organic papers
Acta Cryst.(2005). E61, o2291–o2293 doi:10.1107/S1600536805019513 Jhawaret al. C
28H22O2C6H6
o2291
Acta Crystallographica Section E Structure Reports
Online
ISSN 1600-5368
1,2,3,4-Tetraphenylbutane-1,4-dione benzene
solvate and a brief analysis of the solvated
organic structures in the Cambridge Structural
Database
Amit Jhawar, Ilia A. Guzei,* Vitaliy I. Timokhin and Robert West
Department of Chemistry, University of Wisconsin–Madison, 1101 University Avenue, Madison, WI 53706, USA
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 100 K
Mean(C–C) = 0.003 A˚
Rfactor = 0.054
wRfactor = 0.139
Data-to-parameter ratio = 15.1
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography Printed in Great Britain – all rights reserved
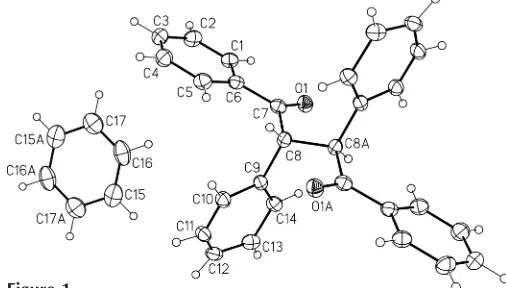
1,2,3,4-Tetraphenylbutane-1,4-dione crystallizes as a benzene solvate in a 1:1 ratio, C28H22O2C6H6. Both molecules reside on crystallographic inversion centers. The carbonyl group of (I) is coplanar with the adjacent phenyl ring within 3.6 (3). A
brief analysis of the organic structures included in the Cambridge Structural Database [Allen (2002). Acta Cryst. B58, 380–388] shows that at least 22.8% of them contain cocrystallized solvent and in 3.8% of these cases the solvent is benzene.
Comment
During the course of our research toward the synthesis of octaphenyl-1,1-spirobisilole (II) and its derivatives, we isolated and spectroscopically and structurally characterized the title compound, (I).
The Si-containing spiro compound (II) and its derivatives may be used for the construction of long-life white-emitting electroluminiscent devices, displays and electric appliances (Fukuda & Genda, 2004; Matsuura et al., 2003). While compound (II) has not been isolated and characterized,
compound (I) was obtained from the reaction between 1,4-dilithiotetraphenylbutadiene and silicon tetrachloride in5% yield by apparent incorporation of ambient oxygen or reaction with water. This compound has been isolated and character-ized spectroscopically (Amat-Guerriet al., 1990; Ceylanet al., 2004) and our spectra (Timokhin et al., 2005) are consistent with the literature data, but the structural analysis is presented here for the first time.
Compound (I) crystallizes as a benzene solvate in a triclinic unit cell with both molecules occupying crystallographic inversion centers. After the final refinement, several peaks of
0.7 e A˚3 were observed in the vicinity of the inversion center occupied by (I). A careful examination of the locations of the peaks indicated a possible second position of the C8– C14 benzyl group, and an alternative refinement accounting for this disorder yielded a9:1 ratio for the disordered group. TheRfactor dropped to 0.042, the height of the residual peaks reduced to 0.2 e A˚3 and the standard uncertainties for bond distances improved negligibly. However, given the need for an idealized geometry for the minor component, and some resultant C—C distances in excess of 1.6 A˚ between the minor component, the ordered part of the molecule and the symmetry-related minor component, we decided to ignore the disorder as it is chemically inconsequential. While the disorder is likely to be real, the results of modeling 11% occupied C atoms are not highly reliable.
The overall molecular geometry is normal, the C O bond distance measuring 1.214 (2) A˚ and the O1 C7—C6—C1 torsion angle being 3.6 (3). In the optimized molecule of (I) at
the PBE1PBE/6–31+G* level of theory, the corresponding values are 1.221 A˚ and 8.83(Frischet al., 2004). A search of
the Cambridge Structural Database (CSD; Version 5.26, May 2005 update; Allen, 2002) for compounds containing Csp3— C( O)—Ph, limited to organic structures with finalRvalues of 0.05 or lower, no disorder and no error, and excluding powder structures and polymers, revealed 282 structures with 337 relevant data (Brunoet al.2002). Among these structures, the average C O bond distance was found to be 1.216 (8) A˚ , a value in excellent agreement with the C O bond distance in
(I). The range for the above O C—C—C torsion angle
among the 337 data spanned from54 to 38. While there are
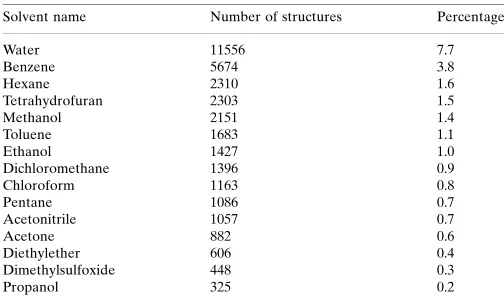
no classical hydrogen bonds present in the structure, the position of the solvent benzene molecule may indicate a weak intermolecular interaction between atom H16 and the C1–C6 phenyl ring. Atom H16 is positioned 3.24 A˚ from the center of the ring, the C16—H16 centroid(C1–C6) angle being 167 (1). Since the structure of the title compound contained a benzene solvent molecule, we analyzed the organic structures reported to the CSD for the presence of cocrystallized solvents in the crystal structure. The search revealed 149417 entries for organic molecules, of which at least 34040 (22.8%) are solvated compounds. Benzene is estimated to be the second most frequently observed cocrystallized solvent. The results of our searches for structures with common solvent molecules are presented in Table 1.
Experimental
For the preparation of 1,4-dilithiotetraphenylbutadiene (Hubelet al., 1964; Braye et al., 1961), diphenylacetylene (3.0 g, 16.8 mmol) was shaken with excess of lithium (0.6 g, 86.5 mmol) in diethyl ether (8 ml) under an argon atmosphere. After an induction period of about 10 min, the reaction mixture became brown–red. Shaking was continued for 2 h. The solution of 1,4-dilithiotetraphenylbutadiene in diethyl ether was filtered and used for further reaction. A suspension of 1,4-dilithiotetraphenylbutadiene in diethyl ether was cannulated in a flask with silicon tetrachloride (0.34 ml, 2.97 mmol) in dioxane (60 ml). The ether was removed by distillation. After 1 h of reflux at 373 K, water (10 ml) and benzene (50 ml) were added, and the solvents were removed by distillation. The yellow–green solid was purified by column chromatography (eluent hexanes–benzene, 0!100% benzene). Compound (I) was isolated as colorless crystals from CDCl3(yield5%, based on the starting diphenylacetylene).
The1H and13C NMR spectra of (I) in solution are consistent with the structure of the product found in the crystal structure and for the NMR spectral data in the literature (Amat-Guerriet al., 1990; Ceylan
et al., 2004). In this reaction, a small amount of hexamethylbenzene was also isolated as pale-yellow crystals from CDCl3/CH2Cl2(yield 3%, based on the starting diphenylacetylene). The presence of hexamethylbenzene was found by the X-ray crystal analysis.
Crystal data
C28H22O2C6H6
Mr= 468.56
Triclinic,P1
a= 8.4629 (13) A˚
b= 8.7530 (13) A˚
c= 9.5926 (14) A˚ = 110.489 (2)
= 94.886 (2)
= 108.300 (2) V= 616.57 (16) A˚3
Z= 1
Dx= 1.262 Mg m 3 Mo-Kradiation Cell parameters from 2387
reflections = 2.3–26.4
= 0.08 mm1
T= 100 (2) K Needle, colorless 0.470.400.30 mm
Data collection
Bruker CCD 1000 area-detector diffractometer
!scans
Absorption correction: multi-scan (SADABS; Bruker, 2003)
Tmin= 0.965,Tmax= 0.977 5010 measured reflections
2468 independent reflections 1972 reflections withI> 2(I)
Rint= 0.016 max= 26.4
h=10!10
k=10!10
l=11!11
organic papers
o2292
Jhawaret al. C [image:2.610.42.295.75.219.2]28H22O2C6H6 Acta Cryst.(2005). E61, o2291–o2293
Figure 1
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.054
wR(F2) = 0.139
S= 1.03 2468 reflections 163 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0528P)2 + 0.5916P]
whereP= (Fo2+ 2Fc2)/3 (/)max< 0.001
max= 0.71 e A˚
3 min=0.33 e A˚
[image:3.610.44.296.252.402.2]3
Table 1
Selected common solvents cocrystallized with organic compounds reported to the CSD.
The percentages are reported relative to all organic structures contained in the CSD.
Solvent name Number of structures Percentage
Water 11556 7.7
Benzene 5674 3.8
Hexane 2310 1.6
Tetrahydrofuran 2303 1.5
Methanol 2151 1.4
Toluene 1683 1.1
Ethanol 1427 1.0
Dichloromethane 1396 0.9
Chloroform 1163 0.8
Pentane 1086 0.7
Acetonitrile 1057 0.7
Acetone 882 0.6
Diethylether 606 0.4
Dimethylsulfoxide 448 0.3
Propanol 325 0.2
All H atoms were refined using a riding model, with C—H = 0.95 (aromatic) and C—H = 1.00 A˚ (other H atom), and withUiso(H) =
1.2Ueq(C).
Data collection:SMART(Bruker, 2003); cell refinement:SAINT
(Bruker, 2003); data reduction:SAINT; program(s) used to solve structure: SHELXTL (Bruker, 2003); program(s) used to refine structure:SHELXTL; molecular graphics:SHELXTL; software used to prepare material for publication:SHELXTL.
We thank the National Science Foundation for financial support.
References
Allen, F. H. (2002).Acta Cryst.B58, 380–388.
Amat-Guerri, F., Rivera-Sagredo, A. & Sanz, J. (1990). J. Photochem. Photobiol. A Chem,55, 87–96.
Braye, E. H., Hubel, W. & Caplier, I. H. (1961).J. Am. Chem. Soc.83, 4406– 4413.
Bruker (2003).SADABS(Version 2.05),SAINT(Version 6.22),SHELXTL
(Version 6.10) andSMART(Version 5.622). Bruker AXS Inc., Madison, Wisconsin, USA.
Bruno, I. J., Cole, J. C., Edgington, P. R., Kessler, M., Macrae, C. F., McCabe, P., Pearson, J. & Taylor, R. (2002).Acta Cryst.B58, 389–397.
Ceylan, M., Gurdere, M. B., Budak, Y., Lazaz, C. & Secen, H. (2004).Synthesis, pp. 1750–1754.
Fukuda, M. & Genda, K. (2004). Japan Patent No. JP 2004253298.
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Montgomery, J. A. Jr, Vreven, T., Kudin, K. N., Burant, J. C., Millam, J. M., Iyengar, S. S., Tomasi, J., Barone, V., Mennucci, B.et al.
(2004). GAUSSIAN03. Revision C.02. Gaussian Inc., Wallingford, CT, USA.
Hubel, K. W., Braye, E. H. & Caplier, I. H. (1964). USA Patent No. 3 151 140. Matsuura, H., Yamada, T. & Kita, H. (2003). Japan Patent No. JP 2004243178. Timokhin, V. I., Guzei, I. A. & West, R. (2005). In preparation.
organic papers
Acta Cryst.(2005). E61, o2291–o2293 Jhawaret al. C
supporting information
sup-1
Acta Cryst. (2005). E61, o2291–o2293
supporting information
Acta Cryst. (2005). E61, o2291–o2293 [https://doi.org/10.1107/S1600536805019513]
1,2,3,4-Tetraphenylbutane-1,4-dione benzene solvate and a brief analysis of the
solvated organic structures in the Cambridge Structural Database
Amit Jhawar, Ilia A. Guzei, Vitaliy I. Timokhin and Robert West
1,2,3,4-Tetraphenylbutane-1,4-dione benzene solvate
Crystal data C28H22O2·C6H6
Mr = 468.56 Triclinic, P1 a = 8.4629 (13) Å b = 8.7530 (13) Å c = 9.5926 (14) Å α = 110.489 (2)° β = 94.886 (2)° γ = 108.300 (2)° V = 616.57 (16) Å3
Z = 1 F(000) = 248 Dx = 1.262 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 2387 reflections θ = 2.3–26.4°
µ = 0.08 mm−1
T = 100 K Needle, colorless 0.47 × 0.40 × 0.30 mm
Data collection
Bruker CCD 1000 area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
ω scans
Absorption correction: multi-scan (SADABS; Bruker, 2003) Tmin = 0.965, Tmax = 0.977
5010 measured reflections 2468 independent reflections 1972 reflections with I > 2σ(I) Rint = 0.016
θmax = 26.4°, θmin = 2.3°
h = −10→10 k = −10→10 l = −11→11
Refinement Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.054
wR(F2) = 0.139
S = 1.04 2468 reflections 163 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained w = 1/[σ2(F
o2) + (0.0528P)2 + 0.5916P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.71 e Å−3
supporting information
sup-2
Acta Cryst. (2005). E61, o2291–o2293
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
supporting information
sup-3
Acta Cryst. (2005). E61, o2291–o2293
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.0324 (7) 0.0223 (7) 0.0296 (7) 0.0109 (6) 0.0124 (6) 0.0125 (6) C1 0.0238 (9) 0.0245 (10) 0.0270 (10) 0.0061 (8) 0.0057 (8) 0.0139 (8) C2 0.0288 (10) 0.0339 (11) 0.0287 (10) 0.0107 (9) 0.0122 (8) 0.0182 (9) C3 0.0352 (11) 0.0314 (11) 0.0237 (10) 0.0154 (9) 0.0093 (8) 0.0115 (8) C4 0.0357 (11) 0.0222 (10) 0.0290 (10) 0.0085 (8) 0.0068 (9) 0.0102 (8) C5 0.0285 (10) 0.0248 (10) 0.0287 (10) 0.0075 (8) 0.0084 (8) 0.0153 (8) C6 0.0222 (9) 0.0246 (9) 0.0239 (9) 0.0110 (7) 0.0070 (7) 0.0136 (8) C7 0.0201 (9) 0.0218 (10) 0.0264 (10) 0.0076 (7) 0.0050 (7) 0.0126 (8) C8 0.0297 (10) 0.0234 (10) 0.0249 (10) 0.0045 (8) 0.0066 (8) 0.0087 (8) C9 0.0247 (10) 0.0249 (10) 0.0371 (11) 0.0104 (8) 0.0113 (8) 0.0165 (8) C10 0.0275 (10) 0.0262 (10) 0.0324 (11) 0.0075 (8) 0.0051 (8) 0.0154 (9) C11 0.0370 (11) 0.0240 (10) 0.0272 (10) 0.0143 (9) 0.0120 (8) 0.0136 (8) C12 0.0322 (10) 0.0366 (11) 0.0313 (10) 0.0224 (9) 0.0146 (8) 0.0221 (9) C13 0.0229 (9) 0.0341 (11) 0.0282 (10) 0.0083 (8) 0.0082 (8) 0.0166 (8) C14 0.0262 (10) 0.0220 (10) 0.0398 (11) 0.0082 (8) 0.0129 (8) 0.0148 (9) C15 0.0417 (13) 0.0317 (12) 0.0401 (12) 0.0034 (10) 0.0161 (10) 0.0109 (10) C16 0.0353 (12) 0.0259 (11) 0.0568 (15) 0.0089 (9) 0.0195 (11) 0.0106 (10) C17 0.0369 (12) 0.0312 (11) 0.0518 (14) 0.0080 (10) 0.0114 (10) 0.0194 (10)
Geometric parameters (Å, º)
O1—C7 1.214 (2) C9—C14 1.399 (3) C1—C2 1.379 (3) C10—C11 1.378 (3) C1—C6 1.399 (3) C10—H10 0.9500 C1—H1 0.9500 C11—C12 1.381 (3) C2—C3 1.390 (3) C11—H11 0.9500 C2—H2 0.9500 C12—C13 1.386 (3) C3—C4 1.386 (3) C12—H12 0.9500 C3—H3 0.9500 C13—C14 1.389 (3) C4—C5 1.380 (3) C13—H13 0.9500 C4—H4 0.9500 C14—H14 0.9500 C5—C6 1.397 (3) C15—C17ii 1.383 (3)
C5—H5 0.9500 C15—C16 1.382 (4) C6—C7 1.494 (3) C15—H15 0.9500 C7—C8 1.535 (3) C16—C17 1.381 (4) C8—C8i 1.527 (4) C16—H16 0.9500
C8—C9 1.541 (3) C17—C15ii 1.383 (3)
C8—H8 1.0000 C17—H17 0.9500 C9—C10 1.383 (3)
supporting information
sup-4
Acta Cryst. (2005). E61, o2291–o2293
C3—C2—H2 119.9 C9—C10—H10 119.6 C4—C3—C2 119.82 (18) C10—C11—C12 120.48 (19) C4—C3—H3 120.1 C10—C11—H11 119.8 C2—C3—H3 120.1 C12—C11—H11 119.8 C5—C4—C3 120.14 (19) C11—C12—C13 119.78 (18) C5—C4—H4 119.9 C11—C12—H12 120.1 C3—C4—H4 119.9 C13—C12—H12 120.1 C4—C5—C6 120.63 (18) C12—C13—C14 119.65 (19) C4—C5—H5 119.7 C12—C13—H13 120.2 C6—C5—H5 119.7 C14—C13—H13 120.2 C5—C6—C1 118.71 (17) C13—C14—C9 120.64 (18) C5—C6—C7 123.45 (16) C13—C14—H14 119.7 C1—C6—C7 117.84 (17) C9—C14—H14 119.7 O1—C7—C6 120.83 (16) C17ii—C15—C16 120.0 (2)
O1—C7—C8 120.40 (17) C17ii—C15—H15 120.0
C6—C7—C8 118.46 (16) C16—C15—H15 120.0 C8i—C8—C7 110.0 (2) C17—C16—C15 120.2 (2)
C8i—C8—C9 111.6 (2) C17—C16—H16 119.9
C7—C8—C9 107.52 (15) C15—C16—H16 119.9 C8i—C8—H8 109.2 C16—C17—C15ii 119.8 (2)
C7—C8—H8 109.2 C16—C17—H17 120.1 C9—C8—H8 109.2 C15ii—C17—H17 120.1
C6—C1—C2—C3 1.0 (3) C6—C7—C8—C9 80.6 (2) C1—C2—C3—C4 −0.3 (3) C8i—C8—C9—C10 123.3 (2)
C2—C3—C4—C5 −0.5 (3) C7—C8—C9—C10 −115.9 (2) C3—C4—C5—C6 0.6 (3) C8i—C8—C9—C14 −59.0 (3)
C4—C5—C6—C1 0.1 (3) C7—C8—C9—C14 61.7 (2) C4—C5—C6—C7 179.70 (18) C14—C9—C10—C11 −2.6 (3) C2—C1—C6—C5 −0.9 (3) C8—C9—C10—C11 175.11 (18) C2—C1—C6—C7 179.50 (17) C9—C10—C11—C12 1.1 (3) C5—C6—C7—O1 −175.90 (18) C10—C11—C12—C13 0.9 (3) C1—C6—C7—O1 3.7 (3) C11—C12—C13—C14 −1.4 (3) C5—C6—C7—C8 10.4 (3) C12—C13—C14—C9 −0.1 (3) C1—C6—C7—C8 −170.00 (17) C10—C9—C14—C13 2.1 (3) O1—C7—C8—C8i 28.6 (3) C8—C9—C14—C13 −175.61 (18)
C6—C7—C8—C8i −157.7 (2) C17ii—C15—C16—C17 −0.1 (4)
O1—C7—C8—C9 −93.1 (2) C15—C16—C17—C15ii 0.1 (4)