I. Abstract
Many biological processes use allosteric regulation to control the rate and selectivity of enzymatic reactions. To achieve similar control in molecular catalysis with simple external additives, a series of aminophenylphosphinite-supported palladium complexes have been prepared and examined in conjunction with alkali metal salts. The first complex contains a pendant aza-crown ether moiety capable of either (a) coordinating cations in the aza-crown-ether pocket or (b) coordinating to palladium with one of the ether donors. The second complex contains a diethylamine moiety, which contains neither a binding pocket for cations, nor ethers capable of binding to palladium. The third scaffold contains a bis(methoxyethyl)amine moiety, which we hypothesized would only support weak cation binding while still providing ether donors for hemilabile palladium coordination. This thesis will present comparative catalysis and binding affinity studies that elucidate the key interactions of cations with pincer-crown ether catalysts.
Graphical Abstract. The targeted comparative catalysis with a system of palladium(II) complexes.
0 or 5 mM LiB(ArF-24)4
CD2Cl2 rt
O PiPr
2
NR2 Pd MeO
12 eq Et2O
B(ArF-20) 4
O PiPr
2
NR2 Pd MeO
B(ArF-20) 4
=
O PiPr
2
N Pd O
O O O
B(ArF-20) 4
MeO O PiPr
2
N Pd O
B(ArF-20) 4
O MeO
O PiPr
2
N Pd
B(ArF-20) 4 MeO
II. Introduction
Hemilabile ligand scaffolds contain a strong donor bound tightly to the metal center, and a weak donor that can reversibly coordinate to the metal center. The weak donor can provide stability in its bound form while also promoting catalytic activity when it dissociates to present an open binding site. Tuning reactivity of hemilabile catalysts allosterically with external additives could have broad implications in small molecule activation chemistry and may provide temporal control over catalytic reactivity.1,2 Examples of tunable hemilability in catalysis include cation-modulated
H2 splitting and controlled and reversible ligation.3 Previous work in the lab has demonstrated
cation-modulated reactivity with iridium pincer-crown ether complexes.1,2 These complexes
contain a strong anchoring pincer backbone with a weak aza-crown ether donor, and upon the addition of a Li+ cation, there is up to a 1000-fold rate enhancement in the isomerization of
allylbenzene to b-methylstyrene.4 The proposed mechanism of ion-controlled catalysis in this
Figure 1. Proposed mechanism of ion-controlled catalysis with an iridium pincer-crown ether catalyst.
This work seeks to expand cation-modulated reactivity to palladium pincer catalysts. The preparation of three palladium aminophosphinite pincer complexes with hemilabile amine ligands of varying structure has been undertaken to further understand cation-modulated reactivity. The targeted palladium(II) complexes are shown in Figure 2.
Figure 2. Palladium aminophosphinite pincer complexes with varying amine donors.
Ir PiPr
2 N O O O O O H + Ph – Ph Ph + – Ph Ir PiPr
2 N O O O O O H M+ M+ M+
O PiPr
2 N Pd O O O O MeO
O PiPr
2
N
Pd O
O
MeO O PiPr
2
N
Pd
B(ArF-20) 4
MeO
O
B(ArF-20) 4
B(ArF-20) 4
Palladium(II) pincer complexes with pendent aza-crown ether and diethylamine ligands, respectively, have been synthesized. Upon halide abstraction, these palladium complexes form cationic species. When the complex contains a crown ether group, the crown stabilizes the complex due to ether ligand donation. Adding cations causes the crown-ether oxygens to dissociate from the metal center, providing an open site for substrate binding during catalysis. When the complex contains a diethylamine in lieu of the aza-crown ether, there is no donor ether ligand available to stabilize open coordination sites, and cations in solution are not expected to have significant effects on the rate of catalysis. This work aimed to synthesize a novel complex with a bis(methoxy)ethylamine ligand that is electronically similar to the crown ether ligand-containing complexes and would also allow for similar ether ligand donation to the Pd center but would not allow for significant cation modulation.
cation-modulated reactivity by elucidating the role of a hemilabile crown ether ligand in tunable catalysis. Comparison of the three complexes provides mechanistic information, including if cation-ligand interactions facilitate ether dissociation and olefin binding, the tunability of catalysis, and the diethylamine-substituted complex with an unprotected metal center will specifically help identify that isomerization is happening at the Pd metal center.
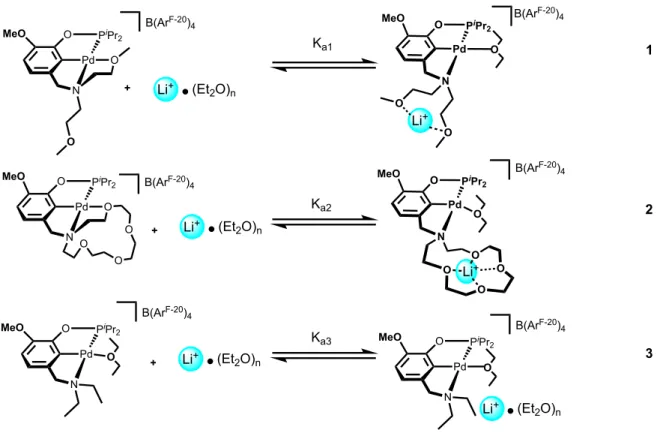
Figure 3. Cation-modulation predictions with the palladium(II) complexes.
O PiPr
2
N
Pd O
B(ArF-20) 4
O MeO
Li+ (Et
2O)n
O PiPr
2 Pd N MeO O O
B(ArF-20) 4
Li+
O Ka1
O PiPr
2 N Pd O O O O
B(ArF-20) 4
MeO O PiPr
2 Pd O N O O O MeO Li+
B(ArF-20) 4
O Li+ (Et
2O)n
Ka2
O PiPr
2
N Pd
B(ArF-20) 4 MeO
O PiPr
2
N Pd
B(ArF-20) 4 MeO
Li+ (Et
2O)n
O
O Li+ (Et
2O)n
Ka3
Prediction: Ka2 > Ka1 > Ka3
1
2
III. Results and Discussion
Synthesis of [(MeO-EtMeONCOP)Pd][B(ArF20) 4]
Synthesis of bis(methoxy)ethylamine ligand precursor
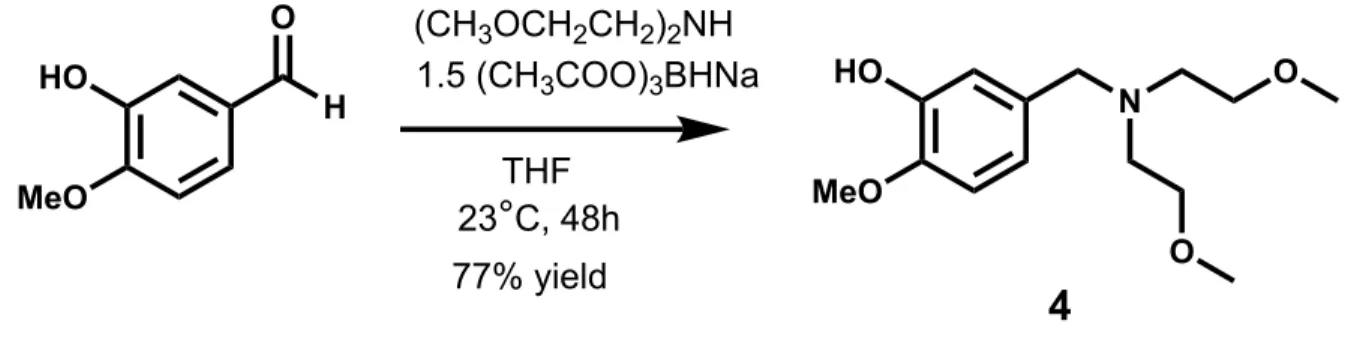
The desired ligand 4, Figure 4, was synthesized at room temperature under an N2 atmosphere
through a reductive amination of 3-hydroxy-4-methoxybenzaldehyde. Following an aqueous bicarbonate workup, the crude product was characterized by 1H NMR spectroscopy. Some of the
aldehyde reduced to an alcohol by-product, which was separated via silica gel chromatography with a single eluent of 60% DCM: 40% ethyl acetate to afford pure product in 77% yield.
Figure 4. Reductive amination to form the bis(methoxy)ethylamine pre-ligand.
Synthesis of (MeO-EtMeONCOP)H
The bis(methoxy)ethylaminophosphinate ligand (MeO-EtMeONCOP)H, 5 (Figure 5), was synthesized
by phosphination of 1 using triethylamine and diisopropylchlorophosphine. An alumina column was sufficient to separate the phosphinated ligand from its hydrolyzed by-product, providing (
MeO-EtMeONCOP)H in 67% yield as a colorless oil. A characteristic singlet in the 31P{1H} NMR
spectrum (δ 155.9) was observed for (MeO-EtMeONCOP)H.
H O HO
MeO
N HO
MeO 1.5 (CH3COO)3BHNa
THF
O
O (CH3OCH2CH2)2NH
4
23°C, 48hFigure 5. Phosphination reaction to form (MeO-EtMeONCOP)H.
Synthesis of (MeO-EtMeONCOP)Pd(Cl)
The neutral complex, (MeO-EtMeONCOP)Pd(Cl), 6, Figure 6, was synthesized through a reflux of
(MeO-EtMeONCOP)H and Pd(cod)Cl
2 in toluene for 48 hours. The yellow-orange solution turned
colorless over the course of the reaction. The crude product was stirred in pentane until fully in solution, filtered, and then the solution was diluted and put in the freezer at -35 °C. The product crystallized out as a yellow crystal in 97% yield. A 31P NMR spectrum showed a resonance at 202
ppm assigned as (MeO-EtMeONCOP)Pd(Cl) (6) in Figure 6.
Figure 6. Reflux reaction to form (MeO-EtMeONCOP)Pd(Cl).
N HO
MeO
O
O NEt3
THF PiPr
2Cl
- HNEt3Cl
N O
MeO
O O P
5 4
-35° rt. 67% yield
N O
MeO
O O P
Toluene
Pd(cod)Cl2
- HCl, COD
Δ
O P
Pd Cl
N MeO
O O
5
6
Synthesis of [(MeO-EtMeONCOP)Pd][B(ArF20)4]
The synthesis of [(MeO-EtMeONCOP)Pd][B(ArF20)
4], 1, Figure 7, was achieved through a chloride
abstraction. When 6 was treated with [Et3Si][BArF4] (generated in situ at room temperature under
a N2 atmosphere), the corresponding cationic species formed, along with triphenylmethane and
chlorosilane by-products. The crude product was washed with pentane and then dissolved in diethyl ether and filtered through a short (1-2 cm) alumina column. After removing the diethyl ether in vacuo, it was washed with more pentane to reveal a white solid in 73% yield, which was characterized by 1H and 31P NMR spectroscopy. A 31P NMR showed a single resonance at 198
ppm, corresponding to an expected upfield shift of a cationic PdII complex, showing no impurities
by 31P-NMR spectroscopy. The 1H NMR showed clean product.
Figure 7. Halide abstraction to form [(MeO-EtMeONCOP)Pd][B(ArF-20)4].
Cation-Modulation Catalysis Predictions and Data
Some palladium(II) complexes are excellent olefin isomerization catalysts.5 Initially, the
isomerization of allylbenzene to b-methylstyrene was utilized as a benchmark reaction to probe cation-modulated reactivity of 1. However, dimers of b-methylstyrene appeared as a side product
O PiPr
2
N
Pd O
O
O PiPr
2
Pd Cl
N
MeO MeO
O O
Et3SiH + [Ph3C][B(ArF-20)4]
C6H5Cl
B(Ar
F-20)
4-Et3SiCl, Ph3CH
23
°
C, 1h73% yield
during isomerization of allylbenzene when LiB(ArF-24)
4 was present is in solution. These products
are proposed to be formed by Friedel-Crafts-type alkylation reactions.6,7 A potential solution is to
use a substrate that lacks an aromatic ring or additional alkenes to prevent the Friedel-Crafts alkylation reactions. The substrate 1-hexene was chosen as it should not be prone to Friedel-Crafts or other carbocationic rearrangement chemistry.
The rates of 1-hexene isomerization, with and without cation additives, were compared across the three palladium pincer complexes to probe the role of cation-ligand interactions in gating substrate binding and controlling reactivity. We predicted 1-hexene isomerization with 1
and 2, would be slow without a cation additive, and that upon the addition of cation additives, there would be a dramatic rate enhancement to isomerization with 2 but only a slight enhancement with
1. We predicted that isomerization with 3 would be rapid without a cation additive and that the addition of a cation would not affect the rate of isomerization.
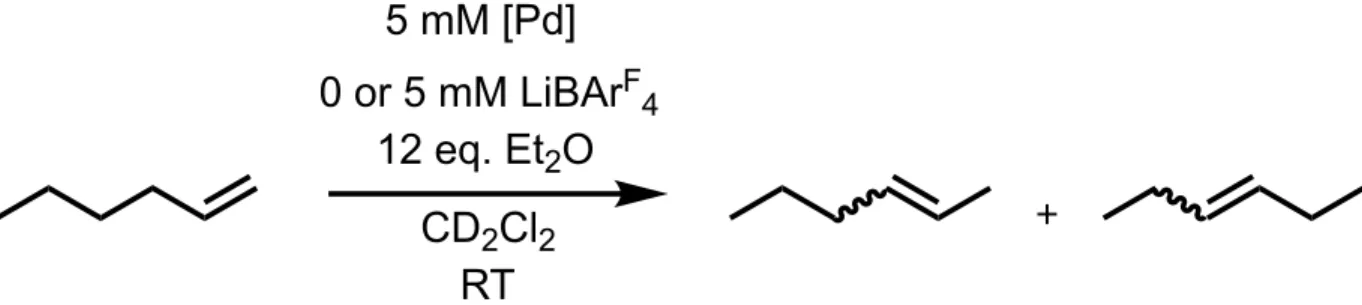
The goal was to compare the catalytic reactivity of the three complexes with and without LiB(ArF-24)4 to explore cation-modulated reactivity, as shown in Figure 8 below. Six reactions were
set up, two with each cationic complex. Three reactions contained 5 mM 1, 2, or 3, respectively, 5 mM LiB(ArF-24)4, 350 mM 1-hexene, 12 equivalents of diethyl ether with respect to the catalyst,
and mesitylene as the internal standard. Three other reactions were run in parallel and set up analogously, but without the added LiB(ArF-24)
4. The reactions were monitored over time via 1H
NMR spectroscopy to track 1-hexene consumption.
Figure 8. General conditions for comparative isomerization reactions of 1-hexene.
5 mM [Pd]
0 or 5 mM LiBAr
F4CD
2Cl
212 eq. Et
2O
RT
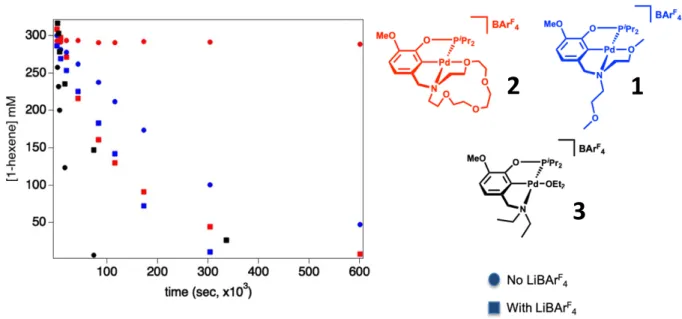
The preliminary isomerization data is shown in Figure 9 below. The disappearance of 1-hexene is plotted versus time for each complex with and without LiB(ArF-24)
4.
Figure 9. Rate comparison of all three palladium (II) complexes.
The preliminary data show that 2, [(MeO-15c5NCOP)Pd][B(ArF-20)
4], catalyzes the
isomerization of 1-hexene in the presence of Li+ cations. When there are no cation additives in
solution, there is very little isomerization, as we see almost no conversion of 1-hexene to the desired products. With complex 1, [(MeO-EtMeONCOP)Pd][B(ArF-20)4], there is a less controllable
gate towards catalysis. When there are no cation additives, 1 still isomerizes 1-hexene, albeit at a slower rate than in the presence of Li+, which enhances the rate of catalysis by approximately a
factor of 1.5. Catalytic reactivity of 1 is tunable by cation additives but the rate enhancement is not as dramatic as with 2. Finally, preliminary data with complex 3, [(MeO-EtNCOP)Pd][B(ArF-20)
4],
indicates that isomerization occurs rapidly without the addition of cations, supporting that isomerization occurs at the palladium metal center. With 3, addition of cations shows a slight
1
2
decrease in activity (attributed to additional Et2O equivalents present when the Li+ salt is added),
indicating that there is no cation rate enhancement for 3.
Since these complexes contain no hydrides, it is proposed that isomerization proceeds through a base-promoted p-allyl mechanism, where the amine of the pincer deprotonates an allylic proton of bound hexene. The proposed mechanism is shown in Figure 10. To further probe the base-mediated p-allyl mechanism, future work will utilize a bulky external base that will bind to the metal center during the isomerization reaction. If the proposed mechanism is dominant, the addition of an external base is expected to speed up the reaction. Additionally, to confirm this mechanism a deuterium labeling experiment could be performed, in which an exclusive 1,3-shift of deuterium in the product, with no deuterium at the second position of the allylic system, would indicate an allyl mechanism.8
O PiPr
2 N Pd O O O O MeO R
O PiPr
2 Pd O N O O O MeO H R H R
O PiPr
2 Pd O N O O O MeO H R
O PiPr
2 Pd O N O O O MeO R
B(ArF-20)
4
B(ArF-20)
4
B(ArF-20)
4
B(ArF-20)
Figure10. Proposed base-mediated allyl mechanism for isomerization.
Investigation of Cation-crown and Cation-bismethoxy Binding
Previous thermodynamic studies in the group have given rise to a quantitative understanding of cation-macrocycle interactions of pincer-crown ether complexes with alkali metal cations. Observing the change in chemical shift of the benzylic linker and crown ether protons as a function of added cations provides a useful handle to monitor cation-crown interactions.9 Previous work
has specifically explored an unblocked Pd(II) neutral chloride complex with LiOTf in acetonitrile solution, as shown in Figure 11. With increasing salt concentration, shifts of the methylene linker resonance were observed via 1H NMR spectroscopy and a binding isotherm was constructed for
various concentrations of palladium (2.5 mM, 5 mM and 10 mM). A binding affinity of 294 M-1
was obtained from this data. This binding affinity is higher than other similar crown ether complexes, including nickel and platinum analogues.10
Figure 11. General titration scheme for unblocked Pd(II) neutral chloride complex in acetonitrile. Initially, we sought to do a similar titration with the neutral palladium chloride complexes with Li(BArF-20)
4 to model the ligand-cation interactions. However, (MeO-EtMeONCOP)Pd(Cl) is
insoluble in acetonitrile, so the titrations were performed instead in CD2Cl2. Solutions containing
10 mM (MeO-EtMeONCOP)Pd(Cl) in CD
2Cl2 were titrated with 0.25, 0.5, 0.75, 1, 1.5, 1.75, and 2
O PiPr
2
Pd Cl
O N
O O
O
CD
3CN
equivalents of LiB(ArF-20)
4 and were monitored via 1H NMR spectroscopy. However, upon the
addition of LiB(ArF-24)
4, a halide abstraction occurred and solely the cationic species (1) was
observed in the titration tubes containing at least one equivalent of LiB(ArF-20)
4, as shown in Figure
12.
Figure 12. Halide abstraction upon the addition of LiB(ArF-20)
4 to (MeO-EtMeONCOP)Pd(Cl).
We wanted to compare this reactivity to that of (Meo-15c5NCOP)Pd(Cl) with Li(BArF-20) 4.
Upon, the addition of two equivalents of LiB(ArF-20)
4 to (Meo-15c5NCOP)Pd(Cl), no halide
abstraction was observed and instead chemical shifts in the methylene linker and crown ether resonances indicative of cation-crown interactions were observed. When 12-crown-4 was added to the solution of (MeO-15c5NCOP)Pd(Cl) and LiB(ArF-20)4, the 1H-NMR spectrum reflected the
original chemical shifts of (MeO-15c5NCOP)Pd(Cl), confirming the change was due to cation-crown
interactions and not a halide abstraction. Comparing the relative binding affinity of both chloride complexes could thus not be achieved in dichloromethane due to halide abstraction of (
MeO-EtMeONCOP)Pd(Cl).
Because the titrations could not be performed with the neutral chloride complexes to explore cation-ligand interactions, we next explored the possibility of performing a titration with the cationic complexes, which would eliminate the possibility of a halide abstraction. This
CH2Cl2
rt.
O PiPr
2
Pd Cl
N MeO
O O
O PiPr
2
N
Pd O
O MeO
B(ArF-20)
4
[Li][B(ArF-20)
approach seemed encouraging because during catalysis with 2, a distinct difference in the crown ether resonances is observed via 1H NMR in reactions containing Li+ compared to reactions with
no cation additive. With 1, there is no observable difference via NMR spectroscopy. However, two equivalents of Li+ were added to 1 to see if there was an observable shift in the 1H NMR and if a
titration was possible. There was no shift in the 1H NMR spectrum. The cationic complex binds
LiB(ArF-20)4 in CD2Cl2 with Ka < 1 M-1, which is the lower detection limit by NMR. This is
important as we need this species to be a weak binder and then to become a stronger binder upon ligand addition.2 However, from this it can be concluded that titrations to look at
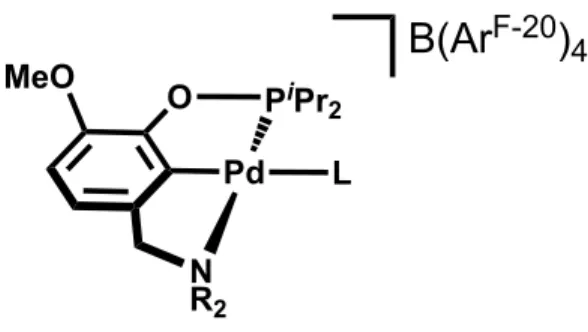
cation-bismethoxy interactions cannot be performed with the neutral chloride or cationic species in dichloromethane. Next, we sought to identify a ligand that binds better than ether to Pd (II) than the hemilabile bis-methoxy ligand in order to measure the binding affinity of that ligand to cations, as shown in Figure 13. We hoped that by binding a neutral ligand trans to the phenyl ring we could facilitate the bismethoxy to bind cations more tightly than when it was bound to the palladium center.
Figure 13. Pd (II) complex with a neutral ligand, L, bound to the metal center.
Our hope was to identify a small, relatively innocent neutral ligand that would bind strongly to palladium and displace the ether donation. Acetonitrile is small and is known to bind very well to palladium, so it was the first ligand that we attempted. Excess acetonitrile was added to 1 in dichloromethane and then the solvent removed, to reveal a new species, [(
MeO-O PiPr2
Pd L
N R2
MeO
B(Ar
EtMeONCOP)Pd(NCMe)][B(ArF-20)
4]. A rough titration was performed to explore if the acetonitrile
complex would allow us to explore cation-bismethoxy interactions. Solutions containing 5 mM
[(MeO-EtMeONCOP)Pd(NCMe)][B(ArF-20)
4] were titrated with 0.5, 1, and 1.5 equivalents of LiB(Ar F-24)
4, as shown in Figure 14.
Figure 14. Titration scheme for [(MeO-EtMeONCOP)Pd(NCMe)][B(ArF-20)
4] with LiB(ArF-24)4 in
dichloromethane.
We monitored the chemical shift of the methylene protons between the bismethoxyethylamine ligand and the phenyl backbone. These shifts were compared to zero equivalents of added salt and were plotted, as shown in Figures 15 and 16. The observable shift suggests there is a measurable binding of Li+ to the bismethoxy ligand. However, more data points are needed in
order to adequately fit the data and extract a binding affinity of cations to the bismethoxy complex. As we are interested in comparing the relative strength of interactions of the two complexes, full titrations with both [(MeO-EtMeONCOP)Pd(NCMe)][ B(ArF-20)
4] and [( MeO-15c5NCOP)Pd(NCMe)][B(ArF-20)
4] will be performed to measure their binding affinities to Li+
cations.
O PiPr
2
Pd N
N MeO
C
CD2Cl2
rt.
0 to 1.5 eq. [Li][B(ArF-24) 4]
O O
B(ArF-20)4
O PiPr
2
Pd N
N MeO
O O
C
B(ArF-20) 4
Figure 15. 1H NMR of methylene linker chemical shift.
Figure 16. Plot of chemical shift with varying amounts of LiB(ArF-24) 4.
4.205 4.21 4.215 4.22 4.225 4.23 4.235 4.24 4.245 4.25 4.255
0 0.002 0.004 0.006 0.008 0.01
δ
(ppm
)
IV. Conclusion
In conclusion, a novel palladium complex, 6, that bears a bis(methoxyethyl)aminophosphinite ligand and its cationic analog, 1, have been prepared. Comparative catalysis of the isomerization of 1-hexene with three palladium(II) complexes has been explored. The aza-crown ether substituted [(MeO-15c5NCOP)Pd][B(ArF-20)4] (2) catalyst is
tunable by cation additives, where the isomerization of 1-hexene proceeds in the presence of Li+,
while in the absence of Li+, catalysis does not occur. The isomerization of 1-hexene by [(
MeO-EtMeONCOP)Pd][B(ArF-20)4] (1) is less influenced by cations in solution. When no cation additives
are present in solution, isomerization still occurs, albeit at a slower rate, and we see a rate enhancement of 1.5 in the presence of Li+. Preliminary data with [(MeO-EtNCOP)Pd][B(ArF-20)
4] (3)
shows rapid isomerization of 1-hexene without cation additives and shows no rate enhancement upon the addition of Li+. The rapid isomerization with 3 without cation additives supports that
isomerization is happening at the palladium metal center.
Future work with comparative catalysis will focus on determining the order in palladium, order in Li+, and the influence of other ligands including diethyl ether in solution to elucidate our
mechanistic hypothesis. We predict that diethyl ether in solution slows down catalysis because it can bind to the metal center, and the rate of catalysis should linearly correlate with the amount of ether in solution. In order to further probe the proposed base-mediated allyl mechanism, external base will be added to the isomerization reaction. If the proposed mechanism is dominant, the addition of an external base is expected to speed up the reaction.
palladium will be titrated with various equivalents of LiB(ArF-24)
4. We hope to extract a binding
affinity from this titration data to compare cation-crown and cation-bismethoxy interactions.
V. Experimental
Synthesis General Considerations. Unless otherwise noted, standard glovebox and vacuum line techniques were utilized to maintain a N2 atmosphere during manipulation of all compounds. A Pure Process Technology solvent system was used to dry and degas organic solvents which were stored over 3 Å molecular sieves. Under standard glovebox operating conditions, pentane, diethyl ether, benzene, and toluene were used without purging, so traces of those solvents were present in the atmosphere and in the solvent bottles. 1H, 31P, and 13C NMR
spectra were recorded on 400, 500, or 600 MHz spectrometers. NMR characterization data are reported at 25 °C. All NMR solvents were purchased from Cambridge Isotopes Laboratories. Chloroform-d (CDCl3), and methylene chloride-d2 (CD2Cl2) were freeze−pump−thaw-degassed
three times, dried by passage through a small column of activated alumina, and stored over 3 Å molecular sieves. 1H and 13C chemical shifts are reported in parts per million relative to residual
protio solvent resonances. All 31P resonances are reported relative to 85% H
3PO4 external standard
(δ 0).
1.5 Hz, 1H), 6.79 (s, 2H), 3.87 (s, 3H), 3.62 (s, 2H), 3.48 (t, J = 6.1 Hz, 4H), 3.33 (s, 6H), 2.73 (t, J = 6.1 Hz, 4H). 13C NMR (151 MHz, Methylene Chloride-d
2) δ 145.75 (d, J = 3.6 Hz), 145.47 (d,
J = 2.1 Hz), 120.37, 115.07 (d, J = 4.8 Hz), 110.34 (d, J = 1.8 Hz), 71.09, 59.07, 58.46, 55.95.
Figure 22. 13C NMR Spectrum (600 MHz) of pre-ligand in CD 2Cl2.
Synthesis of (MeO-EtMeONCOP)H. Under an N
2 atmosphere, 1 (299 mg, 1.11 mmol) was
dissolved in THF (6 mL). Triethylamine (170 μL, 1.21mmol) was added to the solution. In a separate vial, PiPr2Cl (175 μL, 1.10 mmol) was dissolved in THF (6 mL). Both solutions were
cooled to –35° in the freezer. The PiPr
2Cl solution was added slowly to the
MeO-EtMeONCOH/triethylamine solution and the reaction was allowed to warm to room temperature
with stirring. After three hours, the mixture was concentrated under vacuum to a colorless oil and white salt precipitate. The crude oil was dissolved in ether and filtered through an alumina column to yield the product as a colorless oil (285.6 mg, 0.741 mmol, 67% yield). 1H NMR (400 MHz,
Chloroform-d) δ 6.81 (d, J = 8.2 Hz, 1H), 3.84 (s, 3H), 3.64 (s, 2H), 3.48 (t, J = 6.2 Hz, 4H), 3.34 (s, 6H), 2.74 (t, J = 6.2 Hz, 4H), 1.97 (h, J = 7.3 Hz, 2H), 1.33 – 0.99 (m, 15H). 31P NMR (400
MHz, CDCl3) δ 156.52. 13C NMR (151 MHz, Methylene Chloride-d
2) δ 149.45, 148.24 (d, J =
0 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150
13C ppm AS-4-141_13C.2.fid
8.3 Hz), 132.40, 122.19, 120.08 (d, J = 14.8 Hz), 111.79, 71.31, 58.74 (d, J = 77.8 Hz), 55.76, 28.46 (d, J = 18.6 Hz), 18.49 – 15.16 (m).
Figure 23. 1H NMR Spectrum (400 MHz) of bis(methoxy)ethylaminophosphinite ligand in
Figure 24. 31P NMR Spectrum (400 MHz) of bis(methoxy)ethylaminophosphinate in CDCl3.
Figure 25. 13C NMR Spectrum (600 MHz) of bis(methoxy)ethylaminophosphinate in CD 2Cl2. 10
20 30 40 50 60 70 80 90 100 110 120 130 140 150
f1 (ppm)
-200 0 200 400 600 800 1000 1200 1400 1600 1800 2000 2200 2400 2600 2800 3000
Synthesis of (MeO-EtMeONCOP)Pd(Cl). (MeO-EtMeONCOP) (285 mg, 0.739 mmol) and
Pd(COD)Cl2 (195.4 mg, 0.684 mmol) were dissolved in toluene (75 mL). The reaction was
refluxed for 48 hours under a N2 atmosphere. The toluene was removed in vacuo. The crude
product was dissolved in pentane, filtered, diluted, and put in the freezer at -35 °C to yield the product as a pale-yellow crystal (380 mg, 0.722, 97% yield). 1H NMR (500 MHz, CD
2Cl2) δ 6.65
(q, J = 8.2 Hz, 1H), 4.37 (s, 1H), 4.00 (hept, J = 5.3 Hz, 2H), 3.81 (s, 1H), 3.42 (ddd, J = 12.8, 8.1, 3.1 Hz, 1H), 3.28 (s, 2H), 3.09 – 2.97 (m, 1H), 2.44 (qd, J = 13.9, 6.0 Hz, 1H), 1.49 – 1.33 (m, 5H), 1.32 (d, J = 7.0 Hz, 2H), 0.92 (t, J = 6.9 Hz, 0H).31P NMR (500 MHz, CD2Cl2) δ 202.14. 13C
NMR (151 MHz, Methylene Chloride-d2) δ 144.76, 143.96 – 139.84 (m), 116.39, 110.48, 71.39,
67.42, 59.94, 58.58, 56.23, 29.26 (d, J = 25.5 Hz), 17.69 – 14.12 (m).
Figure 26. 1H NMR Spectrum (600 MHz) of (MeO-EtMeONCOP)Pd(Cl) inCD2Cl2.
Figure 27. 31P NMR Spectrum (500 MHz) of (MeO-EtMeONCOP)Pd(Cl) inCD 2Cl2.
Figure 28. 13C NMR Spectrum (600 MHz) of (MeO-EtMeONCOP)Pd(Cl) inCD2Cl2.
Synthesis of [(MeO-EtMeO)NCOP)Pd][B(Ar-F20)
4]. A 20 mL scintillation vial was charged with (MeO-EtMeONCOP)Pd(Cl) (21.8 mg, 0.0414 mmol) and 3 mL C
6H5Cl. In a separate vial, trityl
tetrakis(pentafluorophenyl)borate, [Ph3C][B(ArF-20)4] (37.5 mg, 0.0407 mmol), was dissolved in 2
mL C6H5Cl. To the [Ph3C][B(ArF-20)4] solution was added Et3SiH (6.5 μL, 0.0407 mmol) with
stirring to generate a silylium reagent in situ. After ten minutes, the silylium solution was added to the (MeO-EtMeONCOP)Pd(Cl) solution with stirring. The reaction was allowed to stir for 1 hour
before the solvent was removed under vacuum and washing with pentane. To remove a colored impurity, the product was dissolved in diethyl ether and filtered through an alumina column to yield the product as a white powder (0.0299 mmol, 73% yield).1H NMR (400 MHz, Methylene
Chloride-d2) δ 6.71 (s, 2H), 4.33 (s, 2H), 4.11 – 3.93 (m, 3H), 3.84 (d, J = 4.7 Hz, 6H), 3.52 (s,
11H), 3.36 (d, J = 13.6 Hz, 3H), 2.43 (q, J = 7.1 Hz, 2H), 1.48 – 1.23 (m, 29H). 31P NMR (162
MHz, Methylene Chloride-d2) δ 197.94. 13C NMR (151 MHz, Methylene Chloride-d2) δ 151.18,
Figure 29. 1H NMR Spectrum (400 MHz) of [(MeO-EtMeO)NCOP)Pd][B(ArF-20)4] in CD2Cl2. 0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 8.0 f1 (ppm) -1000 0 1000 2000 3000 4000 5000 6000 7000 8000 9000 10000 11000 12000 13000 14000
KG 1-32 box.1.fid 3/1 alumina column 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240 250 260 270 280 290 300 f1 (ppm) -500 0 500 1000 1500 2000 2500 3000 3500 4000 4500 5000
Figure 30. 31P NMR Spectrum (400 MHz) of [(MeO-EtMeO)NCOP)Pd][B(ArF-20)
4] in CD2Cl2.
Figure 31. 13C NMR Spectrum (600 MHz) of [(MeO-EtMeO)NCOP)Pd][B(ArF-20)
4] in CD2Cl2.
Synthesis of [(MeO-EtMeONCOP)Pd(NCMe)][B(ArF-20)4] In a 4 mL vial, [(
MeO-EtMeONCOP)Pd][B(ArF-20)4] (20.7 mg, 0.0177 mg) was dissolved in dichloromethane (2 mL).
Several drops of acetonitrile were added. The solvent was removed in vacuo and then the product washed with pentane to reveal [(MeO-EtMeONCOP)Pd(NCMe)][B(ArF-20)4] as a white powder (21
mg, 0.0174 mmol, 98.3%). 1H NMR (500 MHz, Methylene Chloride-d
2) δ 6.71 – 6.64 (m, 2H),
4.20 (s, 2H), 3.92 – 3.73 (m, 7H), 3.32 (s, 9H), 3.15 (ddt, J = 14.0, 7.2, 3.7 Hz, 2H), 2.41 (h, J = 7.0 Hz, 2H), 2.28 (s, 3H), 1.41 – 1.18 (m, 16H). 31P NMR (500 MHz, Methylene Chloride-d
2) δ
201.15.
0 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160
13C ppm KG-1-108_13C.2.fid
Figure 32. 1H NMR (500 MHz) of [(MeO-EtMeONCOP)Pd(NCMe)][B(ArF-20)4] in CD2Cl2.
Figure 33. 31P NMR (500 MHz) of [(MeO-EtMeONCOP)Pd(NCMe)][B(ArF-20)4] in CD2Cl2
-0.5 0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5 7.0 7.5 f1 (ppm) -100 0 100 200 300 400 500 600 700 800 900 1000 1100 1200 1300 1400 1500 1600 1700 1800 KG 1-109A.1.fid 4/4 1H 1 5 .5 6 3 .0 9 2 .1 1 2 .1 3 8 .4 3 7 .2 0 2 .0 9 2 .0 0 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240 250 260 270 280 290 300 310 320 330 340 350 360 370 380 f1 (ppm) -40 -20 0 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 KG 1-109A.2.fid 4/4 31P 2 0 1 .1 5
General Procedure for Isomerization of 1-Hexene. The conditions for [(
MeO-EtMeO)NCOP)Pd][B(ArF-20)
4] are given as an example; other reactions were carried out analogously.
A stock solution of [(MeO-EtMeO)NCOP)Pd][B(ArF-20)
4] was prepared by dissolving [(
MeO-EtMeONCOP)Pd][B(ArF-20)
4] (10 mg, 0.0086 mmol) in CH2Cl2 (600 μL). This solution (175 μL)
was transferred to two 4 mL vials and the solvent removed. In a separate 20 mL vial, Li(B(Ar F-24)4 (10 mg, 0.00915 mmol), was dissolved in CH2Cl2 (700 μL); several 190 μL aliquots were
transferred to 4 mL vials and the solvent removed to give known quantities of the lithium salt. A stock solution of diethyl ether (18.6 μL, 0.000177 mmol) and mesitylene (21 μL, 0.00015 mmol) in CD2Cl2 was prepared. A 500 μL aliquot of the stock solution was transferred to the vials
containing Pd and then 1-hexene (31 μL, 0.250 mmol) was added. These solutions were added to the vials containing LiB(ArF-24)
4 and the reaction mixtures were finally added to Teflon-sealed
NMR tubes. The reactions were monitored by 1H NMR for two weeks and the conversion and
yield were determined by integration relative to the internal standard.
General procedure for titration experiments. Conditions for (MeO-EtMeONCOP)Pd(Cl) are
given here, but analogous procedures were used for other complexes. In a glovebox, a 20 mM stock solution of (MeO-EtMeONCOP)Pd(Cl) was prepared by dissolving 31.6 mg (0.06 mmol) in 3
mL of dichloromethane in a 20 mL scintillation vial. To eight 4 mL vials, 250 µL of (
MeO-EtMeONCOP)Pd(Cl) was transferred and the solvent was removed in vacuo to yield a white film.
A 21 mM stock solution of LiB(ArF-20)
4 was prepared by dissolving 54 mg (0.0620 mmol) of
LiB(ArF-20)4 in 3 mL of deuterated dichloromethane in a 20 mL scintillation vial. Aliquots of 0,
60, 120, 180, 240, 365, 425, and 485 µL of LiB(ArF-20)4 stock solution were added to the 4 mL
vials containing (MeO-EtMeONCOP)Pd(Cl), resulting in solutions which contained 0, 0.25, 0.5, 0.75,
amount of dichloromethane-d was added to each vial to reach a total volume of 500 µL and analyzed by 1H NMR (600 MHz).
References
1. Kita, M. R.; Miller, A. J. M.; J. Am. Chem. Soc. 2014, 136, 14519-14529 2. Miller, A. J. M.; Dalton Trans., 2017, 11987-12000
3. Yoo, C.; Dodge, H. M.; Miller, A. J. M.; Chem. Comm. 2019
4. Kita, M. R.; Miller, A. J. M.; Angew. Chem. Int. Ed. 2017, 56, 5498-5502
5. Hassam, M.; Taher, A.; Arnott, G. E.; Green, I. R.; Van Otterlo, W. A. L.; Chemical Reviews.2015, 115 (11), 5462-5569
6. Choi, J. H.; Kwon, J. K.; RajanBabu, T. V.; Lim, H. J.; Adv. Synth. Catal. 2013, 355,
3633-3638
7. Girgor’eva, N. G.; Talipova, R. R.; Korzhova, L.F.; Vosmerikov, A. V.; Kutepov, B.I.; Dzhemilev, U. M.; Russian Chemical Bulletin Int. Ed. 2009, 38, 59-63
8. Biswas, S.; Comments on Inorganic Chemistry. 2015, 35, 301-330.
9. Smith, J. B.; Kerr, S. H.; White, P. S.; Miller, A. J. M.; Organometallics. 2017, 36, 3094-3103.


![Figure 7. Halide abstraction to form [( MeO-EtMeO NCOP)Pd][B(Ar F-20 ) 4 ].](https://thumb-us.123doks.com/thumbv2/123dok_us/8329794.2209343/9.918.111.805.534.810/figure-halide-abstraction-form-meo-etmeo-ncop-pd.webp)

![Figure 14. Titration scheme for [( MeO-EtMeO NCOP)Pd(NCMe)][B(Ar F-20 ) 4 ] with LiB(Ar F-24 ) 4 in dichloromethane](https://thumb-us.123doks.com/thumbv2/123dok_us/8329794.2209343/16.918.101.815.279.505/figure-titration-scheme-meo-etmeo-ncop-ncme-dichloromethane.webp)
