Copyright © 2000, American Society for Microbiology. All Rights Reserved.
Detection of and Discrimination between Gram-Positive and
Gram-Negative Bacteria in Intraocular Samples
by Using Nested PCR
NORA M. CARROLL,
1† EMMA E. M. JAEGER,
1SARAH CHOUDHURY,
1ANTHONY A. S. DUNLOP,
1MELVILLE M. MATHESON,
2PETER ADAMSON,
1NARCISS OKHRAVI,
1*
ANDSUSAN LIGHTMAN
1Department of Clinical Ophthalmology
1and Department of Pathology,
2The Institute of Ophthalmology
and Moorfields Eye Hospital, London EC1V 9EL, United Kingdom
Received 31 August 1999/Returned for modification 25 October 1999/Accepted 22 February 2000
A nested PCR protocol has been developed for the detection of and discrimination between 14 species of
gram-positive and -negative bacteria in samples of ocular fluids. First-round PCR with pan-bacterial
oligo-nucleotide primers, based on conserved sequences of the 16S ribosomal gene, was followed by a
gram-negative-organism-specific PCR, which resulted in a single 985-bp amplification product, and a multiplex PCR which
resulted in two PCR products: a 1,025 bp amplicon (all bacteria) and a 355 bp amplicon (gram-positive
bacteria only). All products were detected by gel electrophoresis. The sensitivity of the assay was between 10
fg and 1 pg of bacterial DNA, depending on the species tested, equivalent to between 24 and 4 live bacteria
spiked in water. The identification was complete in 3.5 h. The molecular techniques were subsequently applied
to four samples of intraocular fluid, (three vitreous and one aqueous) from three patients with clinical signs
of bacterial endophthalmitis (test samples) and two samples of vitreous from a patient with chronic intraocular
inflammation (control samples). In all culture-positive samples (two of three vitreous and one of one aqueous),
a complete concordance was observed between molecular methods and culture results. PCR correctly identified
the gram stain classification of the organisms. The bacterial etiology was also identified in a culture-negative
patient with clinical history and signs highly suggestive of bacterial endophthalmitis. Furthermore, control
samples from a patient with chronic intraocular inflammation remained PCR negative. In summary, this
protocol has demonstrated potential as a rapid diagnostic test in confirming the diagnosis of infection and also
determining the Gram status of bacteria with high specificity and sensitivity.
The advent of DNA amplification by PCR has had a great
impact on the speed and accuracy with which one can identify
a bacterial species or strain. Instead of relying on
time-con-suming and subjective phenotypic tests, it is now possible to
rapidly amplify specific regions of bacterial genomes by PCR
and compare them at the sequence level (30, 34). This has the
advantage of being independent of the state of the organism
(viable or nonviable) and has resulted in the reclassification of
some organisms (24). In addition to the reproducibility of
PCR, it is extremely sensitive, requiring only small numbers of
organisms for analysis. This sensitivity has been exploited as
the basis for a number of tests, including the detection of
pathogens (4, 15, 16, 21, 24) and the determination of
mech-anisms of resistance to specific therapeutic agents (8, 33, 35).
The reported sensitivity of the technique varies, but the
detec-tion by PCR of single organisms or the DNA equivalent to a
single organisms has been reported (3). Nested PCRs are
par-ticularly useful in situations where a high level of sensitivity is
required, as is the case with ocular infections. Use of nested
PCRs in a clinical setting has been hampered by the frequent
incidence of false-positive results, but techniques have been
developed that eliminate this problem (6, 9, 11).
Endophthalmitis is a term referring to severe intraocular
inflammation centered around the vitreous cavity and/or
ante-rior chamber of the eye and may be of infectious origin (caused
by bacteria or fungi). The challenges presented by this
condi-tion to the clinician are considerable, as the severity of the
clinical signs varies greatly according to the time to
presenta-tion, the inoculum size, and the species of the infecting
organ-ism(s) (18, 28). Also, low-grade infections can be difficult to
distinguish from purely inflammatory ocular disease. Ideally,
all cases of infectious endophthalmitis would be culture
proven, but the number of culture-proven cases with typical
signs of infectious endophthalmitis varies greatly from center
to center (2, 18, 28). It is important to establish a diagnosis and
identify the infecting organism, not only because this decides
the further management of the patient but also because it
justifies the treatment given. Confirmation of the diagnosis is
made more difficult by the small volumes of the ocular samples
available for analysis (aqueous, 100 to 150
l; vitreous, 200 to
400
l). The numbers of organisms required to establish an
infection can also be small and may be as low as 14 (31), and
often only a few colonies are cultured by routine
microbiolog-ical methods (usually 40 to 50 CFU). A delay of 24 to 48 h is
usual for routine microbiological processing of the specimens,
although it may take up to 12 days in the case of fastidious
organisms (32). In the absence of a definitive identification of
the causal organism, the clinician must commence therapy on
an empirical basis, using broad-spectrum antimicrobial agents,
because a delay in treatment is often associated with a worse
clinical outcome (12).
Clinical cases which are culture negative and respond to
antibiotic therapy are considered infectious despite the lack of
definitive culture identification. Cultures prove to be negative
for a variety of reasons, such as small sample size,
sequestra-* Corresponding author. Mailing address: Department of Clinical
Ophthalmology, The Institute of Ophthalmology, 11-43 Bath St.,
Lon-don EC1V 9EL, United Kingdom. Phone: 44-(0)171-6086861. Fax:
44-(0)171-6086931. E-mail: [email protected].
† Present address: Department of Medical Biochemistry, University
of Stellenbosch, Tygerberg 7505, South Africa.
1753
on May 15, 2020 by guest
http://jcm.asm.org/
tion of bacteria on solid surfaces (e.g. intraocular lens, lens
remnants, and capsule) leading to low numbers in the liquid
sample, the use of antibiotics prior to sampling, and the
fas-tidious nature of some of the organisms which cause
intraoc-ular infection (6, 22, 28). The use of molecintraoc-ular techniques has,
therefore, been investigated in order to improve the diagnostic
rate and reduce the time to diagnosis. This paper describes an
integrated protocol describing the direct detection in ocular
fluids of pathogens with suspected infective pathology. A
nested PCR approach was developed in which primers based
on the conserved bacterial 16S rRNA gene sequences were
used in the first round of amplification, while a second round
of amplification was able to differentiate between
gram-posi-tive and -negagram-posi-tive pathogens.
MATERIALS AND METHODS
Patient sampling.Intraocular (aqueous and vitreous) sampling was under-taken under sterile conditions. Aqueous sampling was underunder-taken under topical anesthesia, using a 27-gauge (0.33-mm-diameter) needle, and 100 to 200l was aspirated. Vitreous sampling was undertaken as a biopsy through the pars plana. After subconjunctival injection of anesthetic, a vitreous tap was performed using a 23-gauge needle which was inserted through the pars plana 3 mm behind the limbus in aphakic eyes and 4 mm behind the limbus in phakic eyes. A total of 200 to 400l of vitreous was aspirated.
Microbiological assessment.One drop of vitreous was smeared on a slide for Gram and periodic acid-Schiff staining, and the remainder was immediately plated on blood and Sabouraud agar before transport to the microbiology lab-oratory. Plates were incubated under aerobic conditions at 37°C. The cultures were transferred to a 30°C incubator if no growth was apparent after 48 h. In experiments where live bacteria were spiked into PCRs, bacteria were streaked out on blood agar (Biomeriux, Basingstoke, United Kingdom) and isolated colonies were inoculated into 3 ml of brain heart infusion (Biomeriux). A serial 10-fold dilution of overnight cultures was prepared in maximum recovery diluent (Oxoid, Basingstoke, United Kingdom), and aliquots were plated in duplicate for enumeration. Aliquots (5l) of bacterial suspensions were used for PCR.
Bacterial isolates used in this study.Following isolation by culture, bacteria were identified using the API biochemical identification system (API Analytab products, Division of Sherwood Medical, New York, N.Y.). A total of 40 strains of 14 bacterial species were tested (see Table 2). All strains were standard NCTC strains (Public Health Laboratory Service, National Collection of Type Culture, Colindale, London, United Kingdom). Individual strains were stored on beads at ⫺70°C (Mast Diagnostics, Bootle, Merseyside, United Kingdom) and subse-quently cultured on standard media according to the manufacturers’ instructions.
Primer design.The Gram stain-specific primers were designed by creating consensus sequences of a range of common ocular pathogens according to their Gram stain classification and comparing them. The sequences of all primers used in this study are given in Table 1. The gram-positive primer was located between bases 712 and 729 with respect to the sense strand of theEscherichia colirRNA gene sequence and differed from the gram-negative consensus along its length at 5 of its 18 nucleotides, with a 3-nucleotide mismatch at the 3⬘end. Similarly, the gram-negative-organism-specific primer differed from the gram-positive-organ-ism-specific consensus at 8 of its 15 nucleotides, with a 4-nucleotide mismatch at the 3⬘end but was located on the antisense strand. The primers were designed such that differently sized products would be generated, to facilitate an unam-biguous assignment of Gram stain classification. The gram-negative-organism-specific PCR resulted in a single 985-bp amplification product, and the multiplex PCR resulted in two PCR products: a pan-bacterial 1025-bp amplicon and a 355-bp product which was specific to gram-positive bacteria.
Nested PCR.Bacterial DNA was extracted using glass beads and alcohol precipitation, as previously described (8).Taq(AmpliTaq LD; Perkin Elmer,
Warrington, Cheshire, United Kingdom) for the first round of PCR was pre-treated according to the method of Carroll et al. (8). Briefly, prior to PCR amplification the water, buffer, magnesium chloride, andTaqcomponents were mixed and incubated for 30 min at 37°C with 1.0 U of Sau3A1 (Boehringer Mannheim, Lewes, East Sussex, United Kingdom) per U ofTaqpolymerase. The restriction enzyme was subsequently inactivated by incubation at 95°C for 2 min, following which the deoxynucleoside triphosphates (dNTPs), primers, and tem-plate DNA were added and PCR amplification was commenced.Taqfor the second round of amplification was used without pretreatment. PCRs were car-ried out in the proprietary buffers and for the first round of amplification contained a 60M concentration of each deoxynucleoside triphosphate (Phar-macia, Little Chalfont, Buckinghamshire, United Kingdom), 3.0 mM Mg2⫹, 2.5 pmol of each of the primers 16SF and 16SR, and 1 U ofTaqDNA polymerase in a total volume of 25l. The initial denaturation was carried out for 5 min at 94°C, and cycling was performed as follows: 94°C for 10 s, 54.2°C for 10 s, and 72°C for 15 s for 30 cycles (Genius Thermal Cycler; TECHNE, Cambridge, United Kingdom). A second round of amplification used 1l of product from the first round, and a Mg2⫹concentration of 2.5 mM. PCRs specific for gram-negative organisms utilized 5 pmol each of primers NF and N6R. A multiplex PCR which simultaneously detected all species of bacteria and all gram-positive bacteria used 5 pmol each of P2F and NR and 1 pmol of NF. Denaturation was carried out for 5 min at 94°C and cycling was performed at 94°C for 7 s, 60°C for 7 s, and 72°C for 10 s for 30 cycles. Multiple reagent controls from the first round were always subjected to a second round of amplification to control for contam-ination.
PCR of vitreous and aqueous samples.Samples of vitreous and aqueous humors were received either in sterile tubes which had been sealed in the operating theater or in the syringes used to obtain the sample, after the require-ments of the routine diagnostic microbiological service had been fulfilled. Ali-quots (5l) of vitreous and aqueous humors, either neat or diluted 1/10 with sterile water were used in each PCR after they had been heated to 95°C for 2 min to extract the DNA. Samples were subjected to PCR in duplicate. Positive controls containing 10 ng each ofE. coliandStaphylococcus aureusDNA were run for each PCR in neat and diluted vitreous and aqueous humors to check for inhibition of the PCRs by the vitreous. Multiple reagent controls were subjected to two rounds of PCR to control for contamination of reagents.
Electrophoresis and imaging.Following PCR amplification, products were resolved on a 1% agarose–Tris-acetate-EDTA gel and visualized using ethidium bromide under UV illumination, and results were recorded using the UVP Ltd. (Cambridge, United Kingdom) gel documentation system.
[image:2.612.53.294.84.191.2]Sequencing of PCR products.PCR products were electrophoresed on 1% agarose gels, and the bands were excised, extracted using the Qiaquick Gel extraction kit (Qiagen, Crawley, West Sussex, United Kingdom), and sequenced using the fluorescent dye terminator sequencing system (ABI). Sequences were submitted for BLAST searching for similarity to other sequences (1). Consensus sequences were generated and compared using DNASTAR (Madison, Wis.) software.
TABLE 1. Oligonucleotide primers used in this study
Primer
name Primer sequence
Position onE. coli
rRNA gene sequence
(bases)
16SF 5⬘
TTGGAGAGTTTGATCCTGGCTC 3⬘
4–25
16SR 5⬘
ACGTCATCCCCACCTTCCTC 3⬘
1174–1194
NF
5⬘
GGCGGCAKGCCTAAYACATGCAAGT 3⬘
42–66
NR
5⬘
GACGACAGCCATGCASCACCTGT 3⬘
1044–1067
P2F
5⬘
GCGRCTCTCTGGTCTGTA 3⬘
712–729
N6R 5⬘
GGTGCCTTCGGGAAC 3⬘
1013–1027
TABLE 2. Limit of detection of DNA spiked into water of a nested
PCR using Gram-stain-specific bacterial primer pairs
aSpecies (n) Limit of detection with primer pair: N6R-NF P2F-NR
Gram negative
E. coli
(2)
10 fg
NA
K. pneumoniae
(3)
10 fg
NA
S. marcescens
(3)
100 fg
NA
H. influenzae
(2)
10 fg
NA
P. mirabilis
(3)
10 fg
NA
P. aeruginosa
(3)
10 fg
NA
Gram positive
S. aureus
(3)
NA
100 fg
S. epidermidis
(3)
NA
100 fg
S. pyogenes
(2)
NA
100 fg
S. faecalis
(3)
NA
100 pg
S. viridans
(2)
NA
1 pg
S. pneumoniae
(3)
NA
1 pg
P. acnes
(5)
NA
10 ng
B. cereus
(3)
NA
1 pg
aNA, not amplified. Numbers in parentheses indicate the number of strains
tested.
on May 15, 2020 by guest
http://jcm.asm.org/
[image:2.612.311.549.531.711.2]RESULTS
The sensitivity and specificity of the Gram stain-specific
primer pairs was evaluated on a range of common pathogens
and is detailed in Table 2. The sensitivity of the
gram-negative-organism-specific primers was 10 fg of DNA per reaction in all
of the species tested. In contrast, there was a wide variation in
the sensitivity of the gram-positive-organism-specific primer
pair, from 100 fg to 1 pg, reflecting the broad genotypic and
phenotypic diversity of this group. A multiplex PCR was
de-veloped using the primers NF-NR and P2F. The sensitivity of
this PCR was identical to that observed for individual PCRs.
Examples of these PCRs carried out with serial dilutions are
shown in Fig. 1. None of these primer sets amplified human
lymphocyte DNA or genomic DNA from
Candida albicans
or
Aspergillus fumigatus
under the conditions tested. In the
mul-tiplex PCR the 1,025-bp amplicon is the product of the primer
pair NF-NR, which are both universal bacterial primers, while
the 355-bp amplicon is specific for gram-positive bacteria. The
product of the primer pair NF-N6R that is specific for
gram-negative bacteria is 985 bp. Evaluation of the potential of these
primers to amplify DNA from whole bacteria was undertaken
by spiking various numbers of bacteria into water, 5
l of which
was used in the PCRs. The limits of detection (number of
organisms) of the primer pairs for
E. coli
,
Pseudomonas
aerugi-nosa
,
S. aureus
, and
Streptococcus pyogenes
were 5, 24, 4, and 4,
respectively, and the primers were capable of detecting
be-tween 8
⫻
10
2and 4.8
⫻
10
3organisms per ml. The multiplex
and gram-negative-organism-specific PCRs were applied to
four samples of intraocular fluid with suspected infective
pa-thology and two samples from an eye with intraocular
inflam-mation as a control. A comparison was made between the
results obtained by Gram staining, culture, and PCR, and a
summary is given in Table 3. In all culture-positive samples the
results of PCR and culture were 100% concordant. Also, the
results of subsequent DNA sequencing matched the identity of
the bacterium as isolated by culture. Although in this study
PCR was applied to these samples retrospectively, a definitive
result could have been reported 3.5 h after receipt of the
sample.
DISCUSSION
[image:3.612.121.486.76.178.2]The detection by PCR of bacterial DNA from body sites
which are normally sterile, has been used to improve the rate
of microbiological diagnosis for cerebrospinal fluid, synovial
fluid, and vitreous (15, 19, 26). This study has confirmed the
usefulness of molecular techniques in establishing the presence
of infection and has further developed them by determining
the Gram stain status of the infecting bacterium. These
tech-niques were also able to confirm the presence of bacteria in a
patient with culture-negative endophthalmitis, who
demon-strated a clinical history and signs highly suggestive of an
in-fective etiology and who responded well to antibiotic therapy,
thereby providing further evidence of the infective etiology of
the condition. Samples from a patient with a case of chronic
intraocular inflammation served as controls and remained
PCR negative. Gram-positive organisms are isolated from
in-traocular samples in 58 to 96% of cases, e.g.,
Staphylococcus
FIG. 1. Sensitivities of the primer sets were evaluated using dilutions of DNA in water. (A) Multiplex PCR in which the 1,025-bp amplicon is the product of the NF-NR primers (universal bacterial primers) and the 355-bp amplicon is the product of the P2F-NR primers (specific for gram-positive bacteria). The template DNA wasS. aureusNCTC 8532. Lane 1, 10 ng of DNA; lane 2, 1 ng of DNA; lane 3, 100 pg of DNA; lane 4, 10 pg of DNA; lane 5, 1 pg of DNA; lane 6, 100 fg of DNA; lane 7, 10 fg of DNA; lane 8, DNA ladder; lane 9, negative control. (B) Gram-negative-organism-specific PCR in which the 985-bp amplicon is the product of the primers NF-N6R. The template DNA wasE. coliNCTC 10418. Lane 1, 10 ng of DNA; lane 2, 1 ng of DNA; lane 3, 100 pg of DNA; lane 4, 10 pg of DNA; lane 5, 1 pg of DNA; lane 6, 100 fg of DNA; lane 7, 10 fg of DNA; lane 8, DNA ladder; lane 9, negative control.TABLE 3. Summary of the diagnostic tests carried out on intraocular samples
Sampleno.a Initial diagnosis Predisposing condition(s)or surgery Gram strainreaction Culture PCR result
1v
Acute endophthalmitis
Trabeculectomy surgery
Positive
S. pneumoniae
bGram positive
1a
Acute endophthalmitis
Trabeculectomy surgery
Positive
S. pneumoniae
Gram positive
2v
Metastatic endophthalmitis
Staphylococcal osteomyelitis
and septicemia
Positive
S. aureus
c
Gram positive
3v
Chronic endophthalmitis
Cataract surgery
No organisms seen
No growth
Gram positive
4vL
Vitritis secondary to chronic
intraocular inflammation
Not applicable
Not done
No growth
No product
4vR
Vitritis secondary to chronic
intraocular inflammation
Not applicable
Not done
No growth
No product
aAbbreviations: v, vitreous humor; a, aqueous humor; R, right eye; L, left eye.
bThe vitreous sample from this patient’s eye was subcultured from a cloudy brain heart infusion at 24 h and yielded florid growth of streptococci after a further 24 h. cThe vitreous sample from this patient’s eye was culture positive on blood agar, brain heart infusion, cooked meat broth, fluid thioglycolate medium, and R2A agar
after 24 h.
on May 15, 2020 by guest
http://jcm.asm.org/
[image:3.612.57.553.584.695.2]spp. (coagulase-negative staphylococci and
S. aureus
),
Strepto-coccus
spp.,
Bacillus cereus
, and
Propionibacterium acnes
(13,
14, 17, 18, 23). Gram-negative organisms account for a smaller
percentage of culture positive cases, comprising 4 to 29% in
different studies (5, 13, 14, 17, 18, 20, 23). Gram-negative
organisms typically isolated from ocular infections include
E.
coli
,
Proteus mirabilis
,
Serratia marcescens
,
Klebsiella
pneu-moniae
,
Haemophilus influenzae
, and
P. aeruginosa
. Initial
treatment of patients presenting with presumed bacterial
en-dophthalmitis is aided by the Gram staining of samples and is
guided by the results of this rapid test. Compared to infection
with gram-positive bacteria, infections with gram-negative
bac-teria are associated with a greater inflammatory response and
poorer visual prognosis: a reflection of the toxins produced and
the greater virulence of these organisms (20). The Gram stain
status of the infecting bacterium is, therefore, important
be-cause it allows targeted antimicrobial therapy in the later
stages of management and has implications for prognosis and
final visual outcome. In the clinical setting, however, this test is
usually negative (no organisms seen) and, therefore, is only
undertaken when sufficient sample is available for the full array
of culture media to be inoculated. PCR techniques, on the
other hand, only require very small amounts of clinical sample
(5
l) and are not only rapid but sensitive and efficient in
allowing a diagnosis of infection to be made. A prospective
study with larger numbers of clinical samples would be useful
and is now required.
The PCR protocol described in this paper incorporates a
number of safeguards, such that a result can be reported with
certainty. The pretreatment of the
Taq
DNA polymerase
en-sures that false positives due to intrinsic contamination of the
enzyme are avoided. The use of both neat and 1/10 dilutions of
the intraocular fluid for analysis, as well as for positive
con-trols, ensures that a negative PCR result is not due to
inhibi-tion by components of the aqueous and vitreous. Inhibiinhibi-tion of
PCR by ocular fluids has been reported by Wiedbrauk et al.
(36) and was observed in this study (Fig. 2). The effects of
routine dilution on all samples were not tested but were found
to be required in the analysis of 44% of samples in our
subse-quent studies (29; N. Okhravi, P. Adamson, A. Dunlop,
H. M. A. Towler, M. M. Matheson, and S. Lightman,
unpub-lished data). As the inhibition of the reaction was overcome by
diluting the clinical sample in all cases, further studies to
elu-cidate the nature of these inhibitory factors were not
under-taken. Aqueous samples were found to require dilution more
frequently than vitreous samples; therefore, one can assume
the inhibitory factor(s) is present to a greater degree in the
former (29; Okhravi et al., unpublished data).
As the sensitivity of the primers varied with each bacterial
species it was not possible, due to the limited supply of ocular
sample, to test the sensitivity of each reaction with ocular fluids
as well as water. However, other studies in our laboratory have
demonstrated that the sensitivity in water was the same as that
in vitreous as long as two rounds of PCR were used (29).
Although the PCR protocol developed in this study was
devel-oped specifically for ocular samples, it has the potential to be
used in other clinical settings where only small volumes of
clinical samples are available and a high degree of sensitivity is
required.
ACKNOWLEDGMENTS
N.M.C. was supported by Oclyx Ltd. P.A. was supported by Fight for
Sight. N.O. was supported by Wellcome Vision Research Fellowship
045203 and locally organized research funds (no. 221 and 271) from
Moorfields Eye Hospital.
REFERENCES
1.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman.1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res.25:3389–3402. 2.Bazra, M., P. R. Pavan, B. H. Doft, S. R. Wisniewski, L. A. Wilson, D. P. Han,
and S. F. Kelsey.1997. Evaluation of microbiological diagnostic techniques in postoperative endophthalmitis in the endophthalmitis vitrectomy study. Arch. Ophthalmol.115:1142–1150.
3.Bej, A. K., M. H. Mahbubani, R. Miller, J. L. DiCesare, L. Haff, and R. M. Atlas.1990. Multiplex PCR amplification and immobilized capture probes for the detection of bacterial pathogens and indicators in water. Mol. Cell. Probes4:353–365.
4.Berriddge, B. R., J. D. Fuller, J. de Azavedo, D. E. Low, H. Bercovier, and P. F. Frelier.1998. Development of specific nested oligonucleotide PCR primers for the Streptococcus iniae 16S-23S ribosomal DNA intergenic spacer. J. Clin. Microbiol.36:2778–2781.
5.Bohigian, G. M., and R. J. Olk.1986. Factors associated with a poor visual result in endophthalmitis. Am. J. Ophthalmol.101:332–341.
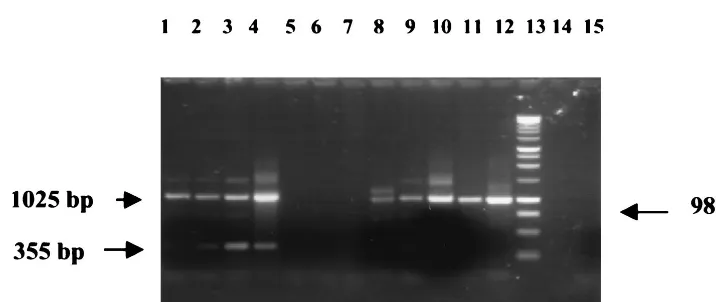
[image:4.612.122.480.68.219.2]6.Busin, M., A. Cusumano, and M. Spitznas.1995. Intraocular lens removal FIG. 2. Results of PCR from a clinical case of culture-positive bacterial endophthalmitis secondary to gram-positive bacteria. The vitreous sample was subjected to PCR as described in the text and electrophoresed on a 1% agarose–TAE gel. Patient sample PCR results appear in duplicate (lanes 3 to 6). Lane 1, multiplex PCR of gram-negative DNA in water (positive control); lane 2, multiplex PCR of gram-positive DNA in neat vitreous (positive control); lanes 3 and 4, multiplex PCR of patient sample (vitreous, diluted 1/10); lanes 5 and 6, multiplex PCR of patient sample (neat vitreous); lane 7, gram-negative-organism-specific PCR of patient sample (vitreous, diluted 1/10); lane 8, Gram negative PCR of gram-negative DNA in water (positive control); lane 9, multiplex PCR of gram-negative DNA in vitreous (positive control); lane 10, multiplex PCR of gram-negative DNA in water (positive control); lane 11, gram-negative-organism-specific PCR of gram-negative DNA in vitreous; lane 12, gram-negative-organism-specific PCR of gram-negative DNA in water; lane 13, DNA ladder; lane 14, negative control (water only, no vitreous); lane 15, negative control (vitreous and water).
on May 15, 2020 by guest
http://jcm.asm.org/
from eyes with chronic low-grade endophthalmitis. J. Cataract Refract. Surg.
21:679–684.
7.Carroll, K. C., R. B. Leonard, P. L. Newcomb Gayman, and D. R. Hillyard.
1996. Rapid detection of the staphylococcal mecA gene from BACTEC blood culture bottles by the polymerase chain reaction. Am. J. Clin. Pathol.
106:600–605.
8.Carroll, N. M., P. Adamson, and N. Okhravi.1999. Elimination of bacterial DNA fromTaqDNA polymerase using restriction endonuclease digestion. J. Clin. Microbiol.37:3402–3404.
9.Cimino, G. D., K. C. Metchette, J. W. Tessman, J. E. Hearst, and S. T. Isaacs.1991. Post-PCR sterilisation: a method to control carryover contam-ination for the polymerase chain reaction. Nucleic Acids Res.19:99–107. 10.Coakley, M., R. P. Ross, and D. Donnelly.1996. Application of the
polymer-ase chain reaction to the rapid analysis of brewery yeast strains. J. Inst. Brewing102:349–354.
11.DeFilippes, F. M.1991. Decontaminating the polymerase chain reaction. BioTechniques10:26–30.
12.Doft, B. H., S. F. Kelsey, S. Wisniewski, D. J. Metz, L. Lobes, J. Rinkoff, M. Davis, and A. Kassof.1994. Treatment of endophthalmitis after cataract extraction. Retina14(4):297–304.
13. Fisch, A., A. Salvanet, T. Prazuck, F. Forestier, L. Gerbaud, G. Coscas, and the French Collaborative Study Group on Endophthalmitis.1991. Epidemi-ology of infective endophthalmitis in France. Lancet338:1373–1376. 14. Forster, R. K., R. L. Abbott, and H. Gelender.1980. Management of
infec-tious endophthalmitis. Ophthalmology87:313–319.
15. Greisen, K., M. Loeffelholz, A. Purohit, and D. Leong.1994. PCR primers and probes for the 16S rRNA gene of most species of pathogenic bacteria, including bacteria found in cerebrospinal fluid. J. Clin. Microbiol.32:335– 351.
16. Harnett, N., Y. P. Lin, and C. Krishnan.1996. Detection of pathogenic
Yersinia enteroliticausing the multiplex polymerase chain reaction. Epide-miol. Infect.117:59–67.
17. Heaven, C. J., P. J. Mann, and D. L. Boase.1992. Endophthalmitis following extracapsular cataract surgery: a review of 32 cases. Br. J. Ophthalmol.
76:419–423.
18. Hughes, D. S., and R. J. Hill.1994. Infectious endophthalmitis after cataract surgery. Br. J. Ophthalmol.78:227–232.
19. Hykin, P. G., K. Tobal, G. McIntyre, M. M. Matheson, H. M. Towler, and S. L. Lightman.1994. The diagnosis of delayed post-operative endoph-thalmitis by polymerase chain reaction of bacterial DNA in vitreous samples. J. Med. Microbiol.40:408–415.
20. Irvine, W. D., H. W. Flynn, D. Miller, and S. C. Pflugfelder.1992. Endoph-thalmitis caused by Gram negative organisms. Arch. Ophthalmol.110:1450– 1454.
21. Jaton, K., R. Sahli, and J. Bille.1992. Development of polymerase chain
reaction assays for detection ofListeria monocytogenesin clinical cerebro-spinal fluid samples. J. Clin. Microbiol.30:1931–1936.
22. Kalicharan, D., W. L. Jongebloed, L. I. Los, and J. G. F. Worst.1992. (An)aerobic bacteria found in the secondary-cataract material. A SEM/TEM study. Doc. Ophthalmol.82(12):125–133.
23. Kattan, H. M., H. W. Flynn, Jr., S. C. Pflugfelder, C. Robertson, and R. K. Forster.1991. Nosocomial endophthalmitis survey. Current incidence of infection after intraocular surgery. Ophthalmology98:227–238.
24. Kawamura, Y., X. Hou, F. Sultana, K. Hirose, M. Miyake, S. Shu, and T. Ezaki.1998. Distribution ofStaphylococcusspp. among human clinical spec-imens and emended description ofStaphylococcus caprae. J. Clin. Microbiol.
36:2038–2042.
25. Lee, S. E., S. Y. Kim, S. J. Kim, H. S. Kim, J. H. Shin, S. H. Choi, S. S. Chung, and J. H. Rhee.1998. Direct identification ofVibrio vulnificusin clinical specimens by nested PCR. J. Clin. Microbiol.36:2887–2892. 26. Mariani, B. D., M. J. Levine, R. E. Booth, Jr., and R. S. Tuan.1995.
Development of a novel, rapid processing protocol for polymerase chain reaction-based detection of bacterial infections in synovial fluids. Mol. Bio-technol.4:227–237.
27. McClellan, K. A., P. J. Bernard, and F. A. Billson.1989. Microbial investi-gations in keratitis at the Sydney Eye Hospital. Aust. N. Z. J. Ophthalmol.
17:413–416.
28. Okhravi, N., H. M. A. Towler, P. Hykin, M. M. Matheson, and S. Lightman.
1997. Assessment of a standard treatment protocol on visual outcome fol-lowing presumed bacterial endophthalmitis. Br. J. Ophthalmol.81:719–725. 29. Okhravi, N., P. Adamson, M. M. Matheson, H. M. A. Towler, and S. Light-man.PCR/RFLP based detection and speciation of bacteria causing endoph-thalmitis. Investig. Ophthalmol. Vis. Sci., in press.
30. Olive, D. M., and P. Bean.1999. Principles and applications of methods for DNA-based typing of microbial organisms. J. Clin. Microbiol.37:1661–1669. 31. Records, R. E., and P. C. Iwen.1989. Experimental bacterial endophthalmitis
following extracapsular lens extraction. Exp. Eye Res.49:729–737. 32. Rogers, N. K., P. D. Fox, B. A. Noble, K. Kerr, and T. Inglis.1994. Aggressive
management of an epidemic of chronic pseudophakic endophthalmitis: re-sults and literature survey. Br. J. Ophthalmol.78:115–119.
33. Smith, I. L., J. M. Cherrington, R. E. Jiles, M. D. Fuller, W. R. Freeman, and S. A. Spector.1997. High levels of resistance ofcytomegalovirusto ganciclovir is associated with alterations in both the UL97 and DNA polymerase genes. J. Infect. Dis.176:69–77.
34. van Belkum, A.1994. DNA fingerprinting of medically important microor-ganisms by use of PCR. Clin. Microbiol. Rev.7:174–184.
35. Visser, M. R., and A. C. Fluit.1995. Amplification methods for the detection of bacterial resistance genes. J. Microbiol. Methods23:105–116. 36. Wiedbrauk, D. L., J. C. Werner, and A. M. Drevon.1995. Inhibition of PCR
by aqueous and vitreous fluid. J. Clin. Microbiol.33:2643–2646.