3-2018
Unveiling the Effects of Linker Substitution in
Suzuki Coupling with Palladium Nanoparticles in
Metal–Organic Frameworks
Xinle Li
Iowa State University and Ames Laboratory
Biying Zhang
Iowa State University and Ames Laboratory, [email protected] Ryan Van Zeeland
Ames Laboratory
Linlin Tang Ames Laboratory
Yuchen Pei
Iowa State University and Ames Laboratory, [email protected]
See next page for additional authors
Follow this and additional works at:https://lib.dr.iastate.edu/ameslab_manuscripts
Part of theNanoscience and Nanotechnology Commons, and theOrganic Chemistry Commons
This Article is brought to you for free and open access by the Ames Laboratory at Iowa State University Digital Repository. It has been accepted for inclusion in Ames Laboratory Accepted Manuscripts by an authorized administrator of Iowa State University Digital Repository. For more information, Recommended Citation
Li, Xinle; Zhang, Biying; Van Zeeland, Ryan; Tang, Linlin; Pei, Yuchen; Goh, Tian Wei; Stanley, Levi M.; and Huang, Wenyu, "Unveiling the Effects of Linker Substitution in Suzuki Coupling with Palladium Nanoparticles in Metal–Organic Frameworks" (2018).Ames Laboratory Accepted Manuscripts. 157.
Palladium Nanoparticles in Metal–Organic Frameworks
Abstract
The establishment of structure–property relationships in heterogeneous catalysis is of prime importance but remains a formidable challenge. Metal–organic frameworks (MOFs) featuring excellent chemical tunability are emerging as an auspicious platform for the atomic-level control of heterogeneous catalysis. Herein, we encapsulate palladium nanoparticles (Pd NPs) in a series of isoreticular mixed-linker MOFs, and the obtained MOF-Pd NPs catalysts were used to unveil the electronic and steric effects of linker substitution on the activity of these catalysts in the Suzuki–Miyaura cross-coupling reactions. Significantly, m-6,6′-Me2bpy-MOF-Pd exhibits a remarkable enhancement in the activity compared to non-functionalized m-bpy-MOF-m-6,6′-Me2bpy-MOF-Pd and m-4,4′-Me2bpy-MOF-Pd. This study unambiguously demonstrates that the stereoelectronic properties of linker units are crucial to the catalytic activity of nanoparticles encapsulated in MOFs. More interestingly, the trend of activity change is consistent with our previous work on catalytic sites generated in situ from Pd(II) coordinated in MOFs bearing the same functional groups, which suggests that both Pd NPs and MOF-Pd(II) catalysts generate similar active centers during Suzuki–Miyaura coupling reactions. This work paves a new avenue to the fabrication of advanced and tunable MOF-based catalysts through rational linker
engineering.
Keywords
Isoreticular metal–organic frameworks, Heterogeneous catalysis, Suzuki–Miyaura cross-coupling, Structure–activity relationship
Disciplines
Chemistry | Nanoscience and Nanotechnology | Organic Chemistry
Authors
Unveiling the Effects of Linker Substitution in Suzuki
Coupling Reaction with Palladium Nanoparticles in
Metal-Organic Frameworks
Xinle Li 1,2, Biying Zhang 1,2, Ryan Van Zeeland 1, Linlin Tang 1, Yuchen Pei 1,2, Zhiyuan Qi 1,2, Tian Wei Goh 1,2, Levi M. Stanley 1,*, and Wenyu Huang 1,2,*
1 Department of Chemistry, Iowa State University, Ames, Iowa 50011, United States;
2 Ames Laboratory, U.S. Department of Energy, Ames, Iowa 50011, United States;
* Correspondence: [email protected]; [email protected]
Abstract: The establishment of structure–property relationships in heterogeneous catalysis is of
prime importance but remains a formidable challenge. Metal-organic frameworks (MOFs)
featuring excellent chemical tunability are emerging as an auspicious platform for the atomic-level
control of heterogeneous catalysis. Herein, we encapsulate palladium nanoparticles (Pd NPs) in a
series of isoreticular mixed-linker MOFs, and the obtained MOF-Pd NPs catalysts were used to
unveil the electronic and steric effects of linker substitution on the activity of these catalysts in the
Suzuki–Miyaura cross-coupling reactions. Significantly, m-6,6´-Me2bpy-MOF-Pd exhibits a
remarkable enhancement in the activity compared to non-functionalized bpy-MOF-Pd and
m-4,4´-Me2bpy-MOF-Pd. This study unambiguously demonstrates that the stereoelectronic
properties of linker units are crucial to the catalytic activity of nanoparticles encapsulated in MOFs.
More interestingly, the trend of activity change is consistent with our previous work on catalytic
sites generated in situ from Pd(II) coordinated in MOFs bearing the same functional groups, which
suggests that both MOF-Pd NPs and MOF-Pd(II) catalysts generate similar active centers during
Suzuki-Miyaura coupling reactions. This work paves a new avenue to the fabrication of advanced
Keywords: isoreticular metal-organic frameworks; heterogeneous catalysis; Suzuki-Miyaura
cross-coupling; structure-activity relationship.
The past decades have witnessed the spectacular rise of research in metal-organic
frameworks (MOFs) due to their multifunctional applications including gas storage, separation,
chemical sensors, drug delivery, and heterogeneous catalysis [1]. MOFs are assembled with metal
ions/clusters and rigid organic linkers and have emerged as an increasingly popular platform for
heterogeneous catalysis due to their ultrahigh surface areas and exceptional
compositional/structural versatility [2]. Of particular interest, MOFs have been rapidly adopted as
novel hosts of nanoparticles (NPs@MOFs) due to multiple merits, such as high surface area,
enhanced stability, and beneficial mass diffusion [3]. Unlike the traditional porous supports
(zeolites and mesoporous silica), the interior chemical environment of MOFs can be chemically
tuned at an atomic level through the thoughtful combination of the building units (metal clusters
and organic linkers) via pre- or post-synthetic modifications [4]. These “programmable” features
of MOFs make them well suited for establishing clear structure–function relationships. It has been
well documented that the manipulation of the functionalities in isoreticular MOFs acts as a potent
approach to control and modulate the properties of parent MOFs for gas sorption capacity and
uptake selectivity, water stability, luminescence, mechanical properties, and catalysis [5-10].
Recently, our group encapsulated Pd NPs in a series of isoreticular UiO-66-R (R = H, NH2, and
OMe) and the alteration of the functional groups on the organic linkers in the UiO-66 lead to
different activities and selectivities in catalytic reactions [11]. This strategy demonstrates that the
atomically precise control of heterogeneous catalytic sites could be achieved via the variation of
in heterogeneous catalysis. However, the systematic studies on the effects of linker substitution in
MOFs on heterogeneous catalysis remain highly underexplored, and yet such studies are poised to
exert a significant impact on the design of efficient MOF-based catalysts [12].
The Suzuki-Miyaura cross-coupling reaction is a privileged transformation that has been
extensively exploited in synthetic organic and materials chemistry [13]. Homogeneous Pd
complexes serve as powerful catalysts but suffer from poor reusability due to catalyst deactivation.
In contrast, heterogeneous Pd catalysts are advantageous due to their facile catalyst recovery and
reuse. Numerous studies have been devoted to the development of heterogeneous catalysts, such
as Pd supported on C [14], zeolite [15], silica [16], magnetic supports (predominately Fe3O4) [17],
graphene oxide [18], and chitosan [19]. Among the abundant heterogeneous Pd catalysts for the
Suzuki-Miyaura cross-coupling reaction, MOF-supported Pd catalysts have received increasing
attention due to their aforementioned distinctive advantages. Considerable progress has been
achieved on the immobilization of metal complexes or NPs in the MOFs for Suzuki-Miyaura
cross-coupling reaction [20-26]. While most of the attention has been concentrated on the catalytic
performances of the catalysts, studies to unveil how the linker substitution in MOFs catalysts can
impact the resultant Suzuki-Miyaura cross-coupling reaction remain rare and to be well explored.
We tackled this challenge by systematically developing Pd NPs encapsulated inside a series
of isoreticular bipyridyl-MOFs (termed m-bpy-MOF-Pd, m-6,6´-Me2bpy-MOF-Pd and
m-4,4´-Me2bpy-MOF-Pd) with the aim to investigate the effect of linker substitution on Suzuki-Miyaura
cross-coupling reaction activity. UiO-67 MOFs lined with 2,2′-bipyridyl moieties were judiciously
m2 g–1) and stabilities. More importantly, the uncoordinated bipyridine moieties in these MOFs
can act as anchoring sites for loading Pd complexes and stabilizing Pd NPs under the reaction
conditions [27, 28]. These isoreticular MOFs are crystalline, porous and robust. Remarkably,
m-6,6´-Me2bpy-MOF-Pd exhibits a dramatic enhancement in the catalytic activity of Suzuki-Miyaura
cross-coupling reaction of iodobenzene compared to m-bpy-MOF-Pd and
m-4,4´-Me2bpy-MOF-Pd. These reactions proceed through cross-coupling of the haloarene and phenylboronic acid
without homocoupling of either reaction partner. Furthermore, m-6,6´-Me2bpy-MOF-Pd is truly a
heterogeneous catalyst evidenced by leaching and recycling tests. This work constitutes a
systematic study of the effect of linker substitution on Pd NPs in catalytic Suzuki-Miyaura
cross-coupling in MOFs.
Scheme 1. Schematic representation of the synthesis of Pd NPs encapsulated in isoreticular
m-bpy-MOFs. Green sphere and octahedra represent Zr clusters, while orange spheres represent Pd
NPs.
We synthesized the substituted 2,2′-bipyridine-5,5′-dicarboxylic acid (H2bpydc)
derivatives, i.e. 4,4′-dimethyl-[2,2′-bipyridine]-5,5′-dicarboxylic acid and
H2bpydc linkers, we prepared mixed-linker MOFs (bpy-MOF, 6,6´-Me2bpy-MOF, and
m-4,4´-Me2bpy-MOF) by solvothermal reaction of ZrCl4 and H2bpydc derivatives (Scheme 1).
Powder X-ray diffraction (PXRD) patterns demonstrate that these MOFs possess isoreticular,
crystalline structure, evidenced by the consistency between as-synthesized MOFs with a simulated
UiO-67 (Figure S1). The exact linker ratio is quantified by 1H NMR after digesting the
[image:7.612.72.545.247.406.2]as-synthesized MOFs using HF in d6-DMSO (Figure S2, Table S1).
Figure 1. TEM images of (a) m-bpy-MOF-Pd; (b) m-6,6´-Me2bpy-MOF-Pd; (c)
m-4,4´-Me2bpy-MOF-Pd.
The encapsulation of Pd NPs in m-MOFs (m-MOF-Pd) was conducted by the
post-synthetic metalation of the MOFs with PdCl2(CH3CN)2 and followed by reduction at 240 °C for 4
hours in a 10% H2/Ar stream (Scheme 1). PXRD studies show that the m-MOF-Pd catalysts remain
intact during the metalation and reduction processes (Figure S3). The Pd content in m-MOF-Pd
was quantitatively determined by inductively coupled plasma-mass spectroscopy (ICP-MS), and
the Pd loadings are 2.8, 2.9, and 3.0 wt.% for bpy-MOF-Pd, 6,6´-Me2bpy-MOF-Pd, and
4,4´-Me2bpy-MOF-Pd, respectively. Transmission electron microscopy (TEM) images of the
2.0 ± 0.3 nm (Figure 1). It is noteworthy that Pd NPs size are slightly larger than the dimension of
the MOF cavities (~1.6 nm), which might be attributed to local defects or deformations of the host
MOFs as usually seen in other NPs@MOFs composites [28]. To further confirm that Pd NPs were
truly confined inside the cavities of bpy-MOFs, m-6,6´-Me2bpy-MOF-Pd and Pd deposited on the
external surface of m-MOF (Pd/m-6,6´-Me2bpy-MOF) catalyst were utilized as catalysts in the
hydrogenation of two probe molecules (i.e. styrene and tetraphenylethylene). Tetraphenylethylene
is too bulky to diffuse through the pore aperture of UiO-67 (6.6 Å) and thus should not be
hydrogenated by the Pd NPs confined inside the MOFs, whereas the small styrene molecule can
readily access the encapsulated Pd NPs and undergo hydrogenation [29]. As shown in Figure S4,
m-6,6´-Me2bpy-MOF-Pd exhibited high activity in styrene hydrogenation but gave no detectable
activity in tetraphenylethylene hydrogenation. In contrast, Pd/m-6,6´-Me2bpy-MOF is active for
both reductions due to the exposed Pd atoms outside the MOF. Taken together, the TEM images
and catalytic hydrogenation of the probe molecules are consistent with the fact that Pd NPs were
encapsulated inside the cavities of the UiO-67 MOFs prepared for this study.
N2 physisorption isotherms of m-MOFs and m-MOF-Pd materials exhibit typical type-I
adsorption behavior, which suggests the microporous nature of the MOF materials (Figure S5).
The Brunauer–Emmett–Teller (BET) surface areas of bpy-MOF, 6,6′-Me2bpy-MOF, and
m-4,4′-Me2bpy-MOF were measured as 2600 m2 g–1, 2300 m2 g–1 and 2200 m2 g–1, respectively
(Table S2). The introduction of Pd NPs leads to decreased BET surface areas of the corresponding
MOFs. The BET surfaces areas of m-bpy-MOF-Pd, m-6,6´-Me2bpy-MOF-Pd, and
m-4,4´-Me2bpy-MOF-Pd decrease to 2200, 2000, and 1800 m2 g–1, presumably due to the incorporation
Using these isoreticular m-bpy-MOFs-Pd catalysts, we aimed to reveal effects of linker
functionality on activities of encapsulated Pd NPs in Suzuki-Miyaura reactions between
iodobenzene and phenylboronic acid. We previously tested MOF stability in a variety of solvents
and base combinations such as DMF/H2O (1:1), EtOH, EtOH/H2O (1:1), toluene/H2O (9:1),
toluene with either potassium carbonate or potassium fluoride as the base [30]. These
m-bpy-MOFs are only stable in pure toluene with potassium carbonate, which was used for the model
[image:9.612.202.413.270.491.2]coupling reaction.
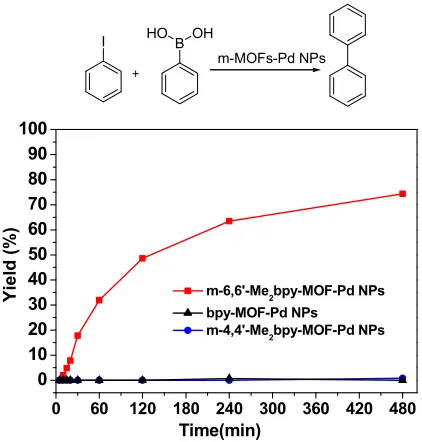
Figure 2. The catalytic difference of m-MOF-Pd in the Suzuki−Miyaura cross-coupling reaction
of iodobenzene. Reaction conditions: iodobenzene (0.1 mmol), phenylboronic acid (0.15 mmol),
K2CO3 (0.2 mmol), toluene (0.5 mL), m-6,6′-Me2bpy-MOF-Pd (1.0 mol % Pd), 85 °C.
In the Suzuki-Miyaura coupling reaction, m-bpy-MOF-Pd, m-4,4´-Me2bpy-MOF-Pd, and
m-6,6´-Me2bpy-MOF-Pd displayed dramatic differences in their catalytic activity (Figure 2). The
m-6,6´-Me2bpy-MOF-Pd exhibited a remarkable enhancement in the activity compared to non-0 60 120 180 240 300 360 420 480
0 10 20 30 40 50 60 70 80 90 100
Yie
ld
(%
)
Time(min)
m-6,6'-Me2bpy-MOF-Pd NPs bpy-MOF-Pd NPs m-4,4'-Me2bpy-MOF-Pd NPs
I HOB OH
functionalized m-bpy-MOF-Pd and m-4,4´-Me2bpy-MOF-Pd, which gave nearly negligible
activity. The neat m-bpy-MOFs without Pd were catalytically inactive, confirming that Pd centers
are the active sites for the Suzuki-Miyaura coupling reaction (Table S3, entry 1). In comparison to
commercial Pd/carbon, m-6,6´-Me2bpy-MOF-Pd exhibited a higher activity (Figure S6). To
exclude the homocoupling of aryl halides or arylboronic acids catalyzed by palladium species, we
performed the Suzuki-Miyaura coupling reaction of iodobenzene and 4-methylphenylboronic acid
with m-6,6´-Me2bpy-MOF-Pd under identical reaction conditions and obtained 4-methyl-1,1
-biphenyl as the only product (Table S3, entry 2). This result shows that biaryl coupling products
are not formed via homocoupling of either the arylboronic acid or the iodoarene coupling partners.
To our surprise, we found that the trend of activity change for Pd NPs encapsulated in
m-bpy-MOF, m-4,4´-Me2bpy-MOF and m-6,6´-Me2bpy-MOF is the same as that demonstrated in
our previous work on catalysts generated from Pd(II) coordinated in m-bpy-MOFs with the same
functional groups [30]. Moreover, the rate constant of the coupling reaction on Pd NPs
encapsulated in m-6,6´-Me2bpy-MOF (0.36 h-1, assuming pseudo-first-order kinetics) is only
slightly lower than that on Pd(II) coordinated in the same MOF (0.45 h-1). Pd(II) coordinated in
m-bpy-MOF and m-4,4´-Me2bpy-MOF also gave negligible activity, which is the same as Pd NPs
encapsulated in the same m-bpy-MOFs. These similarities in activity between Pd NPs and Pd(II)
encapsulated/coordinated in m-bpy-MOFs suggests that the active center for the coupling reaction
could be the same.
The nature of the active center, solid surfaces or molecular species, for Suzuki-Miyaura
coupling reaction when Pd NPs are used as catalysts has long been a topic of debate [32,33]. As
demonstrated previously, Pd NPs can act as a reservoir of molecular species(e.g.,[Pd(Ar)X species
studies suggest that the molecular species, detached from the Pd NPs and coordinated to the
bipyridine units in m-bpy-MOFs is likely the active center in Suzuki-Miyaura coupling reaction
when either Pd NPs encapsulated in MOF or Pd(II) coordinated in m-bpy-MOFs are employed as
the precatalysts. Since m-bpy-MOFs have a significant number of bpy sites available for
coordinating Pd, Pd atoms/ions shed from the surface of Pd NPs can be coordinated to these bpy
sites before they leach out of the m-bpy-MOFs. This speculation is consistent with our leaching
test and shows that upon the removal of the solid catalyst at ~30% conversion, no further increase
in the yield of biphenyl was observed (Figure 3). Moreover, ICP-MS analysis indicates that the
hot filtrate contains a negligible amount of Pd (< 0.1% of added Pd).
Figure 3. Hot filtration test of m-6,6-Me2bpy-MOF-Pd in the Suzuki-Miyaura cross-coupling
reaction. Reaction conditions: iodobenzene (0.1 mmol), phenylboronic acid (0.15 mmol), K2CO3
(0.2 mmol), toluene (0.5 mL), m-6,6′-Me2bpy-MOF-Pd (1.0 mol % Pd), 85 °C for 2 h.
To test the possible deactivation of the catalyst under our reaction conditions, we recycled
the m-6,6´-Me2bpy-MOF-Pd catalyst in coupling reactions at approximately 50% conversion for
0 30 60 90 120
0 10 20 30 40 50
Yi
eld
(
%
)
Time (min)
[image:11.612.201.424.347.520.2]each repeated run. The m-6,6´-Me2bpy-MOF-Pd catalyst can be reused for three consecutive runs
without a significant decrease in the yield of biphenyl (Figure S7). The PXRD analysis of the
recovered catalysts verified that the structural integrity of the bpy-MOFs is retained after the
reaction (Figure S8). However, TEM images of the used m-6,6´-Me2bpy-MOF-Pd catalysts show
significant aggregation (Figure S9). We speculate that the aggregation of Pd NPs after catalysis
might be due to the loss of MOF restriction stemming from the linker dissociation in the MOFs at
elevated reaction temperature [36]. Given that the activity remains consistent during our recycling
tests despite the size increase of the Pd NPs, we speculate that the molecular species (i.e., Pd
atom/ions from the Pd NP reservoir) act as the active center in this system since their concentration
does not change regardless the size of the Pd NPs. This speculation is also consistent with previous
mechanism studies of Pd NPs in cross-coupling reactions [37,38].
In summary, we have developed Pd NPs encapsulated in a series of isoreticular
mixed-linker bipyridyl MOFs as catalysts (m-bpy-MOF-Pd, m-6,6´-Me2bpy-MOF-Pd and
m-4,4´-Me2bpy-MOF-Pd) for Suzuki-Miyaura cross-coupling reactions. The modification of MOF linkers
serves to unveil the electronic and steric effects of the linker substitution in varying their catalytic
properties. Remarkably, the alteration of linker units in MOFs can readily impact the activity of
MOF-Pd catalysts in Suzuki-Miyaura cross-coupling reactions. m-6,6´-Me2bpy-MOF-Pd exhibits
dramatically enhanced activity compared to m-bpy-MOF-Pd and m-4,4´-Me2bpy-MOF-Pd in
model Suzuki-Miyaura cross-coupling reactions due to the steric properties at the bpy-palladium
sites. More interestingly, this trend of activity changes for Pd NPs encapsulated in these
isoreticular m-bpy-MOFs is consistent with our previous work on catalysts generated from Pd(II)
coordinated in these MOFs, which suggest that both Pd NPs and
m-6,6´-Me2bpy-MOF-Pd in the Suzuki-Miyaura cross-coupling reaction is demonstrated by hot
filtration test, ICP-MS analysis of the hot filtrate, and recycling test. The present work not only
highlights the importance of linker engineering for MOF-encapsulated Pd NPs in Suzuki-Miyaura
cross-coupling reactions but also sheds light on a deeper understanding of controlling
heterogeneous catalytic sites through their surrounding chemical environments at the atomic-level
for the development of MOFs-based heterogeneous catalysts.
Acknowledgments
This work is partially supported by NSF grant CHE-1566445. We also thank Ames Laboratory (Royalty
Account) and Iowa State University for startup funds. The Ames Laboratory is operated for the U.S.
Department of Energy by Iowa State University under Contract No. DE-AC02-07CH11358. We thank
Gordon J. Miller for the use of PXRD in his group.
Notes
The authors declare no competing financial interest.
References
[1] Furukawa H, Cordova KE, O’Keeffe M, Yaghi OM (2013) Science 341:1230444.
[2] Chughtai AH, Ahmad N, Younus HA, Laypkov A, Verpoort F (2015) Chem Soc Rev 44: 6804-6849. [3] Dhakshinamoorthy A, Garcia H (2012) Chem Soc Rev 41: 5262-5284.
[4] Kim M, Cahill JF, Fei HH, Prather KA, Cohen SM (2012) J Am Chem Soc 134: 18082-18088. [5] Hendon CH, Tiana D, Fontecave M, Sanchez C, D’arras L, Sassoye C, Rozes L, Mellot-Draznieks C,
Walsh A (2013) J Am Chem Soc 135:10942-10945.
[6] Burtch NC, Jasuja H, Walton KS (2014) Chem Rev 114:10575-10612.
[7] Bauer CA, Timofeeva TV, Settersten TB, Patterson BD, Liu VH, Simmons BA, Allendorf MD (2007) J Am Chem Soc 129:7136-7144.
[8] Tan JC, Cheetham AK (2011) Chem Soc Rev 40:1059-1080.
[9] Vermoortele F, Vandichel M, Van de Voorde B, Ameloot R, Waroquier M, Van Speybroeck V, De Vos DE (2012) Angew Chem Int Ed 51: 4887-4890.
[10] Goh TW, Xiao C, Maligal-Ganesh RV, Li XL, Huang WY (2015) Chem Eng Sci 124: 45-51. [11] Li XL, Goh TW, Li L, Xiao CX, Guo ZY, Zeng XC, Huang WY (2016) ACS Catal 6: 3461-3468. [12] Choi KM, Na K, Somorjai GA, Yaghi OM (2015) J Am Chem Soc 137:7810-7816.
[14] Maegawa T, Kitamura Y, Sako S, Udzu T, Sakurai A, Tanaka A, Kobayashi Y, Endo K, Bora U, Kurita T (2007) Chem Eur J 13: 5937-5943.
[15] Artok L, Bulut H, (2004) Tetrahedron Lett 45: 3881-3884. [16] Sahu D, Das P (2015) RSC Adv 5: 3512-3520.
[17] Sydnes MO (2017) Catalysts 7: 35-49.
[18] Yamamoto SI, Kinoshita H, Hashimoto H, Nishina Y (2014)Nanoscale 6: 6501-6505. [19] Hardy JJE, Hubert S, Macquarrie DJ, Wilson AJ (2004) Green Chem 6: 53-56. [20] Chen LY, Rangan S, Li J, Jiang HF, Li YW (2014) Green Chem 16: 3978-3985. [21] Chen LY, Gao Z, Li YW, (2015) Catal Today 245:122-128.
[22] Pascanu V, Yao Q, Bermejo Gómez A, Gustafsson M, Yun Y, Wan W, Samain L, Zou XD, Martín‐ Matute B (2013) Chem Eur J 19:17483-17493.
[23] Yuan BZ, Pan YY, Li YW, Yin B, Jiang HF (2010) Angew Chem Int Ed 49: 4054-4058.
[24] Carson F, Pascanu V, Bermejo Gómez A, Zhang Y, Platero ‐Prats AE, Zou XD, Martín‐Matute B, (2015) Chem Eur J 21: 10896-10902.
[25] Saha D, Sen R, Maity T, Koner S (2013) Langmuir 29: 3140-3151.
[26] Pascanu V, Hansen PR, Bermejo Gómez A, Ayats C, Platero‐Prats AE, Johansson MJ, Pericàs MÀ, Martín‐Matute B (2015) ChemSusChem 8:123-130.
[27] Manna K, Zhang T, Lin WB (2014) J Am Chem Soc 136: 6566-6569. [28] Lim D, Yoon J, Ryu K, Suh M (2012) Angew Chem Int Ed 124: 9952-9955. [29] Chen LY, Chen HR, Luque R, Li YW (2014) Chem Sci 5: 3708-3714.
[30] Li XL, Van Zeeland R, Maligal-Ganesh RV, Pei YC, Power G, Stanley L, Huang WY (2016) ACS Catal 6: 6324-6328.
[31] Li XL, Guo ZY, Xiao CX, Goh TW, Tesfagaber D, Huang WY (2014) ACS Catal 4:3490-3497. [32] Phan NTS, Van Der Sluys M, Jones CW (2006) Adv Synth Catal 348: 609-679.
[33] Elias WC, Signori AM, Zaramello L, Albuquerque BL, de Oliveira DC, Domingos JB (2017)ACS Catal 7: 1462-1469.
[34] Balanta A, Godard C, Claver C (2011) Chem Soc Rev 40: 4973-4985. [35] Astruc D, Inorg Chem (2007) 46:1884-1894.
[36] Morabito J, Chou LY, Li Z, Manna C, Petroff C, Kyada R, Palomba J, Byers J, Tsung CK (2014) J Am Chem Soc 136: 12540-12543.


![Crystal structure of N [(8E) 12 methyl 14 phenyl 10,13,14,16 tetraazatetracyclo[7 7 0 02,7 011,15]hexadeca 1(16),2,4,6,9,11(15),12 heptaen 8 ylidene]hydroxylamine 1,4 dioxane hemisolvate](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)