Árpád Furka
Combinatorial Chemistry
Árpád Furka
Combinatorial Chemistry
Published by Árpád Furka in electronic form Budapest 2007
Preface
Combinatorial technologies that were invented in 1980s provided a possibility to produce new compounds in practically unlimited number. New strategies and technologies have also been developed that made possible to screen very large number of compounds and to identify useful components of mixtures containing millions of different substances. This dramatically changed the drug discovery process in the pharmaceutical industry and the way the researchers design their experiments. Instead of preparing and examining a single compound, families of new substances are synthesized and screened. In addition, combinatorial thinking and practice proved to be useful in areas outside the pharmaceutical research. Such area are, for example, search for more effective catalysts and materials research.
Combinatorial chemistry became an accepted new branch within chemistry. It is the subject of numerous books, journals, international conferences and university courses. This book is written for university students and young researchers. The author feels it important to make it freely available for all potential readers. For this reason the book will be published exclusively in electronic form that can be downloaded from appropriate Web sites free of charge.
The author wishes to express his appreciation to Dr. József L. Margitfalvi of the Central Chemical Research Institute of the Hungarian Academy of Sciences and Dr. György Kéri of Gedeon Richter Ltd, Budapest for reading parts of the manuscript and for their valuable suggestions.
The mother tong of the author is Hungarian. Despite all efforts the text obviously contains grammatical errors. Correction of these errors, of course, would be important. The help of the readers in this respect would be highly appreciated. If you can help please contact the author by e-mail: [email protected].
Table of Contents
Preface………... v
Table of Contents... vi
1. Introduction………....…... 1
1.1. Birth of the combinatorial approach... 2
1.2. The translated version of the document notarized in 1982... 5
1.3. Publication of the split-mix combinatorial synthesis... 13
References... 14
2. The solid phase synthesis... 15
2.1. Solid supports... 18
2.1.1. Crosslinked polystyrene... 18
2.1.2. Polyethylene glycol (PEG) grafted supports... 19
2.1.3. Inorganic ports... 20
2.1.4. Non-bead form supports... 20
2.2. Linkers, anchors... 20
2.3. Protecting groups... 22
2.3.1. Protection of amino groups... 23
2.3.2. Protection of carboxyl groups... 24
2.3.3. Protection of other functional groups... 24
2.3.4. Coupling reagents for peptide synthesis... 25
2.4. Solid phase synthesis of organic molecules... 26
2.5. Solid phase reagents and scavenger resins in solution phase synthesis... 27
References... 28
3. Parallel synthesis. Synthesis of compound arrays based on saving reaction time... 29
3.1. The parallel synthesis... 30
3.1.1. The multipin metod of Geysen... 31
3.1.2. The SPOT technique of Frank... 32
3.1.3. Other devices for parallel synthesis... 33
3.1.4. Parallel synthetic methods with reduced number of operations... 36
3.1.4.1. Synthesis of oligonucleotides on paper discs... 36
3.1.4.2. The tea-bag synthesis... 37
3.2. The Ugi multicomponent reactions... 37
3.3. Solution phase combinatorial synthesis... 39
3.3.1. Dendrimer supported synthesis... 39
3.3.2. Separations using fluorous tags and fluorous solvents... 40
3.3.3. Application of solid phase reagents... 40
3.3.4. The use of scavengers in solution phase reactions... 41
3.4.1. Automatic parallel synthesizers... 42
3.4.2. Quality control... 45
3.4.3. Parallel purification... 47
3.4.4. Manufacturers of laboratory robots... 48
References... 52
4. Combinatorial synthetic methods... 55
4.1. Combinatorial synthesis on bead-form resin... 55
4.1.1. The split-mix synthesis... 55
4.1.1.1. The key features of the split-mix synthesis... 56
4.1.1.2. Encoding of beads in the synthesis of organic libraries... 61
4.1.1.3. Realization of the split-mix synthesis... 64
4.1.1.4. Automation of the split-mix synthesis... 66
4.1.1.5. Preliminary considerations when planning experiments with peptide libraries... 71
4.1.1.6. Full and partial libraries... 75
4.1.1.7. Unusual partial libraries... 79
4.1.1.8. Binary synthesis using the split-mix procedure... 80
4.1.2. Combinatorial synthesis using amino acid mixtures... 82
4.2. Combinatorial synthesis using soluble support... 83
4.3. Combinatorial synthesis on solid surface... 84
4.4. Combinatorial peptide synthesis by biological methods... 86
4.5. Combinatorial synthesis using macroscopic solid support units... 87
4.5.1. Encoding by attached labels. The radiofrequency and optical encoding methods... 88
4.5.2. Units without labels. Encoding by position in space... 92
4.5.2.1. The Encore technique... 92
4.5.2.2. The String Synthesis... 93
4.5.2.3. String synthesis of cherry picked libraries... 104
4.6. Examples... 110
4.6.1. Split-Mix Synthesis of an encoded benzimidazole library... 110
4.6.2. Synthesis of a 10,000 member piperazine 2-carboxamide . library by Directed Sorting... 115
4.6.3. Synthesis of two libraries on one support... 117
References... 118
5. Screening methods... 121
5.1. High throughput screening of arrays of individual compounds... 122
5.2. Screening of combinatorial libraries. Deconvolution methods... 124
5.2.1.Deconvolution methods for dissolved libraries... 124
5.2.1.1. The iteration method... 124
5.2.1.2. Positional scanning... 129
5.2.1.4. The amino acid tester libraries... 135
5.2.1.5. Other methods for identification of the bioactive component of combinatorial libraries... 138
5.2.1.6. Dynamic combinatorial libraries... 138
5.2.1.7. Examples... 138
5.2.2. Deconvolution methods of libraries tethered to the solid support... 145
5.2.2.1. Screening of combinatorial libraries in tethered form... 146
5.2.2.2. Screening of combinatorial libraries by releasing the content of individual beads intosolution... 147
5.2.2.3. Examples... 148
References... 150
6. Combinatorial methods in materials and catalyst research... 151
6.1. Inorganic materials... 151
6.1.1. Preparation of thin film libraries... 151
6.1.2. Screening... 154
6.2. Heterogeneous catalysts... 155
6.2.1. Fabrication and testing of catalyst libraries... 156
6.2.2. Catalyst library design... 161
6.3. Polymers... 164
References... 168
7. Computational aspects of library design and synthesis... 171
7.1. Software companies... 176
References... 182
1. Introduction
The discovery of new materials played an important role in the history of mankind. Many discovered materials had effect on every day’s life. The impact of some of these materials was so definitive that they gave the name of long historical eras. So bronze gave the name for Bronze Age, for example, and iron for the Iron Age.
The life today is also largely affected by the materials we use. The standard of life could not be the same without semiconductors, insulators, adhesives, synthetic fibers, drugs, pesticides, paints etc. In order to improve our life, more and more useful materials and compounds need to be discovered. The question is how to do that? When we need a new bridge or want to build a skyscraper, for example, first these objects are designed then they are built according to the plans. Can we follow this route when we wish to make a new super conductor or a new drug? Certainly not. Our theoretical knowledge may be sufficient for designing a bridge or a skyscraper but is definitely not enough for designing a new more effective drug or designing a super conductor working at or near room temperature. We do not know exactly how the super conducting or other important properties of materials depend on their structure. The drugs exert their effects by interactions with proteins or other molecules found in living organisms. The rules governing these interactions, however, are largely unknown. The rational design of drugs had some successes. The drug candidates are designed in computers based on the already known three dimensional structures of target proteins. Both the ligand molecule and the protein itself can take up a practically unlimited number of conformations, and that leads to difficulties. The consequence is that mostly the traditional approach is followed: series of compounds are synthesized then the useful drug candidates are identified by trial and error. In practice, thousands of compounds are needed to be prepared and tested in order to find a drug candidate.
In the pharmaceutical research one of the bottlenecks was the synthesis of the very large number of compounds needed in the discovery process. Before 1980 the traditional approach was used. The compounds were prepared one at a time, and their testing were also carried out one by one. In the industry, however, sophisticated methods were developed and applied in order to improve productivity in the mass production of goods. It seems worthwhile to compare the production of compounds to that of automobiles. Compounds are mostly prepared step by step from the starting materials. The automobiles are also assembled from parts. The drug candidates are unique substances all differing from each other. The automobiles are also unique products since they can differ, for example, in their color, in their engines, in their transmission etc. They certainly differ from each other in their locks and keys.
The first car manufacturers in the world were Panhard & Levassor in 1889 and Peugeot in 18911. These French manufacturers did not standardize their car models, each car was different from the other. The first standardized car was the Benz Velo. Benz manufactured 134 identical Velos in 1895.
Ransome Eli Olds invented the basic concept of the assembly line in 1901 that was improved Henry Ford and installed it in his car factory in 1913. As a result, by 1927, 15 million Ford Model Ts had been manufactured. As a result of further improvements and application of
automation, today the streets are full of cars. This shows the power of organizing the process of production and application of automation. These methods that proved to be very successful in industry were not applied at all in the mass production of compounds.
After 1980 the situation began to change. Several innovative papers were published which radically changed our theory and practice in designing and preparing new substances for pharmaceutical research and other areas of application. The new synthetic and screening procedures and, which is also very important, the new way of thinking introduced in these papers founded a rapidly growing new scientific field, Combinatorial Chemistry, revolutionized the pharmaceutical research and are gradually expanding to other areas within and outside chemistry. The new methods were developed in several laboratories. The way of thinking that led to these methods was probably different in all cases. The reasons that lad to the development of the combinatorial synthesis of peptides in the author's laboratory is described below.
1.1. Birth of the combinatorial approach
In 1964/65 I was a post doctoral fellow at the University of Alberta, Canada. I worked on a project led by Professor L. B. Smillie which resulted in determination of the amino acid sequence of a pro-enzyme, chymotrypsinogen-B2. After returning to Budapest, I was wondering from how many sequence possibilities did we choose the right one. Since the protein comprised 245 amino acid residues and any of these positions could be occupied by any of the 20 different natural amino acids, the number of possible sequences, as expressed by a simple formula, amounted to 20245 (=5.65x10318) combinations. This certainly seemed to be a very big number, much-much bigger than, for example, the number of molecules in 1 mole substance (6.02x1023) but in order to really perceive its magnitude it had to be compared to something which was also very big. Finally I found an estimate of the mass of the universe based on an Einstein formula3. I also calculated the mass of a protein mixture in which each sequence variant of the 245 amino acids is represented by only a single molecule.
Estimated mass of the universe: ~1053 kg Mass of the protein mixture: ~10295 kg
The comparison showed that the mass of the protein mixture would exceed that of the universe by more than two hundred orders of magnitude. The number of possible protein sequences also seems striking if it is compared to the estimated number of elementary particles in the visible universe4.
Number of sequences in the mixture: 5.65x10318 Number of elementary particles: ~1088
This was my first meeting with the immense diversity of molecules and the result shocked me.
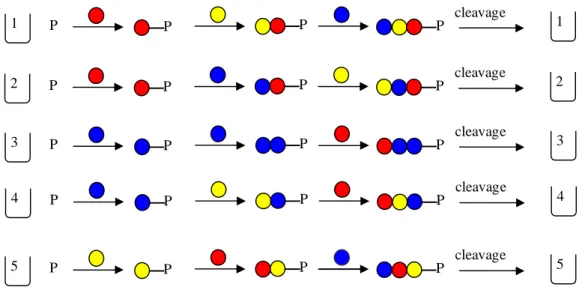
Many years later I applied the same simple formula (20n, where n is the number of amino acid residues) to calculate the number of theoretically possible sequences in peptide families built up from the 20 natural amino acids. Such collections of compounds are named libraries. The results are listed in the second column of Table 1.1.
Although the figures expressing the number of components in peptide libraries were far from being as frightening as the number of the possible protein sequences, they seemed still very large if the possibility of their synthesis was considered. I thought that many useful bioactive peptides could - supposedly - be found among the largely unknown components of the libraries. For this reason the nonexistent peptide libraries reminded me of exceptionally rich gold reefs which await exploitation. Gold can be produced by mining out all the gold containing rock then separating the gold from the useless stone.
Table 1.1. The number of possible peptide sequences. Number of residues Name Number of sequences 2 Dipeptides 400 3 Tripeptides 8,000 4 Tetrapeptides 160,000 5 Pentapeptides 3,200,000 6 Hexapeptides 64,000,000 7 Heptapeptides 1,280,000,000
Exploitation of the peptide libraries could be achieved via the synthesis of all possible sequences followed by screening them against all potential targets. At that time, however, even the synthesis of all, say, pentapeptides seemed absolutely impossible. We usually prepared one peptide at a time mostly by solid phase synthesis (see later the details of this synthetic method) with an elongation rate of one amino acid a day.
Figure 1.1. The optimized synthesis of peptides from three amino acids (A, E and R). The solid support is represented by .
With this rate, the synthesis of all the 3.2 million pentapeptides would have taken 3.2x5=16 million days, that is, 43.8 thousand years of uninterrupted work.
A A A R E E E R R R AA A E R A E R EA RA AE EE RE AR ER RR A E R A E R A E R
The synthesis could have been optimized by reducing to an absolute minimum the number of necessary coupling steps. This can be achieved by using the already prepared peptides as starting materials in the synthesis of the longer ones. This is illustrated in Figure 1.1. A peptide library is prepared by solid phase synthesis using three amino acids (A, E and R). First the amino acids are attached to the solid support (resin). Then the resin containing one of the attached amino acid is divided into three portions and the synthesis is continued with the coupling of one of the amino acids to one of the resin portions and so on. In the first step 3 couplings are carried out, exactly the number of the formed products. In the second step 9 couplings are needed and 9 dipeptides are formed on the resin samples. In general, the number of coupling steps in such an optimized synthesis of a peptide library is the same as the total number of products formed in the whole synthetic process. If the 3.2 million pentapeptides are prepared the number of coupling steps is the sum of amino acids + dipeptides + tripeptides + tetrapeptides + pentapeptides.
20 + 400 + 8,000 + 160,000 + 3,200,000 = 3,368,420
Supposing again the rate of one coupling per day in order to get the necessary time in years, the above figure is divided by 365. The result is 9,228 years. This shows that optimization of the synthesis reduces the time of the synthetic process from 43,800 years to 9,228 years, which is still too long to be realizable.
I considered the accessibility of all peptide sequences to be very important, and around 1980 I began to think about potential solutions for their synthesis. It took only a short time to find one, which would work at least in principle. The idea was also based on the method of solid phase synthesis developed by professor Merrifield5. According to this first idea, the amino acids used in the solid phase preparation of peptides would be replaced by an equimolar mixture of 20 different amino acids in every coupling step of the synthesis. This would lead - at least in principle - to formation of a rapidly growing number of sequences and finally a full peptide library could be cleaved from the support in the form of a mixture. It was clear, however, that in such couplings the products are expected to form in unequal molar quantities as a consequence of the differences in the reactivity of the amino acids. The differences in molarities would be amplified in each successive coupling step leading to a mixture with uncertain composition. I felt that a better solution might exist and I was rethinking the problem again and again. In early spring in 1982 I spent a weekend in a little town in South-East of Hungary forgetting this time the whole diversity problem. To my great surprise, however, next morning I awoke with the perfect solution in my mind. The method based on this idea is known nowadays as the split-mix procedure.
The split-mix method opened the possibility for producing peptide mixtures containing millions of components. Such mixtures, however seemed unacceptable in the conventional drug discovery practice where single compounds were used in pure form. For this reason there was an urgent need to present in addition, an efficient strategy for identification of the bioactive substance that may be present in the complex synthetic mixture. This task, however, looked similar to finding the proverbial needle in a huge haystack. Nevertheless I could develop a theoretical solution in a very short time. I called it “synthetic back searching strategy” which later proved to be in principle identical with the "iteration strategy", published by others.
I was fully aware of the importance of the combinatorial approach in the pharmaceutical research but one of the leading Hungarian pharmaceutical companies I contacted showed no interest at all. In addition, the split-mix method was considered by the patent attorneys only as a potential research tool and for this reason it was judged not to be patentable. They suggested me,
however, to describe the method in a document and - in order to give me some support in potential future priority disputes - notarize it. I did so and the document written in Hungarian - in which the principles of combinatorial chemistry including both synthesis and screening were first clearly explained - was notarized in May, 1982. The photo of the first and last pages of the document is demonstrated in Figure 1.2.
Figure 1.2. The photo of the first and last page of the 1982 document
The 1982 document, as shown in the Figure 1.2, was written in Hungarian. This is the first authentic document in which the principles of combinatorial chemistry are described. The translated version can be seen below.
1.2. The translated version of the document notarized in 1982
STUDY ON POSSIBILITIES OF SYSTEMATIC SEARCHING FOR PHARMACEUTICALLY USEFUL PEPTIDES
Written by Dr. Árpád Furka, university professor Budapest, May 29, 1982
As exemplified, among others, by the peptide hormones discovered so far, the shorter-lengthier peptides take part in a number of important functions in the living organism. It can be supposed, that only a small fraction is known of these biologically active peptides having
potential therapeutic effect. This fact motivates the intensive international and domestic research activity in this field.
Two, in principle different, approaches offer themselves for searching for peptides bearing new biological effects:
1. Isolation of peptides from living organisms based on their previously known biological effects.
2. Preparation of peptides by synthesis with post determination of their biological effects.
Until now the isolation procedure proved to be more effective in spite of the fact that this method is also very laborious. This may be explained by the fact that the number of possible peptides grows rapidly with the number of residues so even the synthesis of all tetrapeptides (160 thousands) seems to be a hopeless task. If we consider the 20 natural amino acids the dependence of the number (Nn) of possible peptides on the number of residues (n) is expressed by the
following formula:
Nn = 20n
If the n-residue peptides are synthesized stepwise and independently, the number of the required synthetic steps (Sn) can be calculated as follows:
Sn= (n-1) 20n
It is noted, that a synthetic step means a complete coupling cycle, that is, in addition to the coupling step itself incorporates the operations connected with the protecting groups, too. With good organization, that is, choosing a systematic synthesis route the number of synthetic steps can be reduced. The minimum number of synthetic steps is:
n i i nS
220
The synthesized peptides are supposed to be submitted to screening tests. Since several tests have to be done on each peptide, the total number of the required screening tests is hopelessly large. If the number of kinds of screening tests is denoted by t, the total number of screening tests is expressed by the following equation:
Tn = t 20 n
Table 1.2 shows the possible number of peptides depending on the number of residues, the number of synthetic steps required for their synthesis, and number of the screening tests, calculating with 10 different tests (t=10). The figures - which are rounded - clearly show, that even the synthesis and testing of all tripeptides would be an almost hopeless venture.
Because of the very large number of possible peptides, the stepwise synthesis of all peptides - even in the case of small ones - is an unrealizable task. The large number of the screening experiments constitutes a further problem. The proposal to be outlined on the next pages will try to somewhat improve this almost hopeless situation.
Table 1.2.
Possible number of peptides (Nn ) containing different number of residues (n),
the number of synthetic steps required for their synthesis (Sn) in an optimized
process, furthermore the number of screening experiments (Tn) calculating
with 10 different screening tests (t=10) (the figures are rounded)
n Nn Sn Tn
2 4 hundred 4 hundred 4 thousand 3 8 thousand 8 thousand 80 thousand 4 160 thousand 168 thousand 2 million 5 3 million 3 million 30 million 6 64 million 67 million 640 million 7 1 billion 1 billion 13 billion 8 25 billion 26 billion 256 billion 9 512 billion 537 billion 5 trillion 10 10 trillion 10 trillion 102 trillion
Systematic search for biologically active small peptides through synthesis and screening of peptide mixtures
The proposal to be outlined here constitutes a research project which makes possible to search for biologically active peptides with much greater chance than before. When I write down this project I'm fully aware of its potential importance in industry. It is also clear, that it's realization is possible only through cooperation of different institutions. Primarily the participation of the pharmaceutical industry is desirable since the investments can be recovered through pharmaceutical industry.
The essence of the proposal is that instead of one by one synthesis of peptides, peptide mixtures should be prepared containing several hundred or several thousand peptides in approximately 1 to 1 molar ratio, and these peptide mixtures should be submitted to screening tests. It will be shown that on this way much labor can be saved both in the synthetic work and in the screening experiments. In the first stage one has to determine whether or not the mixture shows any biological effect. If biological effect is observed, of course, it has to be determined which component (or which components) are responsible for the activity.
Method for synthesis of peptide mixtures
Since not single peptides but rather mixtures of peptides are synthesized, post synthetic purification and removal of by-products are out of question. Because of this, the classical method of synthesis (in solution) can not be used either. In the synthesis of peptide mixtures the solid phase method has to be applied. It is noted here, that in the syntheses not necessarily the 20 amino acids are used. In some cases more than 20 amino acids may be used, for example if - in addition - non-common amino acids are intended to be used as building blocks. Less than 20
amino acids may be used, for example, in decapeptides, since the synthesis of all peptides seems to be unrealistic and have to compromise with the use of fewer kinds of amino acids. Let denote by k the number of the amino acids intended to vary in the i-th position. The numbers of amino acids varied in the C-terminal and N-terminal position are k1 and kn, respectively.
Realization of the synthesis
The resin is divided into k1 equal portions (that is to as many portions as many amino
acids are intended to vary at the C-terminal of peptides). Then each portion of resin is coupled with one of the k1 kinds of amino acids then the amino-protecting group is removed from every
sample. A small quantity is removed from every sample and they are taken aside for later use, then the samples are thoroughly mixed. Then the mixture of aminoacyl resins is divided into
k2 equal portions and each of them is coupled with one of the k2 kinds of protected amino acids
then the amino-protecting groups are removed from each sample. Before mixing, again small samples are removed and taken aside. The mixture of dipeptides is cleaved from a small portion of the mixed resin to use it in biological tests. The rest of the mixed resin is divided into k3 equal
parts and the amino acids intended to occupy the third position are coupled to them. Then the synthesis is likewise continued until the mixture of n-residue peptides is reached.
It is worthwhile to add some notes. As in an ordinary solid phase synthesis, one has to make an effort to achieve good conversion by applying the reagents in excess. Fortunately, however, conversions lower than 100%, or minor unwanted splitting reactions do not cause so serious problems like in ordinary syntheses. The labour requirement could be significantly reduced by using mixtures of properly protected amino acids in acylation reactions. This, however, does not seem to be an acceptable solution because of the differences in the reactivity of the activated amino acids which would lead to the formation of peptides in significantly different concentrations thus causing problems in the screening experiments. Formation of peptides in equal concentrations can only be assured by mechanical mixing of samples followed by dividing into equal portions. This makes possible a complete conversion for every amino acid component. Possibility of acylations with mixtures of several amino acids of identical reactivity might be a matter of further considerations. Smaller differences in reactivities could be compensated by properly selected molar ratios of the amino acid derivatives of the mixture. In the following calculations, however, the possibility of acylations with the mixtures of amino acid derivatives will be left out of considerations.
The number of peptides formed in the synthesis, that is, the number of components in the peptide mixtures - in a general case - can be calculated by the following formula:
Nn = k1.k2 . . . kn-1.kn
If the same number (k) of amino acids are varied in every position
Nn = kn
The number of synthetic steps in the synthesis of a peptide mixture containing Nn peptides
(considering the attachment of the first amino acid to the resin as separate step) is:
If the same number (k) of amino acids are varied in each position,
Sn = nk
The formulae show the advantage of the synthesis of peptide mixtures: the number of the synthetic steps can be calculated by summing the numbers of the varied amino acids, while the number peptides is given by the product of the numbers of the varied amino acids. One example: the synthesis of the mixture of tetrapeptides prepared by varying the 20 kinds of amino acids, needs only 80 synthetic steps! It is noted, that in the same run all shorter peptides - that is the 400 dipeptides and the 8000 tripeptides - are formed, too. The traditional synthesis of these peptides would need 168 400 synthetic steps. A different comparison: in the traditional method with 80 steps only about 30 tetrapeptides can be synthesized.
Screening of peptide mixtures
Peptides mixtures - in the first approximation - are synthesized to determine whether or not they contain biologically active component. It is supposed - although it needs experimental verification - that screening experiments can be made with mixtures, too. This offers great advantage over the traditional method since the number of screening tests is reduced by a factor equal to the number of components of the mixture. For example, the mixture of the 8000 tripeptides can be examined by a single series of tests. If there is active peptide among them, one of the executable t tests gives positive result. If the number of active peptides is more than one, then, of course, more tests may give positive result. In the synthesis of the mixture of n-residue peptides it is wortwhile to test the shorter peptides, too. The synthesis is so designed to allow for this. Taking this requirement into account, and the number of kinds of tests being t, the total number of the executable tests is:
Tn = t(n-1)
Although this equation certainly holds, its realization in practice deserves some notes. There is - without any doubt - an upper limit in the number of components of the peptide mixtures to be submitted to screening tests. It is difficult to estimate this number without experiments. The mixtures may probably contain many thousands of components, and as it can be judged today, the method outlined above is rather limited by possibilities of screening tests than by the number of the required synthetic steps. If there are too many components in the mixture, too large samples have to be applied in the screening experiments to achieve observable effect for a single component. The mixture supposedly contains a number of more or less active analogs and their effect is probably summarized. Nevertheless, an unsurpassable limit in the number of components certainly exists. Therefore in certain cases may prove useful to examine the effect of the n-residue mixtures without final mixing. In other cases the synthesis should be designed so not to surpass the optimal number of components.
"Back-searching" for the active peptide
If the peptide mixture is detected to contain active component, that is, if the mixture shows a new type biological effect, then the further task is the isolation and structure determination of the active peptide followed by its synthesis. Once the mixture containing the
active component or components is in our hand the isolation can be carried out using the effective separation methods, since these make possible to separate the active compound even from thousands of inactive components. It is possible, however, to follow a different method, too. This will be outlined here. This approach to the identification of the active peptides is supposed to be less tedious then the isolation methods, moreover it supplies additional information concerning the structure-effect relationship. Applicability of the method requires a procedure for quantitative determination of activity. For the sake of simplicity let's suppose that the mixture contains a single effective component (besides analogs having the same kind of effect but smaller activity).
Back-searching step No. 1
The experiments are started with the kn samples taken aside in the synthesis of the n
-residue peptides before final mixing. The mixtures of n-residue peptides are cleaved from each resin sample. The mixtures of peptides differ from each other only in the n-th (that is the N-terminal) residue of their component peptides. Each peptide mixture is submitted to a quantitative activity determination. This shows how the activity depends on the terminal amino acid residue, that is, this way we can determine the N-terminal residue of the active peptide, and in addition it will show the effect of its replacement by other amino acid residues. Let's suppose, for example, that the N-terminal residue in the sample showing the highest activity (as well as in the active peptide) is Phe (phenylalanine). It is noted here that if there are several samples showing equally high activity it is practical to choose as the N-terminal residue of the active peptide the cheapest or the synthetically less problematical amino acid. This note holds for the subsequent back-searching steps, too.
Back-searching step No. 2
The experiment is continued with the kn-1 samples taken aside in the synthetic stage of the
(n-1)-residue peptides. The amino acid determined before, that is Phe in our example, is coupled to each sample. Cleavage of the peptides from the support gives kn-1 different peptide mixtures.
Their common feature is that every peptide has Phe in the N-terminal position. By submitting the peptide mixtures to quantitative screening experiments one can determine the amino acid residue occupying position n-1 (that is, the pre-N-terminal position) in the active peptide. This experiment also shows the effect on activity of substitution of this amino acids with other ones. Let's suppose that the pre-aminoterminal amino acid is Arg (arginine). It should be noted that in this back-searching step Phe is coupled to kn-1 samples and the same number (kn-1) of screening
experiments have to be done. Not all of the t kinds of tests are required, only the one proved before to be positive. Consequently the number of the synthetic steps and the number of screening experiments are the same: kn-1. It is also noted that in the previous back-searching step
only screening test are done (their number is kn) synthetic steps are not needed.
Back-searching step No. 3
This, and the subsequent back-searching steps may be realized using two different approaches. The peptides in samples taken aside during the synthesis have to be elongated to contain n residues, in such way, to carry on their N-terminal section the amino acid residues assuring activity. This can be realized on two ways. Either by stepwise coupling with amino acids (in our example with protected Arg then Phe) or by coupling in a single step with a previously
synthesized oligopeptide having the required sequence (in our example Phe.Arg). The required synthetic steps in the two approaches significantly differ. The number of the screening experiments, however, are the same in both cases. Let's turn now to the No. 3. back-searching step.
Stepwise elongation
Let's take the kn-2 samples taken aside in the synthesis of (n-2)-residue peptides. Each
sample is coupled first with protected Arg then with protected Phe. After cleaving the peptides from the support each of the kn-2 peptide mixtures are submitted to activity tests to determine the
amino acid residue occupying the third position counting from the N-terminal end. The number of screening tests to be executed is kn-2. The number of the required synthetic steps is: 2kn-2. The
multiplying factor preceding k is the bigger the shorter are the peptides to be elongated. The numerical value of the factor is equal to the number of amino acids to be coupled with in the elongation process.
Elongation with oligopeptide
A previously synthesized dipeptide (in our example Phe.Arg) is coupled to each of the kn-2
samples taken aside and the process is continued as described above. The number of screening test is also kn-2. The number of synthetic steps (leaving out of consideration the synthesis of the
oligopeptide) is also kn-2. This procedure seems to be more economical. In practice it means that
the active peptide is synthesized in parallel with the screening tests using the classical method started from the N-terminus. Small fractions of the growing peptide are sacrificed in the back-searching steps. This back-back-searching method has the great advantage (in addition to the fact that it needs less synthetic steps) that when the back-searching procedure is finished the active peptide is synthesized, too.
Back-searching of more than one active peptide
In the synthetic peptide mixtures several active peptides may be present, showing different effects. In these cases the number of back-searching steps will be bigger by a factor equal to the number of the differing active peptides. That is, if the number of the active peptides is "a" the values deduced above are multiplied by a. It is noted that the presence in the mixture of peptides having different effects may complicate the back-searching process especially in the case of peptides with opposing effects. This, however, is not treated in details.
The back-searching process ends when the sequences of all active peptides are determined by applying either the oligopeptide or the stepwise elongation method.
Total number of synthetic steps and screening tests summarized for the whole synthetic backsearching process
Number of synthetic steps using oligopeptide elongation
In synthesis:
n i i nk
S
1In back-searching:
1 1 n i i na
k
S
Total in synthesis and back-searching:
1 1)
1
(
n i n i na
k
k
S
If k amino acids are varied in each step: Sn = [n(a + 1) - a]k
Number of synthetic steps using stepwise elongation.
In synthesis:
n i i nk
S
1 In back-searching:
1 1)
1
(
n i i na
n
k
S
Total in synthesis and backsearching:
n i n i i i nk
a
n
k
S
1 1 1)
1
(
If k amino acids are varied in each step:
1 1 n i nnk
ak
i
S
Number of screening tests equally valid using the oligopeptide and stepwise elongation
In synthesis: Tn = t(n-1) In back-searching:
n i i na
k
T
1Total in synthesis and back-searching:
n i i nt
n
a
k
T
1)
1
(
If k amino acids are varied in each step: Tn = t(n-1) + ank
An example: preparation and screening of all pentapeptides
N5 = 320000, n=5 k=20 t=10 a=1
Total number of synthetic steps
Oligopeptide elongation 180 Stepwise elongation 300 Number of tests 140
Extension of the method to other types of compounds
Applicability of the method outlined before is not restricted for only the systematic searching for active peptides. The same principle applies to all other sequential types of compounds, that is, when the compounds belonging to this type of compounds differ from each other only in their building blocks or the sequences of these building blocks. Among them may occur natural compounds like oligosaccharides or oligonucleotides but synthetic products may be taken into account, too. Among these later ones one may think about sequential copolymers or sequential polycondensates. Dr. Árpád Furka university professor File number 36237/1982
I certify this stitched document comprising 14, that is, fourteen pages was subscribed in my presence by Dr. Arpad Furka, university professor, with his own hands.
Budapest, 1982. Nineteen hundred and eighty two, June 15, (fifteen).
Dr. Judit Bokai state notary public
1.3. Publication of the split-mix combinatorial synthesis
The split-mix method was published in 1988 as posters on two international congresses. First on the 14th International Congress of Biochemistry in Prague6, then on the 10th International Symposium of Medicinal Chemistry, Budapest, Hungary7. The manuscript for publication in print was submitted in February 1990 to the International Journal of Peptide and Protein Research. The paper appeared in June 19918. It was unusual, however, that within the 16 month passed between submittance of the manuscript and appearance of the paper four patents were filed on the subject of the split-mix synthesis and the author of one of the patents was the Editor in Chief of the journal where the paper was submitted. Also soon after our paper appeared two other papers were published (in September 1991) on the same topic9,10also with the Editor in Chief among the members of the authors of one of the publications10. More can be read on this subject in a paper published in Periodica Polytechnica Ser. Chem11
http://www.pp.bme.hu/ch/index.html (year 2004) and in the home page of the author of this book12.
References
1. http://inventors.about.com/library/weekly/aacarsassemblya.htm
2. L. B. Smillie, Á. Furka, N. Nagabhushan, K. J. Stevenson, C. O. Parkes Nature 1968, 218, 343.
3. A. Einstein The meaning of relativity, Princeton University Press, 1955, 5th Ed., Princeton, NY, p. 107.
4. A. Linde Scientific American1994, November, p. 48. 5. R. B. Merrifield J. Am. Chem. Soc. 1963, 85, 2149.
6. Á. Furka, F. Sebestyén, M. Asgedom, G. Dibó, In Highlights of Modern Biochemistry, Proceedings of the 14th International Congress of Biochemistry, VSP. Utrecht, The Netherland, 1988, Vol. 5, p 47.
7. Á. Furka, F. Sebestyén, M. Asgedom, G. Dibó Proceedings of the 10th International Symposium of Medicinal Chemistry, Budapest, Hungary, 1988, p 288, Abstract P-168.
8. Á. Furka, F. Sebestyén, M. Asgedom, G. Dibó Int. J. Peptide Protein Res.1991, 37, 487. 9. R. A. Houghten, C. Pinilla, S. E. Blondelle, J. R. . Appel, C. T. Dooley, J. H. Cuervo Nature
1991, 354, 84.
10.K. S. Lam, S. E. Salmon, Hersh E. M, V. J. Hruby, W. M. Kazmierski, R. J. Knapp Nature
1991, 354, 82.
2. The solid phase synthesis
The solid phase synthesis is very important in combinatorial chemistry since most of the combinatorial synthetic procedures are based on this method. The solid phase synthesis was developed by Merrifield1 and demonstrated in the synthesis of peptides.
Peptides and proteins are built up from α-amino acids. The structure of the α-amino acids
is expressed by the following general formula where R is the side chain by which the α-amino acids differ from each other:
Table 2.1. The natural α-amino acids
Name Side chain
-R
Three letter symbol One letter symbol
Alanine -CH3 Ala A
Arginine -(CH2)3NH(C=NH)NH2 Arg R
Asparagine -CH2CONH2 Asn N
Aspartic acid -CH2COOH Asp D
Cysteine -CH2SH Cys C
Glutamine -(CH2)2CONH2 Gln Q
Glutamic acid -(CH2)2COOH Glu E
Glycine -H Gly G
Histidine -CH2(4-imidazolyl) His H
Isoleucine -CH(CH3)CH2CH3 Ile I
Leucine -CH2CH(CH3)2 Leu L
Lysine -(CH2)4NH2 Lys K
Methionine -(CH2)2SCH3 Met M
Phenylalanine -(benzyl) Phe F
Proline Pro P
Serine -CH2OH Ser S
Threonine -CH(CH3)OH Thr T
Tryptophane -CH2(3-indolyl) Trp W
Tyrosine -(4-hydroxybenzyl) Tyr Y
Valine -CH(CH3)2 Val V
The only exception is proline in which the side chain and the amino group form a ring. H2N-CH-COOH
The twenty α-amino acids that are components of proteins are listed in Table 2.1.
In the traditional way, peptides are synthesized in solution from properly protected amino acids.
The carboxyl group of one amino acid is protected (by protecting group B) while the amino group is free. The amino group of the other amino acid is protected (Z) and the carboxyl group is activated (X) in order to make it capable to acylate the other amino acid.
Figure 2.1. Solid phase synthesis of a dipeptide.
1: Attachment of the first N-protected amino acid to the solid support ( ). 2. Removal of the protecting group (Z) from the amino group of the attached amino acid. 3: Coupling the second
N-protected amino acid to the attached one. 4: Cleaving the dipeptide from the solid support and removing the protecting group.
Z-NH-CH-COOX + H2N-CH-COOB Z-NH-CH-CO-NH-CH-COOB
R1 R2 R1 R2 Cl-CH2- Z-NH-CH-COOH + R -CH2- Z-NH-CH-COO R -CH2- NH2-CH-COO R -CH2- NH2-CH-COO R Z-NH-CH-COX + R1 -CH2- NH-CH-COO R Z-NH-CH-CO- R1 NH-CH-COOH + R NH2-CH-CO- R1 1 2 3 4 + N H COOH H
In continuation of the synthesis the amino-protecting group of the dipeptide is removed then acylated with another amino-protected and activated amino acid. The product of every synthetic step of the synthesis is usually isolated from the solution then purified. This is mostly a tedious job.
In the solid phase synthesis the first amino acid is attached by its carboxyl group to a polymer support then the next amino acid is coupled to the already attached one and the rest of the amino acids are coupled sequentially to the attached peptide chain. The solid support used by Merrifield was a styrene-divinylbenzene co-polymer in fine bead form. The resin was functionalyzed by introduction of chloromethyl groups that made possible to attach the first amino acid to the resin.
The solid phase method is demonstrated in Figure 2.1 using the synthesis of a dipeptide as example. In the synthetic step No. 1 the first amino acid is attached to the resin. All functional groups of the amino acid are protected (Z) except the α-carboxyl group. In step No. 2 the
protecting group is removed from the α-amino group of the attached amino acid in order to be able to couple the second amino acid to the first one. The second amino acid is also fully protected except again the α-carboxyl group which is properly activated (by group X). The coupling is realized in step No. 3. The steps No. 2 and No. 3 form a cycle that is sequentially repeated until the attachment of the last amino acid is finished. The closing step of the synthesis (step No. 4 in our example) is the cleavage of the product from the resin which usually involves the cleavage of the protecting groups from the functional groups of the peptide. It seems worthwhile to note that the synthesis can be started with a resin which already contains an attached amino acid. Such resins are commercially available.
Figure 2.2. Reaction vessel for solid phase synthesis
The solid phase reactions can be carried out in a reaction vessel demonstrated in Figure 2.2. The glass or plastic tube has a grid at the bottom that keeps the resin in the vessel when the tap is open. The tube is usually mounted on a laboratory shaker. The resin (containing the attached amino acid) is placed in the reaction vessel then all the multi step operations are carried out in the tube. First the resin is swelled by adding solvent, usually a mixture of dimethyl formamide (DMF) and dichloro methane (DCM), then the group protecting the α-amino group is removed by adding a proper reagent. Following this operation the protected amino acid and the coupling reagent are added in dissolved form. During the coupling reaction, the reaction vessel is shaked. The solid phase reactions are generally slower than the solution phase counterparts and shaking shortens the reaction time. After all of the described operations the resin is thoroughly washed with solvent. The peptide can be elongated by repeating the above cycle. After finishing
Frit Resin
the coupling reactions a mixture of reagents is added which cleaves the peptide from the resin and removes the protecting groups. The synthesized peptide can be recovered from the filtrate.
In the solid phase synthesis the amino acids and the reagents can be added in excess to drive the reactions to completion. The excess of the amino acids and reagents can easily be removed by filtration. The coupling step can even be repeated to ensure complete conversion. The traces of the reagents are removed by repeated washings and the product of coupling remains on the filter in pure form.
As outlined above the elongation of the peptide chain on the support is realized in identical coupling cycles (of course the added protected amino acid may vary from cycle to cycle). This opens the possibility of automation. In fact Professor Merrifield and his colleagues constructed and published an automatic peptide synthesizer2. Today many kinds of solid phase peptide synthesizers are commercially available. In addition, solid phase automatic synthesizers have also been developed for preparation of other kinds of organic compounds, too (see later).
2.1. Solid supports
Since the seminal publication of Merrifield in 19631 different types of solid supports have been developed. The solid support, of course, have to be insoluble in the solvents used in the solid phase synthesis and it is also a requirement not to react with the reagents applied in the synthesis. In the case of most solid supports the reactions take place both inside and at the surface of the solid particles. These supports are mostly used in the form of small resin beads that swell in the solvents applied in the synthesis. The reactions in other kinds of supports take place only at the surface. These supports are used as polymer or glass beads, rods, sheets etc. and (except their surface layer) do not swell in the solvents.
The solid supports are usually composed of two parts: the core and the linker. The starting compound of the synthesis is attached to the support via the linker.
The core ensures the insolubility of the support, determines the swelling properties, while the linker provides the functional group for attachment of the start compound and determines the reaction conditions for the cleavage of the product. The linker itself and the covalent bond formed with the start compound must be stable under the reaction conditions of the synthesis.
2.1.1. Crosslinked polystyrene
Cross-linked polystyrene resins are the most commonly used supports for solid phase synthesis. The polystyrene resins are synthesized from styrene and divinylbenzene by suspension polymerization in the form of small beads. The ratio of divinylbenzene to styrene determines the density of cross links. Higher crosslink density increases the mechanical stability of the beads. Lowering the crosslink density, on the other hand, increases swelling and increases the accessibility of the functional groups buried inside the beads. In practice, mostly 1-2% divinylbenzene is used. Crosslinked polystyrene is very hydrophobic so it swells only in apolar
solvents. Table 2.2 shows the swelling factor (ml/g) of 1% crosslinked polystyrene in different solvents.
Table 2.2. Swelling factor of 1% crosslinked polystyrene
Solvent Swelling factor Solvent Swelling factor
Tetrahydrofurane 5.5 Acetonitrile 4.7
Toluene 5.3 Dimethylformamide 3.5
Dichloromethane 5.2 Methanol 1.8
Dioxane 4.9 Water -
Functional groups can be introduced into the resin by two approaches: either by post-functionalization of the aromatic rings of polystyrene, or by using functionalized styrene in polymerization.
The bead size of the resin is an important factor that has to be considered in solid phase synthesis. The reactions are faster when small beads are used, but application of very small beads may cause problems in filtration. The bead size is characterized either by the diameter of the beads or by the inversely proportional mesh size. In practice most often the 200-400 mesh (35-75 micron) or the 100-200 mesh (75-150 micron) bead sizes are used. The bead size distribution also deserves consideration. A narrow bead size distribution is advantageous. The capacity of the polystyrene beads is around 0.5 mmol/g.
2.1.2. Polyethylene glycol (PEG) grafted supports
PEG-grafted polystyrene has a 1-2% crosslinked polystyrene core and to its aromatic rings, polyethylene glycol chains are covalently attached. Its commercial name is Tentagel3 (Figure 2.3).
Figure 2.3. Structure of PEG-grafted polystyrene (n ~ 70). X is a functional group (Br, OH, SH or NH2) for attachment of the substrate
The advantage of the PEG-grafted polystyrene is that the substrate at the end of a flexible chain is more accessible to reagents. It behaves like being in a solution-like environment. The PEG-chains gives a hydrophilic character to the resin and swells well in water and methanol but poorly in ether or ethanol.
O
O
X n
2.1.3. Inorganic supports
Glass beads with controlled pore size can be manufactured and are commercially available. The glass beads can be functionalized and can be used as supports in solid phase synthesis. The mechanical stability of such glass beads surpass that of the resin beads but do not swell in solvents.
Functionalized ceramics can also be used as supports.
2.1.4. Non-bead form supports
Polymers can be used as supports for solid phase synthesis not only as microscopic beads but also in the form of macroscopic objects if their surface can be functionalized with groups that can serve as anchors to hold the substrate in a reasonable quantity. Using appropriate monomers like styrene or others, polyolefin chains can be grafted by radiation into the surface of the objects and the chains can be functionalized.4 Such grafted macroscopic solid support units as SynPhase crowns and SynPhase lanterns are commercially available in different sizes at Mimotopes, Australia.5
2.2. Linkers, anchors
The initial building block of the compound to be prepared by solid phase synthesis is covalently attached to the solid support via the linker. The linker is a bifunctional molecule. It has one functional group for irreversible attachment to the core resin and a second functional group for forming a reversible covalent bond with the initial building block of the product. The linker that is bound to the resin is called anchor.
The anchor can also be considered as a protecting group of one of the functional groups of the final product and, as such, it determines the reaction conditions by which the product can be cleaved from the support. A large variety of the commercially available resins contain the already built in anchor. A series of selected examples are found below.
Merrifield resin.
The Merrifield resin can be used to attach carboxylic acids to the resin. The product can be cleaved from the resin in carboxylic acid form using HF.
Trityl chloride resin.
The trityl chloride resin is much more reactive than the Merrifield resin. It can be used for attachment of a vide variety of compounds like carboxylic acids, alcohols, phenols, amines,
Resin + Linker Resin Anchor
thiols. The products can be cleaved under mild conditions using a solution of trifluoroacetic acid (TFA) in varying concentrations (2-50%).
Hydroxymethyl resin.
The resin can be applied for attachment of activated carboxylic acids and the cleavage conditions resemble that of the Merrifield resin.
Wang resin.
The resin is used to bind carboxylic acids. The ester linkage formed has a good stability during the solid phase reactions but its cleavage conditions are milder than that of the Merrifield resin. Usually 95% TFA is applied. It is frequently used in peptide synthesis.
Aminomethyl resin.
Carboxylic acids in their activated form can be attached to the resin. Since the formed amide bond is resistant to cleavage, the resin is used when the synthesized products are not cleaved from the support; they are tested in bound form.
Rink amide resin.
The Rink resin is designed to bind carboxylic acids and cleave the product in carboxamide form under mild conditions. The amino group in the resin is usually present in protected form. For attachment of the substrate first the protecting group is removed then it is reacted with the activated carboxylic acid compound. Cleavage of the product in carboxamide form can be performed with dilute (~1%) TFA.
CH2-OH CH2-OH O O-CH3 CH2-NH2 O Cl
Photolabile anchors.
Photolabile anchors have been developed that allow cleavage of the product from the support by irradiation without using any chemical reagents. Such anchors, like the 2-nitro-benzhydrylamine resin below, usually contain nitro group that absorbs UV light.
Traceless anchors.
The initial building block of a multi-step solid phase synthesis needs to have one functional group (in addition to others) for its attachment to the solid support. It may happen that in the end product this group is unnecessary and needs to be removed. For this reason anchors have been developed that can be cleaved without leaving any functionality in the end product at the cleavage site. These traceless anchors usually contain silicon based linkers.
2.3. Protecting groups
If a chemist wants to carry out a reaction on only one functional group of a multi-functional group compound, the reactivity of the rest of the multi-functional groups needs to be suppressed. This can be achieved by application of protecting groups. A protecting group is reversibly attached to the functional group to convert it to a less reactive form. When the protection is no longer needed, the protecting group is cleaved and the original functionality is restored. A large number of protecting groups were developed for use in peptide synthesis since the amino acids are multi-functional compounds. It is an important requirement for a protecting group to be stable under the expected reaction conditions and to be cleavable - if possible - at mild reaction conditions. The stability/cleavage conditions of a protecting group are considered relative to those of the others. Two protecting groups are said to be orthogonal if either of them can be removed without affecting the stability of the other one. Some of the protecting groups most widely used in peptide synthesis are described below.
O CH NH2
OCH3
OCH3
CH NH2
2.3.1. Protection of amino groups
The benzylcarbonyl (Z) group.
Bergmann and Zervas suggested the benzyloxycarbonyl group for amino-protection in peptide synthesis in 1932 and this important protection type is still in use. The Z group can be introduced by the reaction of the amino group containing compound with benzylchloroformate under Schotten-Bauman conditions.
The Z protection is stable under mildly basic conditions and nucleophilic reagents at ambient temperature. Cleavage can be brought about by HBr/AcOH, HBr/TFA or catalytic hydrogenolysis.
The t-butoxycarbonyl (Boc) group.
An alternative choice for amino group protection is the Boc group. Its advantage is that can be removed under milder conditions than the Z group.
The Boc group is completely stable to catalytic hydrogenolysis and as such is orthogonal to the Z group. Basic and nucleophylic reagents are no effect on the Boc group and its removal can be carried out by TFA at room temperature. The most convenient reagent that can be used in the protection reaction is the Boc anhydride (Boc2O).
The 9-fluorenylmethoxycarbonyl (Fmoc) group.
The Fmoc group differs from both Z and Boc groups since it is very stable to acidic reagents. CH2-O-C-Cl O H2N~ CH2-O-C-NH O ~ + Me3C-O-C-NH~ O
H
CH2
-O-C-NH-O
The Fmoc group can be removed under basic conditions. Usually 20% piperidine dissolved in DMF is used as reagent. One of the reagents for introducing the Fmoc group is the FmocCl.
2.3.2. Protection of carboxyl groups
Carboxyl groups are most often protected by converting them to benzyl esters or t-butyl esters.
The benzyl esters are cleaved by saponification, HBr/AcOH, HF and catalytic hydrogenation but not by TFA. Their response to acids is similar to that of the Z groups but somewhat less sensitive.
The t-butyl esters, unlike benzyl esters, are stable to bases or nucleophilic attack. The properties of t-butyl esters are somewhat similar to those of the Boc groups although they are less sensitive to acidolysis. They can be cleaved by TFA.
2.3.3. Protection of other functional groups
The alcoholic and phenolic hydroxyl groups are protected by converting them to benzyl ether or t-butyl ether. The former protecting group can be cleaved by HF, HBr/AcOH or by catalytic hydrogenolysis and the latter one by TFA.
Thiol groups can also be protected by benzyl ether formation or by tritylation.
The guanidino group (present in arginine) can be protected by nitration or by arylsulphonyl groups. The nitro group resists HBr/AcOH and can be cleaved by liquid HF. Among the arylsulphonyl groups the tosyl (Tos) group can be cleaved by liquid HF or sodium in in liquid ammonia. Two other arylsulfonyl groups are more sensitive to acidic conditions. The 2,2,5,7,8-pentamethylchroman-6-sulphonyl (Pmc) group can be cleaved by TFA under conditions
~COO-CH2- ~COO C
CH3
CH3
CH3
similar to the removal of the Boc group. The 4-methoxy-2,3,6-trimethylbenzenesulphonyl (Mtr) group is also cleaved by TFA but is less sensitive and requires a few hours for cleavage.
The amide groups (in side chains of asparagine and glutamine) can be protected by tritylation. The trityl protecting group is stable to base, catalytic hydrogenolysis, very mild acid but is cleaved with TFA. It is used in conjunction with the Fmoc amino group protection strategy. The NH group of the imidazole ring (in the side chain of histidine) is protected in conjunction with the Fmoc strategy by tritylation. The trityl protecting group can be removed by TFA at room temperature.
The indole ring (in tryptophane) can be protected by Boc group that can be removed by TFA.
2.3.4. Coupling reagents for peptide synthesis
In the coupling reactions of peptide synthesis the carboxyl group of the acylating amino acid is activated. Care should be taken in selecting the activation method to avoid racemization. One of the choises is 1,3-diisopropylcarbodiimide (DIC) with addition of N-hydroxybenztriazole (HOBt) in order to reduce racemization.
NH-C-NH~ O O Me Me Me SO2 Me Me NH-C-NH~ O Me Me Me Me SO2 Pmc protection Mtr protection NH-C-NH~ O O2
N-Protection by nitro group
N
N CPh3
Protection by trityl group
Protection by Boc group
N Boc
1,3-Diisopropylcarbodiimide (DIC) N-Hydroxybenztriazole (HOBt)
Another very often used coupling reagent is O-benztriazole-N,N,N’,N’-tetramethyl-uronium-hexafluoro-phosphate (HBTU) that is known not to cause racemization.
N N N O N CH3 H3C N CH3 CH3 PF6 _ + O-benztriazole-N,N,N’,N’-tetramethyl-uronium-hexafluoro-phosphate (HBTU)
One of the bases applied in the coupling reactions is N,N'-Diisopropylethylamine (DIPEA).
N,N'-Diisopropylethylamine (DIPEA)
2.4. Solid phase synthesis of organic molecules
The vast number of reactions developed for the synthesis of organic molecules were optimized for solution phase. In the decades following its introduction by Merrifield,1 of the solid phase method was mainly used in peptide chemistry. Except the studies of Lezenoff,6-9 Camps,10 Frechet11-13 , Crowley and Rapoport,14 little experience has been accumulated in its application in the synthesis of organic molecules. The advent of combinatorial chemistry, however, induced radical changes and initiated a fast expanding research in the field. The classes of chemical reactions developed for solid phase as a result of this research are showed below:
Anchoring reactions
Amide bond forming reactions Aromatic substitutions Condensation reactions N=C=N CH CH H3C H3C CH3 CH3 N N N N OH
Cycloaddition reactions Organometallic reactions Michael additions
Heterocyclic forming reactions Multi-component reactions Olefin forming reactions Oxidation reactions Reduction reactions Substitution reactions
Protection/deprotection reactions Cleavage from supports
Other types of solid phase reactions
Excellent compilations of these reactions were prepared by Hermkers et al.15,16, Á. Furka17 and W. M. Bennett.18
2.5. Solid phase reagents and scavenger resins in solution phase synthesis
Solid phase additives are successfully applied in many solution phase synthetic reactions. In solid phase reactions the substrate is bound to the solid phase carrier and the reagents are in solution. In solution phase reactions both the substrates and the reagents are in solution. In some solution phase reactions, however, the reagent is bound to resin. The advantage of such reagents is that the by products of the reagent remains bound to the resin and can be easily removed from the reaction mixture by filtration. One example is the polymer bound HOBt that is used in amide formatting reactions.
N N N
OH
Polymer bound HOBt
More solid phase reagents and examples of their applications are found in the already mentioned compilations.15-18
Different types of resins can also be used in solution phase reactions for removal of the excess of reagents, substrates or by products. Those resins that can be used for such purposes are named scavenger resins. One example is formylpolystyrene19-20 which is used for removal of primary amines from reaction mixtures.