week of uracil selection followed by 1
week of positive-negative selection on 5-fluoroorotic acid–hy-gromycin plates, 10 colonies were randomly selected and
ex-amined for MS8 allelic replacement. Analysis by PCR and
Southern blotting showed that 3 of the 10 colonies werems8
knockout mutants. Figure 7 shows PCR analysis of two knock-out mutants. Analysis with two sets of primers (set 1, Brp plus Bfp; set 2, Brp2 plus Bfp) showed that the knockout mutants yielded a band 2.1 kb larger than that of the parental strain,
consistent with replacement of the single-copyMS8gene with
the knockout construct (i.e., replacement of 0.8 kb of MS8
sequence with the 2.9-kbhphmarker). To confirm the loss of
the native locus, DNA from the parental strain and two
knock-out mutants was digested withBamHI and examined by
South-ern blotting, as shown in Fig. 8. A radiolabeled probe
repre-senting the 0.8 kb ofMS8sequence deleted from the knockout
construct hybridized to a 5.4-kb band only in the parental
strain. In contrast, anhphprobe hybridized to a 7.6-kb band
only in the knockout mutants, as predicted for allelic
replace-ment ofMS8.
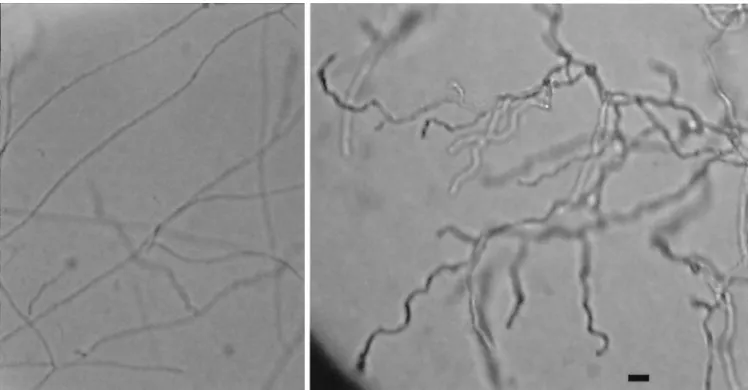
ms8knockout mutants form aberrant mycelia.ms8
knock-out mutants shifted to 25°C showed no significant phenotypic changes early in the Y-M transformation. Germ tube emer-gence and elongation occurred in both the parental strain and knockout mutants at approximately the same time and rate. After several days of growth on HMM/u plates, however, the
mycelia of thems8knockouts were substantially different from
those of the parental strain, as shown in Fig. 9. Colonies of the knockout mutants were less than half the diameter of normal colonies, had a brownish granular appearance, and produced a deep-red pigment particularly remarkable upon viewing the back of the agar plate. Microscopic examination showed that the hyphae of knockout mutants were nearly 40% wider (0.32
versus 0.23m) than normal, as shown in Fig. 10. Whereas
hyphae in the parental strain generally gave rise to long, straight filaments, those in the knockout mutants gave rise to much shorter branches, with a dramatic “zig-zag” twisted ap-pearance.
To ensure that the aberrant mycelial morphology of thems8
knockouts was the result of loss of function ofMS8, we
trans-FIG. 5. Forced expression ofMS8in yeast. Yeast was transformed with the linearized pXTU3 vector and selected on HMM plates. Total RNA was isolated and analyzed by Northern blotting. Each lane contained 20g of RNA. The image on the left shows RNA from normal yeast and mold. The image on the right shows RNA from six independent transformants (S1 to S6). The blot was probed with radiolabeledMS8cDNA.
FIG. 6. Map ofMS8knockout vector. A map of the native locus (TheMS8open reading frame is indicated by the large open box) is shown at the top, and the knockout construct is at the bottom. PCR primer sites are indicated by arrows. Approximately 0.8 kb ofMS8coding sequence was replaced with the 2.9-kb hygromycin marker (hph). Approximately 2 kb of 5⬘flanking sequence and 1.5 kb of 3⬘flanking sequence were included. This linear construct was used to replace theTEF1-MS8sequence (XhoI toApaI) in pXTU3 to form the knockout vector pXTU4⌬.
on September 8, 2020 by guest
http://ec.asm.org/
formed the knockout mutants with pXTU3 (MS8cDNA driven
by theTEF1promoter) and pXTU3g (anMS8genomic
frag-ment with the native promoter) to complefrag-ment thems8
mu-tation. Knockout mutants were transformed with pXTU3 and pXTU3g and plated on HMM plates to maintain the plasmids by uracil selection. Colonies grown at 25°C demonstrated
nor-mal colony and microscopic morphologies, as shown for the parental strain in the left-hand images of Fig. 9 and 10. In
contrast, ms8 knockout mutants transformed with pXTU1
(which has theUra5marker but noMS8sequence) still
exhib-ited the aberrant morphology, as shown for thems8knockouts
in the right-hand images of Fig. 9 and 10. Thus, the aberrant phenotype was not incidental to the experimental protocol but
due toMS8loss of function.
DISCUSSION
The most obvious environmental cue for the M-Y shift after spores or mold fragments are inhaled is the upshift to 37°C. Laboratory cultures shifted from 25 to 37°C mimic this process
in vitro, and nearly all studies ofH. capsulatumdimorphism in
the literature have used a simple temperature shift to favor mold (25°C) or yeast (37°C). Although the temperature shift is
clearly an important dimorphism signal inH. capsulatum, it is
not necessary or sufficient for the morphological transition. Under certain conditions, yeast can be grown at the “nonper-missive” temperature of 25°C (10) and mold can be grown at 37°C (10, 14, 15). Presumably, the temperature shift is the natural trigger for induction of a gene regulation cascade which results in the dramatic differentiation process from a multicellular form to a unicellular form or vice versa. Clearly, the dimorphic shift is associated with dramatic changes in gene regulation, as demonstrated by the numerous differentially reg-ulated genes found in subtracted-library and differential-dis-play experiments in our laboratory and others (6, 13, 18).
TheMS8gene was interrupted by a single intron with typical
GT-5⬘and AG-3⬘termini (Fig. 1). A putative splice box
match-ing the filamentous-fungus consensus RCTRAC (GCTAAC)
was found starting 27 nt upstream of the 3⬘end of the intron.
Comparison of several cDNA clones revealed thatMS8had
two poly(A) sites, as do many of theH. capsulatumgenes we
FIG. 7. PCR analysis of knockout mutants. Lanes A, DNA from parental strain G184ASura5-11;lanes B and C, DNA from putative knockout mutants. Primer set 1, Brp and Bfp; primer set 2, Brp2 and Bfp. Replacement of the native locus should yield a band 2.1 kb larger than that in the parental strain.
FIG. 8. Southern blot analysis of knockout mutants. DNA was ex-tracted from the G184ASura5-11parental strain (lanes A) and two putative knockout mutants (lanes B and C), cut withBamHI, separated on a 0.7% gel, and blotted onto a nylon membrane. The left-hand image shows the blot hybridized with a radiolabeled probe represent-ing the 0.8 kb ofMS8sequence removed in the knockout vector. The right-hand image shows the same blot stripped and probed with a radiolabeledhphmarker.
FIG. 9. Colony morphology of G184AS ura5-11 parental strain (left) andms8knockout mutant (right). The colonies are shown on HMM/u agar after 6 weeks of growth at 25°C. The photographs were taken from the same plate and with the same lighting conditions and magnification. Bar, 5 mm.
on September 8, 2020 by guest
http://ec.asm.org/
have isolated. The most distal poly(A) site was preceded by a canonical poly(A) signal sequence, AATAAA, starting 26 nt upstream. The second poly(A) site results in a transcript 42 nt shorter and is not preceded by an obvious poly(A) signal se-quence. The sequence surrounding the putative translational start site (boldface and underlined), TCATCATGTC, showed high similarity to the Kozak sequence [TCA(C/A)(A/C)ATG (G/T)C] seen in filamentous fungi (1). Primer extension exper-iments showed three closely spaced transcriptional start sites of similar strengths 228, 220, and 217 nt upstream of the transla-tional start site. Each transcriptransla-tional start was the A in a CAY motif. A consensus TATA box (TATATAA) was located start-ing 44 nt upstream of the first transcriptional start site. The
predictedMS8protein was 203 amino acids long with an
ex-pected mass of 21.3 kDa and a pI of 6.77. Particularly striking
was the abundance of two amino acids in the putative MS8
protein. Over 40% of MS8p was composed of only two amino acids, glycine (22%) and glutamine (19%). GenBank similarity searches revealed only one significant match in the database: a
glycine-rich protein from the plant-pathogenic fungus
Colleto-trichum gloeosporioides(accession no. U94186; BLAST
simi-larity, 10⫺21). TheC. gloeosporioidesprotein is similar to MS8p
in mass (22 kDa), G and Q content (24 and 19%, respectively), and cysteine content (0%) but is significantly more basic (pI, 9.18) than MS8p. Unfortunately, the function of this protein in
Colletotrichumis also unknown.
Expression of MS8 is not detected on Northern blots of
RNA from the yeast morphotype but is quite abundant in mold cells (18). During the 25°C Y-M transition, expression was first detected at 11 h (Fig. 3), corresponding to the emergence of germ tubes from the yeast. As the germ tubes elongated, the
level of theMS8transcript increased dramatically, particularly
on day 3, which corresponded to hyphal branch formation in
most of the cells.MS8transcript levels remained high in
ac-tively growing mold cultures.
H. capsulatum has inefficient homologous recombination, and only very recently has allelic replacement become feasible inHistoplasma. Our knockout experiments with a slight
mod-ification of the positive-negative selection methodology of Seb-ghati et al. (16) showed that nearly one-third of the colonies on
the final selection plates werems8knockouts. This is similar to
the frequency reported for their knockout of the calcium
bind-ing protein gene (CBP1). This positive-negative selection
method now makes it possible to isolateH. capsulatum
knock-out mutants in less than a month.
IfMS8was essential for formation of mold cells, we would
expectms8knockout mutants to be unable to form mycelia.
Clearly,MS8is not an absolute requirement for mold
forma-tion, since the knockout mutants did form hyphae (albeit of
aberrant shape and size). The presence of a functionalMS8
gene is also not required for the shift back to the yeast
mor-photype, since ms8 knockout mutants growing as mold can
shift to apparently normal yeast at 37°C (not shown). The
identity of the red pigment produced by thems8mutants is not
known. Red-pigmented variants ofH. capsulatum have been
reported, one isolated from a canebrake (12) and one from biopsy material from a human immunodeficiency virus-positive patient (17). The identity of the pigment produced by these variants is also not known. Although it is possible that the
pigments produced by the variants and thems8knockout
mu-tants are related, we have no data at this time to allow a conclusion.
Clearly, it is not essential thatMS8be silent to maintain the
yeast morphotype, since yeast forced to express high levels of
theMS8transcript (Fig. 5) still grew as yeast at 37°C.MS8is
required, however, for normal hypha formation.ms8knockout
mutants formed aberrant short, twisted hyphae and gave rise to small, pigmented, slow-growing mold colonies (Fig. 9). The function of MS8p within the cell is unknown. Examination of the putative MS8p with various proteomics structural analysis programs (http://www.expasy.ch/tools) revealed primarily that the protein is expected to be quite flexible and hydrophilic, with two short regions of alpha helix in the C terminus. The lack of diagnostic structural features and the BLAST similarity match to a single protein whose function is also unknown make it impossible to postulate a particular function for this protein.
FIG. 10. Microscopic morphology of G184ASura5-11parental strain (left) andms8knockout (right) stained with lactophenol cotton blue and photographed at the same magnification. Bar, 2m.
on September 8, 2020 by guest
http://ec.asm.org/
future work, we plan to examine the cellular localization of
MS8p withGFP-MS8fusions to help elucidate the role of this
protein in mold formation.
ACKNOWLEDGMENT
This work was supported in part by grant AI49357 from the National Institutes of Health.
REFERENCES
1.Ballance, D. J.1986. Sequences important for gene expression in filamentous fungi. Yeast2:229–236.
2.Gurr, S. J., S. E. Unkles, and J. R. Kinghorn.1987. The structure and organization of nuclear genes of filamentous fungi, p. 93–139.InJ. R. Kinghorn (ed.), Gene structure in eukaryotic microbes. IRL Press, Oxford, United Kingdom.
3.Jones, D. H., and S. C. Winistorfer.1993. Genome walking with 2- to 4-kb steps using panhandle PCR. PCR Methods Appl.2:197–203.
4.Keath, E. J., and F. E. Abidi.1994. Molecular cloning and sequence analysis ofyps-3, a yeast-phase-specific gene in the dimorphic fungal pathogen His-toplasma capsulatum. Microbiology140:759–767.
5.Keath, E. J., G. S. Kobayashi, and G. Medoff.1992. Typing ofHistoplasma capsulatumby restriction fragment length polymorphisms in a nuclear gene. J. Clin. Microbiol.30:2104–2107.
6.Keath, E. J., A. A. Painter, G. S. Kobayashi, and G. Medoff.1989. Variable expression of a yeast-phase-specific gene inHistoplasma capsulatumstrains differing in thermotolerance and virulence. Infect. Immun.57:1384–1390. 7.Klimpel, K. R., and W. E. Goldman.1987. Isolation and characterization of
spontaneous avirulent variants ofHistoplasma capsulatum. Infect. Immun.
55:528–533.
8.Majumdar, D., Y. J. Avissar, and J. H. Wyche.1991. Simultaneous and rapid
yashi, and L. Carratu.1986. Irreversible block of the mycelial-to-yeast phase transition ofHistoplasma capsulatum. Science231:476–479.
12.Morris, P. R., A. A. Terreni, and A. F. DiSalvo.1986. Red-pigmented His-toplasma capsulatum—an unusual variant. J. Med. Vet. Mycol.24:231–233. 13.Retallack, D. M., and J. P. Woods.1999. Molecular epidemiology, patho-genesis, and genetics of the dimorphic fungusHistoplasma capsulatum. Mi-crobes Infect.1:817–825.
14.Rippon, J. W.1968. Monitored environment system to control cell growth, morphology, and metabolic rate in fungi by oxidation-reduction potentials. Appl. Microbiol.16:114–121.
15.Sacco, M., B. Maresca, B. V. Kumar, G. S. Kobayashi, and G. Medoff.1981. Temperature- and cyclic nucleotide-induced phase transitions of His-toplasma capsulatum. J. Bacteriol.146:117–120.
16.Sebghati, T. S., J. T. Engle, and W. E. Goldman.2000. Intracellular para-sitism byHistoplasma capsulatum: fungal virulence and calcium dependence. Science290:1368–1372.
17.Staib, F., and G. Grosse.1996. Brown-red pigment formation by the mycelial phase of a clinical isolate ofHistoplasma capsulatumon Staib agar. A pre-liminary report. Zentbl. Bakteriol.283:515–521.
18.Tian, X., and G. Shearer.2001. Cloning and analysis of mold-specific genes in the dimorphic fungusHistoplasma capsulatum. Gene275:107–114. 19.Unkles, S. E.1992. Gene organization in industrial filamentous fungi, p.
29–53.InJ. R. Kinghorn and G. Turner (ed.), Applied molecular genetics of filamentous fungi. Blackie, Glasgow, United Kingdom.
20.Woods, J. P., and W. E. Goldman.1993. Autonomous replication of foreign DNA inHistoplasma capsulatum: role of native telomeric sequences. J. Bacteriol.175:636–641.
21.Woods, J. P., E. L. Heinecke, and W. E. Goldman.1998. Electrotransforma-tion and expression of bacterial genes encoding hygromycin phosphotrans-ferase and beta-galactosidase in the pathogenic fungusHistoplasma capsu-latum. Infect. Immun.66:1697–1707.
22.Worsham, P. L., and W. E. Goldman.1988. Selection and characterization of
ura5mutants ofHistoplasma capsulatum. Mol. Gen. Genet.214:348–352.