Inner-sphere
versus
outer-sphere coordination of BF4
-
in a
NHC-gold(I) complex
Richard M. P. Veenboer,
†Alba Collado,
‡Stéphanie Dupuy,
†Tomas Lebl,
†Laura Falivene,
§Luigi
Cavallo,
§David B. Cordes,
†Alexandra M. Z. Slawin,
†Catherine S. J. Cazin
¶and Steven P. Nolan
*¶¤† EaStCHEM, School of Chemistry, University of St Andrews, North Haugh, St Andrews, KY16 9ST, United Kingdom. ‡ EaStCHEM, School of Chemistry, University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh, EH9 3FJ, United Kingdom.
§ Physical Sciences and Engineering Division, King Abdullah University of Science and Technology, Thuwal, Saudi Arabia ¶ Department of Inorganic and Physical Chemistry, Ghent University, Building S3, Krijgslaan 281, 9000 Gent, Belgium. ¤ Department of Chemistry, College of Science, King Saud University, P. O. Box 2455, Riyadh 11451, Saudi Arabia. Supporting Information Placeholder
ABSTRACT: The role of counterions in chemistry mediated by gold complexes stretches much further than merely providing charge balance to cationic gold species. Interplay between their basicities and coordination strengths influences interactions with both the gold center and substrates in catalysis. Actual monogold(I) active species are generally believed to be mono-coordinated species, formed from the abstraction or the decoordination of a second ligand from precursor complexes, but only little experimental evidence exists to underpin the existence of these transient species. The formation of a bench-stable neutral IPrCl -gold(I) tetrafluoroborate complex is herein reported. Experimental studies by X-ray diffraction analysis, NMR spectroscopy and theoretical studies by DFT calculations were conducted to determine the composition, structure, and behavior of this complex. The absence of an auxiliary ligand resulted in inner-sphere coordination of the counterion in the solid state. In solution, an equilibrium between two conformations was found with the counterion occupying inner-sphere and outer-sphere positions, respectively. Stoichiometric and catalytic reactivity studies with the tetrafluoroborate complex have been conducted. These confirmed the lability of the inner-sphere coordinating counterion that gives the IPrCl-gold(I) fragment similar behavior to related systems.
INTRODUCTION
The use of cationic gold(I) complexes in homogenous catalysis continues to develop at an impressive rate.1 Detailed studies are being conducted to gauge the influence of different ligands (L, L’) and (coordinating) counterions (X) in complexes of general composition [Au(L)(L’)][X] (I) or [Au(L)(X)] (II) on their catalytic activity (Scheme 1).2 Both
N-heterocyclic carbenes (NHCs) and phosphines are widely used as ancillary ligands (L) for these cationic gold catalysts.3 The most frequently encountered auxiliary ligands (L’) coordinate through nitrogen atoms and include nitriles, amines and pyridines.4 The choice of the counterion (X) has been traditionally based on the availability of silver(I) salts AgX (X = OTf, OTs, BF4, PF6, SbF6) that are used to abstract chloride from gold chloride precursors to form cationic gold(I),5 but with the declining use of these reagents,6 and the development of silver-free systems,7 the use of counterions such as phosphate,8 triflate9 and borates10 has been explored as well. Scheme 1.Species involved in gold-catalyzed addition reactions to alkynes. [Au] = Au(L).
Interaction of the counterion with the gold center has repeatedly been found to play a pivotal role in governing the efficiency of catalysis and studies of the cornerstone reactions of gold(I) catalysis, addition of (oxygen-based) nucleophiles to
alkynes, have provided a useful conceptual framework (Scheme 1).11 Decoordination of the auxiliary ligand L’ from a species [Au(L)(L’)][X] (I) would convert the outer-sphere ion-pair to [Au(L)(X)] (II), an inner-sphere ion-pair. In the presence of water12 or alcohol13 and alkyne, the relative basicity of the nucleophile (HOR) and X- governs the equilibrium between [Au(X)]·HOR (III) and catalytically inactive [Au(L)(OR)] (IV) and HX. Sufficiently labile counterions would bring III and alkyne substrate in equilibrium with the alkyne-coordinated outer-sphere ion-pair [Au(L)(alkyne)][X]·HOR (V), the starting point for catalysis. The outer-sphere counterion would now be available as a proton-shuttle to assist in deprotonation of the nucleophile and later in protodeauration to release the organic addition product.14
Scheme 2.Formation of unstabilized species [Au(IPr)(solvent)][X].
With OR2 = THF (A), Et2O (B). X = PF6, BF4. Ar =
2,6-iPr
2C6H3.
Active gold(I) catalysts are often postulated to be monocoordinated outer-sphere ion-pairs with an unspecified second ligand L’, [Au(L)(L’)][X] (VI).15 These structures with an “empty coordination site” on gold are frequently proposed based on the known low dissociation energies of the
I II
IV
V III
VIII
counterions used,12,16 without spectroscopic or structural data to support these claims.17 For these putative species, the second coordination site on the gold(I) center should actually be occupied by any weak ligand such counterions as in [Au(L)(X)] (II) or solvents as in [Au(L)(solvent)][X] (VII).18 Two examples of gold(I) complexes with weakly coordinating ether ligands, [Au(L)(OR2)][X] (VIII) have been studied previously: [Au(IPr)(THF)][PF6]19 and [Au(IPr)(OEt2)][BF4]20 (Scheme 3.2). These species were formed through anion metathesis of [Au(IPrCl)(Cl)] (1) and protonolysis of [Au(IPrCl)(OH)] (2) with silver salt and acid, respectively.NMR studies revealed that [Au(IPr)(OEt2)][BF4] (type VIII) existed in equilibrium with [Au(IPr)(L)][BF4] (of type VI) in CD2Cl2 at 203 K,20 although this latter species would be indistinguishable from [Au(IPr)(CH2Cl2)][BF4] (of general type VII). The low ligand strength of the BF4- anion compared to the solvent molecules (Et2O and CD2Cl2) would prevent its inner-sphere coordination and [Au(IPr)(FBF3)] (type II) would not be likely to exist in solution (Scheme 3). Scheme 3.Hypothetical equilibria of [Au(NHC)(L)]+, BF
4
-and L’ in solution.
[image:2.612.318.534.426.602.2]Attempts to isolate the NHC-gold(I) ether complexes (VIII) or other “monocoordinated” cationic complexes [Au(NHC)(L)][X] (VI) bearing either NHC ligands (e.g.
[Au(IPr)][BF4],20 [Au(ItBu)][BF4]21 and [Au(ITrop)][BArF24]22) or phosphine ligands (e.g. [Au(PPh3)][X], X = BF4, PF6 or SbF6)23 have been described to lead to rapid decomposition. Straub and co-workers have reported the formation of [Au(IPr**)][BArF24].1024 Aldridge and coworkers have explored the abstraction of hydride from less sterically encumbered [Au(6-Dipp)(H)] by B(C6F5)3.25 The transient species [Au(6-Dipp)(HB(C6F5)3)] proved highly unstable and evolved to [Au(6-Dipp)(C6F5)] and borane HB(C6F5)2.
In contrast to the unstable monocoordinated complexes [Au(L)][X] (VI), various examples of stable dicoordinated gold(I) complexes with neutral auxiliary ligands, [Au(L)(L’)][X] (I), have been studied. Examples include those with coordinating molecules of acetonitrile such as [Au(IPr)(NCCH3)][PF6]26 and [Au(PR3)(NCCH3)][SbF6]27 or those with coordinating molecules of substrate such as [Au(IPr)(3-hexyne)][BF4],28 [Au(PR3)(alkene)], [Au(PR3 )-(alkyne)]29,30 or [Au(PR
3)(allene)].31,32 Structural data obtained from X-ray diffraction studies unambiguously confirmed the outer-sphere coordination of the counterions in these complexes and 19F,1H-HOESY and 1H-DOSY NMR experiments have been used to further assess the dynamic behavior of the ion-pairs in solution.28
Examples of inner-sphere coordination of BF4- anions exists for transition-metals other than gold, with literature examples
for cobalt,33 nickel,34 copper,35 zinc,36 rhodium,37 silver,38 and platinum.39 The few reported complexes [M(L)
n(X) n] that bear
at least one NHC ligand are non-coinage metal ones, with palladium (n = 1, X = FBF3),40 ruthenium (n = 2, X = FBF3)41 and indium (n = 1, X = FSbF5)42 centers instead. Monocoordinated neutral complexes of copper(I) and silver(I) with an anionic aryl ligand, [M(2,4,6-Ph3C6H2)] (M = Cu, Ag), have been characterized in the solid state,43 but to the best of our knowledge, evidence for monocoordinated complexes of gold(I) is lacking.
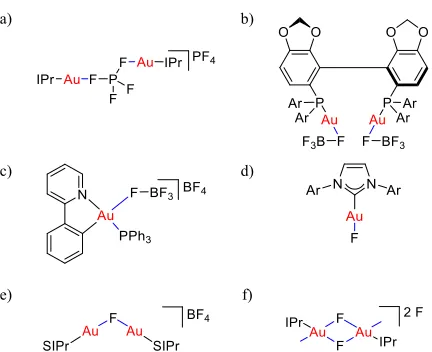
Despite the challenges associated with the isolation of [Au(L)(X)] (II) or [Au(L)][X] (VI) with weakly coordinating counterions (X), various cationic gold(I) species with a 1:1 ratio of [Au(L)]+ and X- without auxiliary ligand L’ have been isolated (Figure 1). For example, attempts to grow crystals of norbonadiene stabilized complex [Au(IPr)(nbd)][PF6] have been described to produce [{Au(IPr)}2(µ-PF4)][PF4] instead (Figure 1a).19 This remarkable complex contains both inner- and outer-sphere PF4- fragments. The digold(I) complexes [{Au(X)}2(DTBM-Segphos)] (X = FBF3, OClO3, Figure 1b)44 and a (C^N)-cyclometalated gold(III) congener (Figure 1c)45 constitute other examples of inner-sphere coordination of the counterions. Coordination of these counterions in the inner coordination sphere of gold proceeds via the formation of an apparent gold-halide bond.46 Indeed, gold-fluorine bond distances (2.0 to 2.3 Å) measured in solid-state structures are comparable to the ones obtained in gold(I) complexes [Au(NHC)(F)] (Figure 1d, 2.071(2) Å)47 and [{Au(SIPr)}2(µ -F)][BF4] (Figure 1e, 2.060(1) Å)48 and gold(III) complexes bearing either NHC ([Au(IPr)(CH3)(µ-F)]2[F]2, Figure 1f, 2.034(3) and 2.124(3) Å),49 phosphine (2.024(5) Å)50 or pincer ligands (2.264(3) Å).51
Figure 1. Selected gold(I) complexes.
a) b)
c) d)
e) f)
Au-F bond distances (Å): a) [{Au(IPr)}2(µ-PF4)][PF4], 2.055(4)
and 2.042(4);19 b) [{Au(FBF
3)}2(DTBM-Segphos)], 2.101(15)
and 2.095(15), Ar = 3,5-di-tert-butyl-4-methoxyphenyl;44 c) [Au(C^N)(PPh3)(FBF3)][BF4], 2.095(4).45 d) [Au(NHC)(F)],
NHC = IPr, 2.071(2);47 NHC = SIPr, 2.0281(17);52 e) [{Au(SIPr)}2(µ-F)][BF4], 2.060(1);48 f) [Au(IPr)(CH3)(µ-F)]2[F]2,
2.034(3) and 2.124(3);49
None of the reported gold complexes containing inner-sphere coordinating counterions (Figure 1a-c) has been studied in great detail. The observation of [{Au(IPr)}2(µ-PF4)][PF4]19 VII
II V
and [{Au(FBF3)}2(DTBM-Segphos)]44 in the solid state suggested that judicious choice of a NHC ligand and fluorinated counterion would permit access to a “monocoordinated” species [Au(NHC)(X)] (of type II) or [Au(NHC)(L)][X] (of type VI) without a specified auxiliary ligand L’.
RESULTS AND DISCUSSION Synthesis of [Au(IPrCl)(FBF
3)]/[Au(IPrCl)(L)][BF4] (3). Based on our experience with NHC-monogold19,53 and (hydroxide bridged) NHC-digold54 complexes containing BF4
counterions, the synthesis of
[Au(NHC)(FBF3)]/[Au(NHC)(L)][BF4]/ (type II/VI) was targeted. The synthesis was first approached by performing metathesis reactions of [Au(NHC)(Cl)] with AgBF4 in CH2Cl2, a poorly coordinating solvent. The use of various NHC ligands (e.g. IPr, IAd, ItBu, IDD, SIPr, IPentCl and IMesCl) did not allow isolation of well-defined complexes, and the formation of [Au(IMes)2][BF4]55 was observed in the reaction from [Au(IMes)(Cl)].56, 57 Starting from [Au(IPrCl)(Cl)] (1) however, an air- and moisture-stable solid product [Au(IPrCl )-(FBF3)]/[Au(IPrCl)(L)][BF4] (3) was obtained (Scheme 4, reaction A).58 No trace of the dicoordinated cationic side-product [Au(IPrCl)
2][BF4] was observed. Complex 3 could also be accessed from the recently developed NHC-gold(I) acetonyl [Au(IPrCl)(CH2COCH3)] (4) complex to avoid the use of silver salts (Scheme 4, reaction B).59
Scheme 4.Synthetic access to 3.
Ar = 2,6-iPr
2C6H3.
Complex 3 proved stable in CDCl3 solution and decomposition was not observed after 15 days at room temperature according to 1H and 19F{1H}-NMR analyses. The solution remained clear and the formation of precipitate was not observed, indicating that decomposition to gold(0) had not occurred.60 Additionally, 3 was found to be soluble in C
6D6, toluene-d8 and DMSO-d6. Solutions of 3 in C6D6, or toluene-d8 could be heated to 120 °C without observable decomposition in 1H and 19F{1H}-NMR spectra.61 Heating of CDCl
3 or DMSO-d6 solutions to 60 °C, however, resulted in decomposition. The formation of [Au(IPrCl)(Cl)] (1) in the former solvent was attributed to release of Cl- from chloroform. In the latter solvent, the well-defined species that formed was tentatively assigned to [Au(IPrCl)
2][BF4].
IR and X-ray studies of 3 in the solid state. The presence of a BF4- ion in the isolated complex was initially supported by the observation of a characteristic B-F stretching band (at 1059 cm-1) in the solid-state IR spectrum of 3.62 The diffraction study confirmed the expected stoichiometry and charge balance in 3 (Figure 2).63 One of the fluorine atoms of the BF4 unit pointed to the gold atom of the IPrCl-Au fragment, while another fluorine points to a chlorine atom of the backbone of a second IPrCl-Au fragment. The gold-fluorine
distance of 2.027(7) Å was significantly shorter than the sum of the Van der Waals radii of 3.13 Å for gold and fluorine atoms64 and similar to that of the previously reported complexes with gold-fluorine linkages (Figure 1). In the solid state, 3 was thus found to be best described as inner-sphere complex[Au(IPrCl)(FBF
3)] (3-in), rather than as outer-sphere complex [Au(IPrCl)(L)][BF
4] (3-out). The BF4 moiety was found to be no longer tetrahedral and the B1-F1 distance (1.495(18) Å) is larger than that of the other B-F bonds (average of 1.38 Å). The carbenic carbon-gold distance of 1.960(9) Å is close to that of [Au(IPrCl)(NTf2)] (15) (1.967(4) Å)7c and similar to that of other cationic NHC-gold complexes.26 Torsion angles C3-N2-C1-Au1 and C4-N5-C1-Au1 of -179.1(6)° and 179.6(6)°, different angles N2-C1-C4-N5-C1-Au1 and N5-C1-Au1 of 128.5(6)° and 125.8(6)° as well as different torsion angles C1-N2-C6-C7 and C1-N5-C18-C19 of 88.8(10)° and -79.6(11)° are all indicative of a slightly unsymmetrically twisted geometry of the IPrCl ligand. A co-crystallized molecule of acetone was found in the crystal lattice, but large distances of the constituting atoms to 3 (at least 3 Å) indicated that interaction with the gold complex were absent. An alternative assignment of [Au(IPrCl)(FBF
3)] (3-in) as isoelectronic hydroxyfluoroborate complex [Au(IPrCl)(OH)(BF3)] (5-in) could be refuted.61
Figure 2. Solid-state structure of 3·((CH3)2CO). One fragment of [Au(IPrCl)(FBF
3)] (3-in) from the unit cell is shown. Thermal ellipsoids are set at the 50% probability level. Hydrogen atoms, a molecule of acetone and the minor component of disorder in F2, F3 and F4 are omitted for clarity.
NMR studies of 3 in solution. The behavior and reactivity of 3 in CDCl3 solution were studied by means of NMR spectroscopy at ambient temperature (295 K). 1H-NMR and 19F{1H}-NMR spectra of isolated 3 and of crystals 3·((CH3)2CO) matched, confirming that the crystals had the same composition as the isolated material. The 1H-NMR spectra showed two sets of isopropyl methyl groups with non-equivalent intensities, indicative of an equilibrium between different species. Similarly, two distinct sets of signals were present in both 19F{1H}-NMR and 11B-NMR spectra (Figure 3),65 suggesting that the BF
4- occupied different sites in solution as in [Au(IPrCl)(FBF
3)] (3-in) and [Au(IPrCl)(L)][BF4] (3-out). No other signals (e.g. that expected for gold fluoride) were detected.
1
a)
[image:4.612.66.282.47.303.2]b)
Figure 3. NMR spectra of 3 in CDCl3 (4 mM) at ambient temperature. a) 19F{1H}-NMR spectrum. Inset shows area between -149.6 and -149.8 ppm. b) 11B-NMR spectrum in CDCl3 (12 mM) at ambient temperature.
Both sets of fluorine signals displayed the characteristic split with 1:4 ratio expected from the natural abundance of 10B:11B. Furthermore, characteristic splitting patterns for heteronuclear coupling to these nuclei with spin 3 and 3/2 (5.2 Hz), respectively, in the signals at lower field confirms bonding between fluorine and boron. Likewise, in the 11 B-NMR spectrum, a broadened set of signals was observed for the signals around -0.7 ppm (about 4.7 Hz) while the resonance at -1.4 ppm appeared as a sharp singlet (Figure 3b). Observation of coupling for the lower field signals and not for the higher field signals supported the assignment of the signals around -1.4 and -0.7 ppm to outer and inner sphere coordination, respectively.66 Additional coupling was not observed in addition to the heteronuclear F-B coupling. Similar chemical shifts of the signals at higher field in both 11
B-NMR (-1.4 ppm) and 19F{1H}-NMR (-153.2 ppm) spectra (Figure 3) to those of [Au(IPr)(NCCH3)][BF4] (1.1 ppm and -153.7 ppm, in CDCl3 at ambient temperature)19 also supported the assignment of these signals to outer-sphere coordinating BF4- (3-out). Absence of an auxiliary ligand L’ in the CDCl3 solution of 3 would suggest that 3-out would actually exist as solvent-separated ion-pair [Au(IPrCl)(CDCl
3)][BF4] (6) (Scheme 3).
The carbenic carbon 13C NMR chemical shifts are indicative of the environment around [Au(NHC)(L)] fragments and have been used to predict the Lewis acidity of gold complexes.67,68 Correspondingly, a shift from 175.1 ppm (in CDCl3) for [Au(IPrCl)(Cl)] (1)69 to higher fields of 166.4 ppm (in CDCl
3) for [Au(IPrCl)(NCCH3)][BF4] (7), 163.0 ppm (in CD2Cl2) for [{Au(IPrCl)}
2(µ-OH)][BF4] (8) or 168.4 ppm (in CDCl3) for [Au(IPrCl)(NTf
2)] (15)70 agreed nicely with the known Lewis
and acidity of the latter three cationic complexes.22,53,71
Carbenic 13C chemical shifts of [Au(NHC)(Cl)] (for SIPr, δ =
196.4 ppm in CD2Cl2)72 shift only by about 2-5% when compared to the corresponding [Au(NHC)(F)] (for IPr, δ = 172.7,73 for SIPr, δ = 185.9,52 both in CD2Cl2). While not determined experimentally, a carbenic 13C chemical shift for [Au(IPrCl)(F)] would thus be expected to reside in the range of 166 to 172 ppm. The carbenic 13C chemical shift of 3 appeared at 163.2 ppm and indicated that this complex would be more Lewis acidic than 7 consistent with a gold-complex containing a loosely bound BF4- ion rather than one with tightly bound fluoride as in [Au(IPrCl)(F)].
Dynamic behavior of 3 in solution. The presumed existence of 3 in CDCl3 as a mixture of species with inner-sphere coordinating and outer-inner-sphere coordinating BF4- anions raised the question as to whether these species existed as monomeric entities and whether they would interconvert.The presence of chlorine substituents in the IPrCl ruled out the option to perform 19F,1H-HOESY NMR spectroscopy that might have supported the outer-sphere coordination of BF4 -(close to the NHC backbone) in [Au(IPrCl)(L)][BF
4] (3-out) or [Au(IPrCl)(CDCl3)][BF4] (6). 1H-DOSY NMR experiments were performed to permit assignment of the single set of IPrCl proton resonances in the 1H-NMR spectrum to either a monomeric or multinuclear species.61 Based on the resonance of the C-H isopropyl signal (at 1.29 ppm), a diffusion constant of 8.87×1010 m2s-1 was measured (in CDCl3), which was in the same order of magnitude as the previously reported for [Au(IPr)(OH)] (8.86×1010 m2s-1, in THF-d
8).74 These values correspond to a hydrodynamic radius of 7.0 Å, clearly in agreement with mononuclear NHC-gold species.
The presumed reversible displacement of BF4- from [Au(IPrCl)(FBF
3)] (3-in) by a solvent molecule in a CDCl3 solution of 3 to form solvent-separated ion-pair [Au(IPrCl)(CDCl
3)][BF4] (6) was studied by variable temperature 19F-EXSY NMR experiments. Cross-peaks between the signals around -149 ppm and -152 ppm, confirmed the reversibility of this process and both the associated thermodynamic and kinetic parameters were determined from relative signal intensities at different temperatures (Table 1). The Van 't Hoff analysis gave negative values for ΔH and ΔS (Table 1, entries 1-2), corresponding to a Gibbs free energy change ΔG of only 1.0 kcal·mol−1 at 298.15 K. These values were indicative of slightly more favorable inner-sphere coordination of BF4- than outer-sphere
in lieu of a stronger auxiliary ligand. The negative entropy of activation could be explained by the higher order in the state with two solvated ions [Au(IPrCl)]+ and BF
Table 1.Thermodynamic and kinetic parameters for the equilibrium between 3-in and 6.[a]
Entry Direction Parameter Value Units
1 k1
ΔH −22.8±2.2 kcal/mol
2 k1
ΔS −5.7±1.0 kcal/mol/K
3 k1
ΔH‡ 29.9±4.0 kcal/mol
4 k1
ΔS‡ 40.8±6.0 kcal/mol/K
5 k-1
ΔH‡ 28.6±7.0 kcal/mol
6 k-1
ΔS‡ 38.6±11.0 kcal/mol/K
[a] Obtained from measurements of 3 in CDCl3 at 12 mM concentration over a temperature range of 285-310 K (with 5 K intervals).
Stoichiometric reactivity of 3. The reactivity of 3 was studied by subjecting it to different substrates that are known to coordinate to electron-deficient gold(I) centers (Scheme 5).61 Acetonitrile was found to smoothly displace the BF
4- to form [Au(IPrCl)(NCCH3)][BF4] (7) (Scheme 5, reaction A). The formation of [{Au(IPrCl)}
2(µ-OH)][BF4] (8) from hydrolysis of 3 was studied (Scheme 5, reaction B). The reaction of metal tetrafluoroborate complexes (e.g. of Mn, Re, W, Cr, Fe, Os, Pt, Zr, Ti) with water had been described to form metal-aqua complexes [M(L)n(OH2)m][BF4],15b and the intermediacy of aquo-species [Au(NHC)(OH2)][BF4] with subsequent formation of {Au(NHC)}2(µ-OH)][BF4] has been computed to be a favorable pathway.14 As expected from this precedent, an equilibrium situation was observed between 3 and 8 when water was added to a sample of 3 in CDCl3.75 When 3 was mixed with [Au(IPrCl)(OH)] (2),7b complex 8 formed predominantly, consistent with the 2:1 stoichiometry of gold to BF4- in the mixture.
Scheme 5.Displacement of BF4- from 3.
Coordination of arenes to cationic phosphine-gold,27 and NHC-gold complexes has previously been achieved as in [Au(L)(η-arene)][X]76 and [Au(IPr)(π-Ph-C≡C-Ph)][BF4]
complex has been demonstrated to be an air stable compound.77 The facile displacement of BF
4- from 3 (by neutral solvent molecules) prompted us to test whether η -arene and π-alkyne complexes could be formed from reactions of 3 as well. The expected species [Au(IPrCl)(η -benzene)][BF4] (10) was found to be in equilibrium with 3-in when 3 was dissolved in benzene (Scheme 5, reaction C). The signal in the 19F{1H}-NMR spectrum assigned to 10, however, was not observed when a stoichiometric amount of benzene was added to a solution of 3 in CDCl3. Addition of a stoichiometric amount of diphenylacetylene (11) to a CDCl3 solution of 3, however, did not form [Au(IPrCl)(
π-Ph-C≡ C-Ph)][BF4] (12) (Scheme 5, reaction D). A protonolysis reaction of 4 with in HBF4·OEt2 the presence of 11 in CDCl3 gave a mixture that appeared to contain 12, but it was found to rapidly decompose to 1,2-diphenylethan-1-one (14), demonstrating the higher reactivity of the [Au(IPrCl)]+ fragment when compared to the [Au(IPr)]+ analogue.
Catalytic activity of [Au(IPrCl)][BF
4]. The predicted high Lewis acidity of 3 (from the 13C-NMR chemical shift of the carbene) and its facile conversion to both acetonitrile complex [Au(IPrCl)(NCCH3)][BF4] (7) and digold hydroxide complex [{Au(IPrCl)}
2(µ-OH)][BF4] (8), suggested that it could be used as a catalyst in transformations that are known to be mediated by those complexes. Various reactions that proceed through π -activation of triple bonds or by σ-activation of benzylic alcohols were selected: alkyne hydration, hydroalkoxylation with various alcohols and dehydrative etherification. Next to comparison of the activities of complexes 3, 7 and 8 with BF4 -counterions the neutral triflimide complex [Au(IPrCl)(NTf
2)] (15) was also tested to allow for comparison of results of complexes with different anions.7a,23 Catalyst system AgBF4/[Au(IPrCl)(Cl)] (1) was included in the benchmarking studies to provide an example of an in situ generated species.61 Catalytic activity of complex 3 in all these transformations confirmed the catalytic relevance of this gold(I) species with an inner-sphere coordinating counterion. Additional stoichiometric reactions of 3 seemed to indicate that BF4- was a better ligand for [Au(IPrCl)]+ than alcohols and species [Au(IPrCl)(OHR)][BF4] were not observed.
Computational modelling studies. To support the experimental observations, modelling studies using DFT implemented in the Gaussian09 package78 with the BP86 GGA functional79 were conducted.61 Benchmarking studies established that the important consideration of solvent effects with the charged species involved through use of the PCM solvation model for CH2Cl2 permitted predictions to be made with an accuracy of approximately 5 kcal·mol-1. Negative Gibbs free energy values of -10.0 and -9.1 kcal·mol-1 for displacement of BF4- from 3-in by acetonitrile or [Au(IPrCl)(OH)] (2) to form [Au(IPrCl)(NCCH
3)][BF4] (7)or [{Au(IPrCl)}
2(µ-OH)][BF4] (8) were in agreement with the experimental observations (Scheme 5). Small absolute Gibbs free energy values (< 1.0 kcal·mol-1) for reactions of 3-in with diphenylacetylene (11) or methanol could not be used to support the corresponding experimental results. A large positive Gibbs free energy value of 6.5 kcal·mol-1 for displacement of BF4- from 3-in by phenol agreed with the experimental result.
10 12
8 7
3
3-in 6
Scheme 6.Reactions of 3-in with various compounds.
[a] Association of two molecules of 3-in..[b] Dissociation of BF 4
-from 3-in. Free energy changes (PCM, dichloromethane) are given in kcal·mol-1 in parentheses. Shaded areas highlight
spontaneous reactions (green), non-spontaneous reactions (red) and those were firm assignment could not be made (gray).
Favorable existence of 3 as [Au(IPrCl)(FBF
3)] (3-in) rather than as outer-sphere species such as [Au(IPrCl)(L)][BF
4] (3 -out) or solvent-separated ion-pair [Au(IPrCl)(CH2Cl2)][BF4] (9) also followed from positive calculated Gibbs free energy values of 9.8 and 6.8 kcal·mol-1.80 This first value was nearly as large as the calculated Gibbs free energy value for association of two equivalents of 3-in to form [{Au(IPrCl)}
2(BF4)][BF4] (13) of 10.4 kcal·mol-1. Even though dissociation of BF4- from 3-in remained disfavored, coordination of a solvent molecule incurred some stability relative to 3-out with an empty coordination site on gold. These data were in qualitative agreement with the experimental results that predicted a slightly unfavorable reaction of 3-in to [Au(IPrCl)(CDCl3)][BF4] (6) (∆G = 1.0 kcal·mol-1) and the existence of the [Au(IPrCl)]+ fragment as a monomeric species in solution.
CONCLUSIONS
Having capitalized on the apparently remarkably more stable gold(I) complexes bearing the IPrCl ligand compared to those bearing other NHC ligands, a stable species containing the [Au(IPrCl)]+ fragment was found to exist in the presence of a tetrafluoroborate counterion as a stable species [Au(IPrCl )-(FBF3)]/[Au(IPrCl)(L)][BF4] (3) without the need for an auxiliary stabilizing ligand. Even though this complex resembled a neutral gold-fluoride in the solid state, the BF4 -anion remained labile and complex 3 could be considered an equilibrium between neutral and cationic gold species in solution. A series of NMR experiments and computational modelling studies have unveiled a process that interconverts inner-sphere and outer-sphere coordinating counterion in these two conformations and confirmed the favorable coordination of solvent molecules in the coordinatively unsaturated complex where the BF4- anion resided in the outer-sphere. Both stoichiometric and catalytic studies have shown that the [Au(IPrCl)]+ fragment in complex 3 retained a similar behavior to that is known for other gold(I) complexes that bear that IPrCl ligand. The ability of a weakly coordinating counterion to coordinate to a cationic gold(I) fragment in the absence of better auxiliary ligands should be considered in the design of future catalyst systems. Future studies will be devoted to gaining an understanding of the stability of the complex bearing the IPrCl ligand relative to congeners bearing other NHC ligands. The mechanism of activation of different types of substrates such as alkynes and alcohols by complex 3 is subject of extended investigations as well.
EXPERIMENTAL SECTION
Procedure for synthesis of 3. To a stirred solution of [Au(IPrCl)(CH
2COCH3)] (4) in dichloromethane (1.4 M) was added,
at room temperature in air, a solution of HBF4·OEt2 (50% in diethyl
ether, 1.05 equivalents). After 30 minutes, the solution was filtered over MgSO4 with additional dichloromethane (3 times initial reaction
volume). Part of the solvent was removed under vacuum (about halve of the initial reaction volume remained) and the product was precipitated by addition of pentane (about the initial reaction volume). The product was collected by filtration, washed with additional pentane (about twice the initial reaction volume) and dried under high vacuum to give a microcrystalline solid in quantitative yield. 1
H-NMR (500 MHz, CDCl3): δ 7.60 (t, 3J(H,H) = 7. 7 Hz, 2H; p-PhC),
7.36 (d, 3J(H,H) = 7.7 Hz, 4H; m-PhC), 2.39 (h, 3J(H,H) = 6.8 Hz,
4H; CH), 1.34 (d, 3J(H,H) = 6.8 Hz, 12H; CH3), 1.27 (d, 3J(H,H) =
6.8 Hz, 12H; CH3). 13C{1H}-NMR (126 MHz, CDCl
3): δ 163.2 (1C; Ccarb), 146.1 (4C; o-PhC), 132.1 (4C; i-PhC), 130.8 (2C; p-PhC), 124.9 (4C; m-PhC), 119.6 (2C; C), 29.3 (4C; CH), 24.5 (4C; CH3),
23.7 (4C; CH3). 11B-NMR (128 MHz, CDCl3): δ -0.70 (br), -1.40 (s). 19F{1H}-NMR (470 MHz, CDCl
3): δ -149.6 (br), -149.7 (q, 1J(F,B) =
5.2 Hz), -153.2 (s), -153.3 (s). Anal. calcd for C29H37AuBCl2F4N3: C,
43.75; H, 4.62; N, 3.78. Found: C, 43.64; H, 4.70; N, 3.74. FTIR (ATR): ṽ = 1059 cm-1 (B-F).
ASSOCIATED CONTENT Supporting Information
General information, crystallographic information, characterization data for stoichiometric and catalytic experiments, DFT calculations and Cartesian coordinates for calculated structures. This material is available free of charge via the Internet at http://pubs.acs.org. CCDC 1514529 (3) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
ACKNOWLEDGMENT
The European Research Council (ERC) and the Engineering and Physical Sciences Research Council (EPSRC) are gratefully acknowledged for their support. S. P. N. and L. C. thank King Abdullah University of Science and Technology (KAUST) for support. Dr. Filippo Stella is thanked for his help with NMR experiments. This publication is based upon work supported by the King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) under Award No. OSR-2015-CCF-1974-03.
11
AUTHOR INFORMATION
Corresponding Author
* E-mail: [email protected]. Tel: +32 (0) 9264 4458. Fax: +32 (0) 9264 4983.
Note
The authors declare no competing financial interests.
ORCID
Richard M. P. Veenboer: 0000-0002-4878-580X Alba Collado: 0000-0001-6215-1822
Tomas Lebl: 0000-0002-0269-3221
Laura Falivene: 0000-0003-1509-6191 Luigi Cavallo: 0000-0002-1398-338X David B. Cordes: 0000-0002-5366-9168 Alexandra M. Z. Slawin: 0000-0002-9527-6418 Catherine S. J. Cazin: 0000-0002-9094-8131 Steven P. Nolan: 0000-0001-9024-2035
ABBREVIATIONS
BArF24 = tetrakis(3,5-bis(trifluoromethyl)phenyl)borate; COSY =
correlation spectroscopy; 6-Dipp = 1,3-bis(2,6-diisopropylphenyl)-3,4,5,6-tetrahydropyrimidinylidene; DOSY = diffusion-ordered spectroscopy; DTBM-Segphos: 5,5’-bis[di(3,5-di-tert-butyl-4-methoxyphenyl)phosphino]-4,4′
-bi-1,3-benzodioxole; EXSY = exchange spectroscopy; HOESY = heteronuclear NOESY; IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene; ItBu =
1,3-ditert-butylimidazol-2-ylidene IPrCl: 4,5
dichloro-1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene; IPr** = 1,3-bis{2,6- bis[bis-(4-tert-butylphenyl)methyl]-4-methylphenyl}-2,3-dihydro-1H-imidazol-2-ylidene; IMe = 1,3-dimethylimidazol-2-ylidene; IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene. ITrop = 1,3-bis(5H-dibenzo[a,d]cyclohepten-5-yl)imidazol-2-ylidene; nbd = norbornadiene; NMR = nuclear magnetic resonance; NHC = N-hetereocyclic carbene; NOESY = NOE spectroscopy; NOE = nuclear Overhauser effect; SIPr: 1,3-bis-(2,6-diisopropylphenyl)imidazolin-2-ylidene; Tf = trifluoromethane-sulfonate; TFA = trifluoroacetic acid; Ts = tosylate.
REFERENCES
1. (a) Hashmi, A. S. K., Gold Bull. 2004, 37, 51-65; (b) Hashmi, A. S. K.; Hutchings, G. J., Angew. Chem. Int. Ed. 2006,45, 7896-7936; (c) Hashmi, A. S. K., Chem. Rev. 2007,107, 3180-3211; (d) Raubenheimer, H. G.; Cronje, S., Chem. Soc. Rev. 2008,37, 1998-2011; (e) Lin, J. C. Y.; Huang, R. T. W.; Lee, C. S.; Bhattacharyya, A.; Hwang, W. S.; Lin, I. J. B., Chem. Rev. 2009,109, 3561-3598; (f) Rudolph, M.; Hashmi, A. S. K., Chem. Soc. Rev. 2012, 41, 2448-2462; (g) Leyva-Pérez, A.; Corma, A., Angew. Chem. Int. Ed. 2012,
51, 614-635; (h) Raubenheimer, H. G.; Schmidbaur, H., J. Chem.
Educ. 2014,91, 2024-2036; (i) Hashmi, A. S. K., Acc. Chem. Res.
2014,47, 864-876; (j) Friend, C. M.; Hashmi, A. S. K., Acc. Chem.
Res. 2014,47, 729-730; (k) Dorel, R.; Echavarren, A. M., Chem. Rev.
2015,115, 9028-9072; (l) Pflästerer, D.; Hashmi, A. S. K., Chem.
Soc. Rev. 2016,45, 1331-1367.
2. (a) Zuccaccia, D.; Belpassi, L.; Rocchigiani, L.; Tarantelli, F.; Macchioni, A., Inorg. Chem. 2010,49, 3080-3082; (b) Biasiolo, L.; Del Zotto, A.; Zuccaccia, D., Organometallics 2015,34, 1759-1765.
3. (a) Marion, N.; Nolan, S. P., Chem. Soc. Rev. 2008,37, 1776-1782; (b) Gorin, D. J.; Sherry, B. D.; Toste, F. D., Chem. Rev.
2008,108, 3351-3378; (c) Díez-González, S.; Marion, N.; Nolan, S.
P., Chem. Rev. 2009,109, 3612-3676; (d) Gaillard, S.; Cazin, C. S. J.;
Nolan, S. P., Acc. Chem. Res. 2011,45, 778-787.
4. Nun, P.; Gaillard, S.; Slawin, A. M. Z.; Nolan, S. P., Chem.
Commun. 2010,46, 9113.
5. (a) Peone, J.; Vaska, L., Angew. Chem. Int. Ed. 1971,10, 511-512; (b) Bond, G. C., Gold Bull. 1972, 5, 11-13; (c) Ito, Y.; Sawamura, M.; Hayashi, T., J. Am. Chem. Soc. 1986,108, 6405-6406; (d) Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N., Chem. Lett.
1987,16, 405-408; (e) Preisenberger, M.; Schier, A.; Schmidbaur, H.,
J. Chem. Soc., Dalton Trans. 1999, 1645-1650.
6. Jia, M.; Bandini, M., ACS Catal. 2015,5, 1638-1652. 7. (a) Ricard, L.; Gagosz, F., Organometallics 2007, 26, 4704-4707; (b) Gaillard, S.; Slawin, A. M. Z.; Nolan, S. P., Chem.
Commun. 2010,46, 2742-2744; (c) Patrick, S. R.; Gómez-Suárez, A.;
Slawin, A. M. Z.; Nolan, S. P., Organometallics 2013,33, 421-424; (d) Ramón, R. S.; Gaillard, S.; Poater, A.; Cavallo, L.; Slawin, A. M. Z.; Nolan, S. P., Chem. Eur. J. 2011,17, 1238-1246.
8. Gaillard, S.; Rix, D.; Slawin, A. M. Z.; Lacour, J.; Nolan, S. P., Dalton Trans. 2012,41, 8235.
9. (a) Hirner, J. J.; Faizi, D. J.; Blum, S. A., J. Am. Chem. Soc.
2014,136, 4740-4745; (b) Jaroschik, F.; Simonneau, A.; Lemière, G.; Cariou, K.; Agenet, N.; Amouri, H.; Aubert, C.; Goddard, J.-P.; Lesage, D.; Malacria, M.; Gimbert, Y.; Gandon, V.; Fensterbank, L.,
ACS Catal. 2016,6, 5146-5160.
10. Weber, S. G.; Zahner, D.; Rominger, F.; Straub, B. F.,
Chem. Commun. 2012,48, 11325.
11. Biasiolo, L.; Trinchillo, M.; Belanzoni, P.; Belpassi, L.; Busico, V.; Ciancaleoni, G.; D'Amora, A.; Macchioni, A.; Tarantelli, F.; Zuccaccia, D., Chem. Eur. J. 2014,20, 14594-14598.
12. Trinchillo, M.; Belanzoni, P.; Belpassi, L.; Biasiolo, L.; Busico, V.; D’Amora, A.; D’Amore, L.; Del Zotto, A.; Tarantelli, F.; Tuzi, A.; Zuccaccia, D., Organometallics 2016,35, 641-654. 13. Ciancaleoni, G.; Belpassi, L.; Zuccaccia, D.; Tarantelli, F.; Belanzoni, P., ACS Catal. 2015,5, 803-814.
14. Gatto, M.; Belanzoni, P.; Belpassi, L.; Biasiolo, L.; Del Zotto, A.; Tarantelli, F.; Zuccaccia, D., ACS Catal. 2016, 7363-7376. 15. (a) Rosenthal, M. R., J. Chem. Educ. 1973,50, 331; (b) Beck, W.; Suenkel, K., Chem. Rev. 1988,88, 1405-1421; (c) Strauss, S. H., Chem. Rev. 1993,93, 927-942; (d) Macchioni, A., Chem. Rev.
2005,105, 2039-2074.
16. Kovács, G.; Ujaque, G.; Lledós, A., J. Am. Chem. Soc.
2008,130, 853-864.
17. (a) Hashmi, A. S. K., Angew. Chem. Int. Ed. 2010,49, 5232-5241; (b) Ranieri, B.; Escofet, I.; Echavarren, A. M., Org.
Biomol. Chem. 2015,13, 7103-7118.
18. Bartolomé, C.; Ramiro, Z.; Peñas-Defrutos, M. N.; Espinet,
P., ACS Catal. 2016, 6537-6545.
19. de Frémont, P.; Marion, N.; Nolan, S. P., J. Organomet.
Chem. 2009,694, 551-560.
20. Gaillard, S.; Bosson, J.; Ramón, R. S.; Nun, P.; Slawin, A. M. Z.; Nolan, S. P., Chem. Eur. J. 2010,16, 13729-13740.
21. Baker, M. V.; Barnard, P. J.; Brayshaw, S. K.; Hickey, J. L.; Skelton, B. W.; White, A. H., Dalton Trans. 2005, 37-43. 22. Brill, M.; Collado, A.; Cordes, D. B.; Slawin, A. M. Z.; Vogt, M.; Grützmacher, H.; Nolan, S. P., Organometallics 2015,34, 263-274.
23. Mézailles, N.; Ricard, L.; Gagosz, F., Org. Lett. 2005,7, 4133-4136.
24. The NMR studies showed that BArF24- remained
outer-sphere and stabilization of the cationic gold center was described to originate from the interaction of the encapsulating ligand instead. 25. Phillips, N.; Dodson, T.; Tirfoin, R.; Bates, J. I.; Aldridge,
S., Chem. Eur. J. 2014,20, 16721-16731.
26. de Frémont, P.; Stevens, E. D.; Fructos, M. R.; Mar Díaz-Requejo, M.; Pérez, P. J.; Nolan, S. P., Chem. Commun. 2006, 2045-2047.
27. Herrero-Gómez, E.; Nieto-Oberhuber, C.; López, S.; Benet-Buchholz, J.; Echavarren, A. M., Angew. Chem. 2006, 118, 5581-5585.
28. (a) Akana, J. A.; Bhattacharyya, K. X.; Müller, P.; Sadighi, J. P., J. Am. Chem. Soc. 2007,129, 7736-7737; (b) Ciancaleoni, G.; Biasiolo, L.; Bistoni, G.; Macchioni, A.; Tarantelli, F.; Zuccaccia, D.; Belpassi, L., Organometallics 2013,32, 4444-4447.
29. Shapiro, N. D.; Toste, F. D., Proc. Natl. Acad. Sci. 2008,
105, 2779-2782.
30. The alkyne tethered phosphine ligand, diphenyl(4-(triisopropylsilyl)but-3-yn-1-yl)phosphane, coordinates to two distinct gold centers, producing an A-frame type complex. The alkene tethered phosphine ligand, (4-methylpent-3-en-1-yl)diphenylphosphane, also coordinates to two distinct gold centeres, producing linkage polymers.
31. Brown, T. J.; Sugie, A.; Dickens, M. G.; Widenhoefer, R.
A., Organometallics 2010,29, 4207-4209.
32. PR3 = (2-biphenyl)di-tert-butylphosphine, allene = 3-methylbuta-1,2-diene.
33. Flörke, U.; Hoffmann, A.; Henkel, G., CCDC 1452942: Experimental Crystal Structure Determination, 2016, DOI: 10.5517/cc1krx3b.
34. Herbert, D. E.; Lara, N. C.; Agapie, T., Chem. Eur. J. 2013,
19, 16453-16460.
35. Rossin, A.; Tuci, G.; Giambastiani, G.; Peruzzini, M.,
ChemPlusChem 2014,79, 406-412.
36. Döring, A.; Flörke, U.; Hoffmann, A.; Jones, M. D.; Kuckling, D.; Michaelis de Vasconcellos, J.; Herres-Pawlis, S., Z.
Anorg. Allg. Chem. 2015,641, 2147-2156.
37. Hiyam Salem, David Milstein, Linda JW Shimon CCDC 668208: Experimental Crystal Structure Determination, 2007, DOI: 10.5517/ccdc.csd.ccqfb3k
38. Maier, J. M.; Li, P.; Hwang, J.; Smith, M. D.; Shimizu, K.
D., J. Am. Chem. Soc. 2015,137, 8014-8017.
39. Friedemann, R.; Seppelt, K., Eur. J. Inorg. Chem. 2013,
2013, 1197-1206.
40. Chernyshova, E. S.; Goddard, R.; Pörschke, K.-R.,
Organometallics 2007,26, 3236-3251.
41. Fawcett, J.; Harding, D. A. J.; Hope, E. G.; Singh, K.; Solan, G. A., Dalton Trans. 2009, 6861.
42. Michelet, B.; Colard-Itté, J.-R.; Thiery, G.; Guillot, R.; Bour, C.; Gandon, V., Chem. Commun. 2015,51, 7401-7404. 43. Lingnau, R.; Strähle, J., Angew. Chem. Int. Ed. 1988,27, 436-436.
44. Abadie, M.-A.; Trivelli, X.; Medina, F.; Capet, F.; Roussel, P.; Agbossou-Niedercorn, F.; Michon, C., ChemCatChem 2014, 6, 2235-2239.
45. Tlahuext-Aca, A.; Hopkinson, M. N.; Daniliuc, C. G.; Glorius, F., Chem. Eur. J. 2016,22, 11587-11592.
46. Krossing, I.; Raabe, I., Chem. Eur. J. 2004,10, 5017-5030. 47. Mankad, N. P.; Toste, F. D., Chem. Sci. 2012,3, 72-76. 48. Wyss, C. M.; Tate, B. K.; Bacsa, J.; Wieliczko, M.; Sadighi, J. P., Polyhedron 2014,84, 87-95.
49. Mankad, N. P.; Toste, F. D., J. Am. Chem. Soc. 2010,132, 12859-12861.
50. Winston, M. S.; Wolf, W. J.; Toste, F. D., J. Am. Chem.
Soc. 2015,137, 7921-7928.
51. Kumar, R.; Linden, A.; Nevado, C., Angew. Chem. Int. Ed.
2015,54, 14287-14290.
52. Laitar, D. S.; Müller, P.; Gray, T. G.; Sadighi, J. P.,
Organometallics 2005,24, 4503-4505.
53. Veenboer, R. M. P.; Nolan, S. P., Green Chem. 2015,17, 3819-3825.
54. (a) Gómez-Suárez, A.; Oonishi, Y.; Meiries, S.; Nolan, S.
P., Organometallics 2013,32, 1106-1111; (b) Biannic, B.; Aponick,
A., Eur. J. Org. Chem. 2011,2011, 6605-6617; (c) Veenboer, R. M.
P.; Dupuy, S.; Nolan, S. P., ACS Catal. 2015, 5, 1330-1334; (d) Dupuy, S.; Gasperini, D.; Nolan, S. P., ACS Catal. 2015,5, 6918-6921.
55. Möhlmann, L.; Wendt, O. F.; Johnson, M. T., Acta
Crystallogr. Sect. E 2011,67, m719-m720.
56. (a) Marion, N.; Ramón, R. S.; Nolan, S. P., J. Am. Chem.
Soc. 2009,131, 448-449; (b) Marion, N.; Gealageas, R.; Nolan, S. P.,
Org. Lett. 2007,9, 2653-2656.
57. Such complexes are known to be catalytically inactive and their facile formation under these general conditions explains why IMes ligands are only rarely used in homogeneous catalysis compared to the slightly larger IPr ligand.
58. Cordón, J.; López-de-Luzuriaga, J. M.; Monge, M.,
Organometallics 2016,35, 732-740.
59. Gasperini, D.; Collado, A.; Goméz-Suárez, A.; Cordes, D. B.; Slawin, A. M. Z.; Nolan, S. P., Chem. Eur. J. 2015,21, 5403-5412.
60. Kumar, M.; Jasinski, J.; Hammond, G. B.; Xu, B., Chem.
Eur. J. 2014,20, 3113-3119.
61. Full results and details are given in the Electronic Supporting Information.
62. Amster, R. L.; Taylor, R. C., Spectrochim. Acta 1964,20, 1487-1502.
63. Hydrolysis of BF4- anions to afford HOBF3 units is known.
In order to rule out the formation of this species, further analyses were carried out. See ESI for a detailed discussion.
64. Bondi, A., J. Phys. Chem. 1964,68, 441-451.
65. No additonal splitting of signals was detected in non-proton decoupled fluorine-NMR spectra.
66. It could be assumed that the more symmetrical environment of the boron nucleus in the outer coordination sphere would impede the quadrupole relaxation that enabled the observation of hetero-nuclear coupling in the inner-sphere coordinating counterion.
67. (a) Herrmann, W. A.; Runte, O.; Artus, G., J. Organomet.
Chem. 1995,501, C1-C4; (b) de Frémont, P.; Singh, R.; Stevens, E.
D.; Petersen, J. L.; Nolan, S. P., Organometallics 2007,26, 1376-1385; (c) Tapu, D.; Dixon, D. A.; Roe, C., Chem. Rev. 2009,109, 3385-3407.
68. Upfield shifts indicate a lower electron density at the carbenic carbon atom associated with a higher net charge transfer of the ligand to the gold atom as dictated by the electron deficiency of the more Lewis acidic gold atom.
69. Gaillard, S.; Slawin, A. M. Z.; Bonura, A. T.; Stevens, E. D.; Nolan, S. P., Organometallics 2010,29, 394-402.
70. Müller, R. S. R. Homogeneous gold catalysts: development of applications for gold(I) catalysts bearing N-heterocyclic carbene ligands. University of St Andrews, 2011.
71. Gómez-Suárez, A.; Gasperini, D.; Vummaleti, S. V. C.; Poater, A.; Cavallo, L.; Nolan, S. P., ACS Catal. 2014,4, 2701-2705. 72. Collado, A.; Gómez-Suárez, A.; Martin, A. R.; Slawin, A. M. Z.; Nolan, S. P., Chem. Commun. 2013,49, 5541-5543.
73. Meiries, S.; Nolan, S., Synlett 2014,25, 393-398.
74. Brulé, E.; Gaillard, S.; Rager, M.-N.; Roisnel, T.; Guérineau, V.; Nolan, S. P.; Thomas, C. M., Organometallics 2011,
30, 2650-2653.
75. Formation of 8 would coincide with formation of one equivalent of HBF4 would form from deprotonation of water by the
second equivalent of BF4- in 3. For dedicated experiments that show a
shift of this equilibrium in the presence of excess HBF4, see: S.
Gaillard, J. Bosson, R. S. Ramón, P. Nun, A. M. Z. Slawin and S. P. Nolan, Chem. Eur. J.2010, 16, 13729-13740.
76. (a) Fu, M.-C.; Shang, R.; Cheng, W.-M.; Fu, Y., ACS
Catal. 2016,6, 2501-2505; (b) Zuccaccia, D.; Belpassi, L.; Tarantelli,
F.; Macchioni, A., J. Am. Chem. Soc. 2009,131, 3170-3171.
77. Oonishi, Y.; Gómez-Suárez, A.; Martin, A. R.; Nolan, S.
P., Angew. Chem. Int. Ed. 2013,52, 9767-9771.
Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.;
Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A., Gaussian09 Revision B.01.
79. (a) Perdew, J. P., Phys. Rev. B 1986,33, 8822-8824; (b) Perdew, J. P., Phys. Rev. B 1986,34, 7406-7406; (c) Becke, A. D., J.
Chem. Phys. 1988,88, 2547-2553.
80. The energy of 3-out has been calculated as the sum of separate [Au(IPrCl)]+ and BF
4- fragments.
Study of the new bench-stable NHC-gold(I) tetrafluoroborate complex, [Au(IPrCl)][BF
![Figure 2. Solid-state structure of 3·((CH3)2CO). One fragment of [Au(IPrCl)(FBF3)] (3-in) from the unit cell is shown](https://thumb-us.123doks.com/thumbv2/123dok_us/8747528.389114/3.612.65.304.367.458/figure-solid-state-structure-fragment-iprcl-fbf-shown.webp)

![Table 1. Thermodynamic and kinetic parameters for the equilibrium between 3-in and 6.[a]](https://thumb-us.123doks.com/thumbv2/123dok_us/8747528.389114/5.612.65.281.511.688/table-thermodynamic-kinetic-parameters-equilibrium.webp)
