ContentslistsavailableatScienceDirect
Current
Plant
Biology
j ourn a l h o m e pa g e :w w w . e l s e v i e r . c o m / l o c a t e / c p b
Computational
approaches
to
identify
regulators
of
plant
stress
response
using
high-throughput
gene
expression
data
Alexandr
Koryachko
a,
Anna
Matthiadis
b,
Joel
J.
Ducoste
c,
James
Tuck
a,
Terri
A.
Long
b,∗,
Cranos
Williams
a,∗∗aElectricalandComputerEngineering,NorthCarolinaStateUniversity,Raleigh,NC,USA bPlantandMicrobialBiology,NorthCarolinaStateUniversity,Raleigh,NC,USA
cCivil,Construction,andEnvironmentalEngineering,NorthCarolinaStateUniversity,Raleigh,NC,USA
a
r
t
i
c
l
e
i
n
f
o
Articlehistory: Received4April2015 Accepted28April2015 Keywords: Stressresponse Transcriptionfactors Generegulatorynetworks AlgorithmsArabidopsisthaliana
a
b
s
t
r
a
c
t
Insightintobiologicalstressregulatorypathwayscanbederivedfromhigh-throughputtranscriptomic datausingcomputationalalgorithms.Thesealgorithmscanbeintegratedintoacomputationalapproach toprovidespecifictestablepredictionsthatanswerbiologicalquestionsofinterest.Thisreview conceptu-allyorganizesawidevarietyofdevelopedalgorithmsintoaclassificationsystembasedondesiredtypeof outputpredictions.Thisclassificationisthenusedasastructuretodescribecompletedapproachesinthe literature,withafocusonprojectgoals,overallpathofimplementedalgorithms,andbiologicalinsight gained.Thesealgorithmsandapproachesareintroducedmainlyinthecontextofresearchonthemodel plantspeciesArabidopsisthalianaunderstressconditions,thoughthenatureofcomputationaltechniques makestheseapproacheseasilyapplicabletoawiderangeofspecies,datatypes,andconditions.
©2015TheAuthors.PublishedbyElsevierB.V.ThisisanopenaccessarticleundertheCCBY-NC-ND license(http://creativecommons.org/licenses/by-nc-nd/4.0/).
1. Introduction
Plantsaresessileorganismssubjecttoconstantlychanging envi-ronments.Theabilitytorespondtotheseenvironmentalchanges, therefore,iskeytoplantadaptationandsurvival.Anoverallgoal of plantabiotic stressresearch is todevelop anunderstanding ofthemolecularcomponentsofasingleorcombinatorialstress response and show how these components interact, enabling directedgeneticmanipulationsthatcanenhancestresstolerance
[1].Transcriptionfactorsareoneofthefirstcategoriesofgenes activatedinresponsetoastress[2].Transcriptionfactoractivity canleadtoalterationsinactivityandaccumulationofdownstream transcriptionfactorsandproteinsthatmodulateplantmorphology andmolecularcomposition.Inthisway,manipulationofthe activ-ityofjustonetranscriptionfactororasmallfamilyoftranscription factorscanlead toalterationsin a transcriptionalcascadewith dramaticoutcomes.Thisstrategyisthebasisforboth evolution-aryadaptationaswellasgeneticmanipulationofstressresponses
[3,2,4].Despiteawidespreadfocusonstress-inducedtranscription
factorsandarecentbreadthofhighthroughputdata,successful
∗Correspondingauthor.Tel.:+19195150478. ∗∗Co-correspondingauthor.
E-mailaddresses:[email protected](T.A.Long),[email protected] (C.Williams).
geneticmanipulationsoftranscriptionfactorsincropplantsthat improvestresstolerancearelimited.Identificationof transcriptio-nalregulatorsinthemodelspeciesArabidopsisthalianaisafirst steptothesearchforcandidatesforgeneticmodificationincrop species,yetanumberoflimitationsexistinthisidentification.First, with5–10%ofplantgenomesreportedtocodefortranscription factors,thenumberofcandidategenestostudyisextensive[5–7]. Second,itisdifficulttopredicttheeffectsofonetranscription fac-torin isolationandevenmoredifficulttopredictcombinatorial effectsoftranscriptionfactorsactingonthesametargetsor act-ingincomplexes.Finally,thewaysinwhichtranscriptionfactor activityismodulatedinresponsetoonestressoracombination ofstressesarecomplex.Inotherwords,ahugenumberofpossible experimentsexisttotesttheeffectofcombinationsoftranscription factorsundercombinationsofstresses.Computationalapproaches playacriticalroleintheresearchprocessbyproducinga setof testablepredictions,thuslimitingthespaceofexperimentsneeded toyieldabetterunderstandingofthecascadingresponses result-ingfromstress.These predictionsrange frominvolvementof a transcriptionfactorin astressresponsetodetaileddescriptions oftranscriptionfactorand targetgeneinteractiondynamics.An increasingabundanceofcomputationalapproachesnecessitatesa carefulevaluationoftheutilityandapplicationofthesetools.
Inthisreview,wesummarizeandorganizealgorithmsinvolved incurrentandpromisingcomputationalapproachesinto5 cate-gories(“Types”)basedonthetypeofinferenceanalgorithmaims http://dx.doi.org/10.1016/j.cpb.2015.04.001
2214-6628/©2015TheAuthors.PublishedbyElsevierB.V.ThisisanopenaccessarticleundertheCCBY-NC-NDlicense(http://creativecommons.org/licenses/by-nc-nd/4. 0/).
toobtainfromabiologicaldataset.Wefocusonalgorithmsthat canoperateongeneexpressiondata,asthisiscurrentlythemost availableformofhighthroughputdata.Wethendemonstratehow algorithmsfromdifferentcategorieshavebeencombinedinthe scopeofcomputationalapproachestoachievespecificobjectives associatedwithplantstressresponse.Additionalexamplesof algo-rithmsthatfallintotheproposedclassificationsystemcanbefound eitherinplantrelated[8–10]orgeneralcomputationalapproach
[11–15]reviewarticles.Webuildonthesereviewsbyorganizing
algorithmsbasedontheirutilityandhighlightinghowinferences achievedbyalgorithmsinmultiplecategoriescanbesystematically combinedtoachieveabetterunderstandingofthetranscriptional cascadeinvolvedinstressresponse.We thendiscusshowthese algorithmshavebeenutilizedinrecentstudiestoachievea cer-tainobjective.Inthisway,researchersaimingtoacquirespecific predictionsfromanexpressiondatasetcanmoreefficientlychoose appropriatealgorithms.
2. Background
Environmental conditions in agricultural settings are highly variable, leading to suboptimal crop yields and survival rates
[16].Thefrequencyandintensityofenvironmentalextremes, par-ticular drought, heat, and pests are expected to increase with climatechange[1,17,16].Alargenumberofstressresponse stud-iesarefocusedonelucidatingtranscriptionalcascadesregulating responsestoindividualandcombinedstresses.Transcription fac-tors thatplay important rolesin modulating suchcascadesare candidatesforgeneticengineeringapproachesandareworthyof intensivestudy.
Geneexpressionanalysisisawidelyproposedmeansofbringing a greater understandingto allabiotic stress responsesfor sev-eralreasons.Alargenumberofgeneshavealteredexpressionin responsetostressandthesealterationsplayanimportantrolein adaptation[18,19].Expressiondataisalsorelativelycheap.Because ofthis,high-throughputgeneexpressiondatasetshavebeen gen-eratedandarepubliclyavailableforamultitudeofstresses,both bioticandabiotic,withexamplesinA.thalianaincludingbutnot limitedtopathogeninfection[20–22],cold[23],pH[24],salt[25], light [26],and nutrient [25,27–30]stress.Though thesestudies arecomparableintheory,afewlargestudieshaveattemptedto mitigatethe effects ofvariations in experimentalsetupby col-lecting expression data under different stresses imposed with otherwiseidenticalgrowthconditions[18]orundercombinations ofstresses[31–34].Analysesoftheseconcurrentand combinato-rialexperimentsinparticularhave revealeddistinctpatternsof differentstressresponsesalongwithsomecommonfeatures, infer-ringthatbothgeneralandspecificstressresponsepathwaysexist. For example, analysisof theAtGenExpress databaseof concur-rentstressapplicationindicatesthatsomeabioticstressesresult insustainedgeneexpressionalterationsand othersintransient alterations[18].Asetofearly-andcommonlyinducedgenes, rep-resentingthesocalledPlantCoreEnvironmentalStressResponse (PCESR),includestranscriptionfactors,indicating thata general stressresponsemaybetranscription factormediatedand likely occurs earlyin stress response cascades [18,35]. Combinatorial studiesindicatethatgenesrespondingtocombinedstressesare oftendistinctfromthoserespondingtoindividualstresses, high-lighting a need for both more studies of this type as well as computational methods to attempt to predict these emergent behaviors[31–34].Despitetheseextensiveanalyses,limiteddirect predictionsconcerningstresspathwayshavebeenmadeand val-idated.Themajorityofdetailedcharacterizationsoftranscription factors,includingdirect promoterbindingandinfluenceon tar-getgene expression,are theresultof traditional studies.These
studiesaretimeandcostintensive.Furthermore,sincemanykey regulatorshavebeenfoundthroughphenotypicmutantscreens, subtleyetimportantphenotypesandgenescaneasilybemissed. Redundancyisexpectedincriticalregulatorymechanisms[36],and predictionsconcerningwhichregulators ormutantstocombine in a geneticengineering strategywould beextremely valuable. Arecent increaseinalgorithm developmentand utilizationwill helptoincreasethepredictivepowerinavailabledatasetssothat regulatorsandcombinatorialregulatorymechanismsbeyondthe “lowhanging fruit” canbeidentified.In thefollowing sections, wedescribeandorganizesetsofalgorithmsandimplementations thereofinexperimentalapproaches,aimingtobringattentionto thebenefitoftheseapproachesandfacilitatefutureincreasesin frequencyandstrengthofcomputationalbiologystudies.
3. Classificationofinferencealgorithms
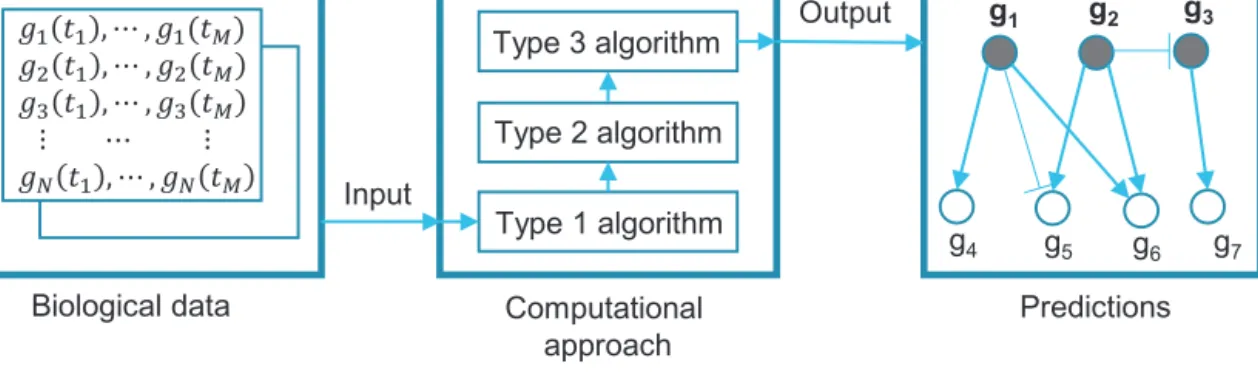
Manycomputationalalgorithmshavebeendevelopedfor ana-lyzinggeneexpressiondata.Wefocushereonalgorithmscapable of identifyingstress related genes,groupinggenes byfunction, inferringconnectionsbetweengenes,estimatinggeneinteraction direction and type, and predicting gene expression states and values ininterconnectedregulatorynetworks.Thesealgorithms differincomplexityandimpliedassumptions,butcanbeclassified basedonfunctionality.Wecategorizethesealgorithmsin5distinct groupsbasedonthetypeofinsighttheyprovidetoabiological pro-cessofinterest.Dependingonresearchobjectives,thesealgorithms caneitherbeusedseparatelyorasapartofasystematic compu-tationalapproachwhereinferencesfromalgorithmsofonetype canbeusedasinputforalgorithmsofanothertype.Forexample,a computationalapproachdesignedtopredictgeneinteractionsand theirtypebasedontimecoursemicroarraydatacanbecomprised of3algorithmsofdifferenttypesthatsequentiallyprocessinput datatoobtainadesiredoutput(Fig.1).
Thealgorithmsdescribedcanbeappliedtotranscriptomicdata obtainedatMtimepointsortreatments(tj,j=1,...,M)foraset ofNgenes(gi,i=1,...,N).Examplesofsuchdatasetsincludethe globalabioticstressexpressiondatabaseAtGenExpress[18].This databaseincludesdatasetsformultipleabioticstresstreatments thatareobtainedforN≈24,000genesatM=7timepointsusing AffymetrixATH1GeneChipmicroarrayanalysis.Hence,theactivity ofeachgenecanberepresentedbyasetofnumbersgi(t1),gi(t2),
...,gi(tM),formingapatternthatisusedbyalgorithmstomake inferences.
Type1algorithmsattempttocapturegenesthatarerelevantto a particular condition. Techniques for determining differentially expressedgenes are an example of algorithmsfalling into this category[37].Differentialexpressiontechniquesworkby assum-ing that significant changein transcript levelsof a given gene understressconditionrelativetoitsactivityundernormal condi-tionsindicatesthatthegeneplaysaroleinthestressresponse. Thisassumption disregardsposttranscriptional modificationsas alternatemeansofgeneproductregulation.Sincetranscript mea-surementprecisioncanvaryfromoneexperimentalapproachto another,statisticaltestsareoftenappliedtodeterminethe sig-nificanceofthechangeintranscriptlevels.Student’st-testfor2 treatmentsorANOVAforasetoftreatmentsarecommonlyapplied to deduce statistical significance. Other differential expression inferencealgorithmsweredevelopedforlargescaleexperimental techniquessuchasmicroarrays,forwhichthecorrelationbetween within-arrayreplicatescan betakeninto consideration [38],or RNA-Seq, forwhichcountbasedstatistics aremoreappropriate
[39,40].
Type 2algorithmsaimto identifyrelationshipsbetween genes. These algorithms work by assuming that genes with “similar”
Fig.1.Conceptualviewoftheinformationflowinacomputationalapproach.Biologicaldataisusedtoidentifygenesofinterest(Type1algorithm),inferconnections betweenthesegenes(Type2algorithm),andpredicttypesoftheseconnections(Type3algorithm).
expressionpatternsareco-regulatedorarepartofthesame regula-torypathway[13].Techniqueslikeco-expressionanalysis[41–45]
fallintothiscategory.
Common metrics that have been used to assess similarities betweengenesbasedontheirexpressionpatternsincludePearson correlation coefficient [42,46–48], Spearman correlation coeffi-cient [49–51], partial correlation coefficient [52–54], Euclidean distance[55,56],andmutualinformation[57–59].Thesemetrics typicallyrepresenta quantifiedmeasurethatestablishesa pair-wisecomparisonbetweentheexpressionlevelsoftwogenes,g1 and g2, acrosstime points or experimentaltreatments. Kumari etal.[60]presentedastudythatevaluatedtheutilityofSpearman rankcorrelation,WeightedRankCorrelation,Kendall,HoeffdingsD measure,Theil-Sen,RankTheil-Sen,DistanceCovariance,and Pear-soncorrelationcoefficientontranscriptionaldatafordetermining geneassociation.TheauthorsfoundthatSpearman,Hoeffding,and Kendallcorrelationcoefficients weremore effectivein identify-ingrelatedpathwaygenesthanothers.Incontrast,Maetal.[61]
claimthatbasedonmanualinspectionoftheexpressionpatterns ofseveralpairsofTF-targetgenes,theGinicorrelationcoefficient cancompensatefortheshortcomingsofthePearson,Spearman, Kendall,andTukeysbiweightcorrelationsindetectingtransient regulatoryrelationshipsbetweentranscription factorsandtheir targets.Metricssuchasareabetweenexpressioncurves[62],Z -score[63],andothersappearintheliteraturebuthavenotbeen extensivelyevaluated.
Relationshipsbetweenindividualgenesoracrossestablished groupsofgenescanbegeneratedbasedonthesesimilaritymetrics. Atypicalprocedureforestimatingrelationshipsbetween individ-ualgenesistosetathresholdvalueandassignconnectionsbetween geneswhosepairwisesimilarity valueishigherthan aselected threshold[62,58].Thestatisticalsignificanceofthesimilaritycan alsobetakenintoconsiderationwhenestablishingaconnection
[57].Groupsofsimilarlybehavinggenesareinmostcasesidentified usingclusteringalgorithms.Clusteringalgorithmsapplysimilarity metricstoisolategroupsofco-expressedgenes.k-means cluster-ing[64],theMarkovClusteralgorithm[65,66],biclustering[67], self-organizingmaps[68],hierarchicalclustering[69],and affin-itypropagation[70]areexamplesofclusteringalgorithmsapplied totranscriptomicdata.Martinetal.[64]appliedk-means cluster-ing,hierarchicalclustering,andself-organizingmapstotimeseries transcriptomicdatafrommice.Theresultssuggestedthatk-means wasabletoconveycomparablegroupingtohierarchicalclustering, andself-organizingmaps(morethan80%agreement)while main-taininglessofacomputationalloadthanotherapproaches.Freyand Dueck[70]showedthattheaffinitypropagationalgorithmyields morecompactclusterscomparedtok-meansintermsofthesum ofinterclusterdistanceswhichmightimplytighterrelationships betweengenesinthesamecluster.
Clustering has also been used to reduce the complexity of buildingtranscriptionalnetworksbyreducing highdimensional networkswithmanygenestolowerdimensionalnetworksof clus-tersof genesor “metagenes”,which represent groups ofgenes withsimilarexpressionactivity.Theexpressionpatternofa meta-gene may be defined as thecluster average or the expression pattern of the gene with the highest sum of similarities with itsclustermembers.Somealgorithmshaveextracted metagene expressionpatternsfirstbyapplyingprincipalcomponentanalysis (PCA)orsingularvaluedecomposition(SVD)totheoverall expres-siondataset.Theclustersarethenassembledbasedonsimilarities betweengeneandmetageneexpressionpatterns[71,72].
Type3algorithmsaimtoinfercausalrelationshipsbetweengenes. Causalinferenceproceduresareoftenbasedontheassumptionthat achangeinonegene(g1)willresultina subsequentchangein anothergene(g2)atsomelatertimeifg1activatesorinhibitsg2
[73–77].Thus,theapproachissimilartoco-expressionanalysisin
thatitaimstofindgeneswithsimilartemporalexpression pat-terns.Thekeydifferencedistinguishingthisapproachfromthose inType2istheassumptionthatthesesimilaritieswilloccurata delay,allowingforinferenceonthedirectionofregulation(which genecomesfirstinaregulatorycascade)inadditiontoa relation-shipconnection.TheequationforPearsoncorrelationcoefficient, forexample,canbemodifiedtoassessthistemporalcharacteristic byincorporatingatimedelay.Eq.(1)reflectssimilarityatthedelay ofonetimeunit.Thealgorithmscapturetheregulationdelayfor apairofgenesbyselectingthetimeunitdurationthatmaximizes thecorrelationcoefficient[75].
g1→g2 =
M−1 j=1 (g1(tj)−g¯1)(g2(tj+1)−g¯2) M−1 j=1 (g1(tj)−g¯1)2 M−1 j=1 (g2(tj+1)−g¯2)2 , where g¯1= 1 M−1 M−1 j=1 g1(tj), g¯2= 1 M−1 M−1 j=1 g2(tj+1) (1)Twosets ofsimilarity values,each correspondingtoa range of delaysforacertaindirectionofshift,arecalculatedtoassessthe strength anddirectionality ofconnectionin each pairofgenes. Smallsimilarityvalues,correspondingtoalowprobabilityof reg-ulation,canberemoved,leavingtheremaininghighconfidence connectionstocharacterizegenesthathavepotentialcausal rela-tionships.Approachesthatusemodificationsofthemetricin(1)
havebeeneffectiveforsingledatasetswith50and27timepoints andsamplingintervalsof20min[75]andfor acollectionof18 datasetswith7timepointsineachandsamplingintervalsranging from0.5to12h[78].Othersampletimesmayberelevant depend-ingonthefeaturesthatexistinthedata.
Anotherclassofalgorithmsthatinferregulatoryinteractions betweengenesisBayesiannetworks[79,80].Bayesiannetworks arecapableofinferringregulatoryconnectionsfromtimecourse andnon-timecoursedata.Thesealgorithmsattempttofindcausal connectionsbasedonBayes’rulebyexplicitlychoosinganetwork structure that best describesexperimental data. The algorithm considersanetworkofgeneregulationsasasetofdependencies wheretheprobabilityofexpressionofatargetisconditionedon theexpressionofitsregulator.Theseregulationsaredescribedas conditionalprobabilities.Algorithmsthentrytofinda network structurethatbestdescribesthedatabasedonascoringfunction. Identificationofthenetworkstructureisacomputationally inten-siveproblem.Complexitygrowsexponentiallywithanincreasing numberofnodes[11].Forexample,around1018different topolo-giesariseforanetworkofonly10genes[12].Thus,mostofthe approachesusingBayesiannetworksconcentrateonasmallsubset ofgenes(typicallywhensomeportionofageneregulatorynetwork isalreadyknown)oremploysub-optimalbutlesscomputationally intensesolutionstohandlelargernetworks[11].
DynamicBayesianNetworks(DBN)[81,82]incorporate order-inginformationintimecoursedatatoallowforfeedbackloops(not allowedinstandardBayesiannetworks).Thesefeedbackloopsare allowedbytreatingexpressionofthesamegeneatdifferenttime pointsasdifferentnodes.Nodescorrespondingtothesamegene arecombinedafterthestructureinferenceprocedure.This algo-rithmleadstoanincreaseincomplexitysincethenumberofnodes involvedinstructureinferenceroutineisaproductofthenumber ofgenesandthenumberoftimepoints.
Type4algorithmsaimtoinfercombinationsofregulator expres-sionstatesthatarenecessarytoresultinaparticularstateoftarget. Thesealgorithms canbeconceptualizedasa search fora func-tionalrelationshipbetweenatargetanditsregulator(s)(gi=f(g1,
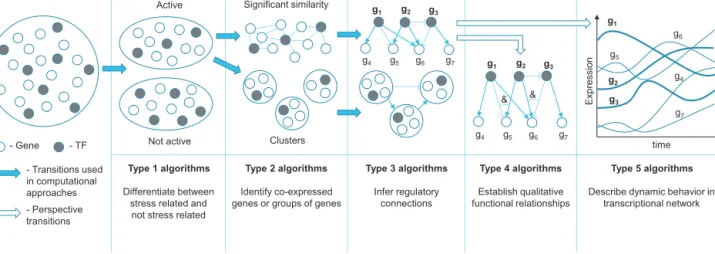
g2,...,gN)).Inthiscase,aqualitativemeasureofgenebehavior canbeused,withgeneexpressionvalues representedaseither highorlow, activeor inactive,or“ON”or“OFF”tosimplifythe problem.An“ON”stateofonlyacoupleregulatorsmaysufficeto upregulatetheexpressionofthetarget.Thisqualitativeassumption allowstheuseofBooleannetworks[83]inType4inference prob-lems.ExpressionvaluesinBooleannetworkinferenceapproaches arediscretizedmostlyintwostates,representinganactivitylevel ateachtimepoint[84–86].Regulatoryconnectioninference algo-rithmstrytofindabinaryfunctionthatcomputesthenextstateof agenebasedonacombinationofothergenes’statesusingsimple Booleanoperations,e.g.AND(&)ifmorethanoneregulatorshould haveacertainstatetoinfluenceacommontarget,OR(|)ifanyof theregulatorstatessufficeforthesamepurpose,andNOT(¬)inthe caseofrepression(Fig.2).Thegoalofthisapproachistofindthe simplestfunctionforeachgene,whichisthefunctionthatdepends onthefewestregulatorgenespossible.
Adirectapproach tofindthesimplestBooleanfunctionthat satisfiesagivendatasetistocompareallpossiblefunctions capa-bleofgeneratingtheobservedexpressionpattern.Thenumberof Booleanfunctionsthatcanrepresenttheexpressionactivityofa generegulatedbyasmanyasntranscriptionfactorsis22n
[87],
makingtheproblemcomputationallyinfeasibleforalarge(more than10)numberofgenes.Somealgorithmsusepriorknowledge toconfinethenumber of genestoanalyze.Othersrelyon net-workstructuresinferredbyothertypesofalgorithmstoconfine thenumber,type and directionalityofpossibleregulatory rela-tionshipsbetweenindividualgenesorgroupsofgenes.Another factorconstrainingtheuseofBooleannetworksinwholegenome datasetanalysisisthesmallnumberofsamples(timepoints) asso-ciatedwithmostdatasets.Thesesmallsamplesizestypicallydo notprovidethediversityneededtouniquelydefinerelationships acrossalargenumberofindividualgenes.Forexample,for5time points,which isthemedian numberin typicalgeneexpression
Fig.2.Booleannetworkrepresentationingraphicalandfunctionalforms. Combi-nationsoftranscriptionfactorsg1,g2,andg3influenceexpressionofeachotherand targetgenes.Thestateofg6,forexample,isinfluencedbyacombinationofg2and g3orbyg1alone.
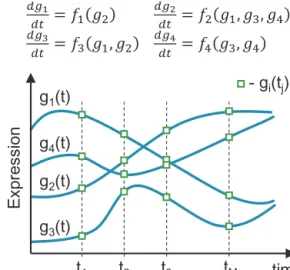
Fig.3. Type5algorithmsoutputintermsofthesystemofODEsandpredictedgene expressiondynamics(gi(t))basedonexperimentalvalues(gi(tj)).Inthisexample,
theexpressionpatternofeachgeneisinfluencedbytheexpressionofatleastone othergene,withsomegenes(g4)influencedbytheirownexpression(feedback loop).
datasets[88],thenumberofgeneswithdistinctBoolean expres-sionpatternsislimitedtoonly25=32.Anyattemptedanalysisof morethan32geneswithsuchadatasetwouldresultinatleast 2geneswithidenticalbehaviorwhichwouldlimitresolutionto groupsofgenes(e.g.metagenes)asopposedtoindividualgenes.
Type5algorithmsaimtodescribedynamicbehaviorina trans-criptionalnetwork.Theresultingnetworkrepresentationallowsfor thereconstructionofcontinuouschangesintranscriptsovertime (Fig.3).Ordinarydifferentialequationsarecommonlyusedto cap-turethedynamicsassociatedwithgeneexpressionchanges[89]. Theseequationsallowfortheestimationofgeneexpression val-uesatanygiventimepointeitherbetweensamples(interpolation) orbeyondthelastcollectedsample(extrapolation)[29].Whena generegulatorynetworkisrepresentedintermsoflinear differ-entialequations,theinstantaneouschangeinexpressionofagene isrelatedtothesumofweightedexpressionvaluesofinfluencing genes: dgi dt = N
k=1 aikgk, (2)whereaikrepresentinfluencecoefficients.Coefficientsforlinear differentialequationsareofteninferredusingtheLeastAbsolute ShrinkageandSelectionOperator(LASSO)algorithm[90],a modi-ficationofthelinearregressionapproach.WhenLASSOisusedfor ODEinferencepurposes,thechangesinexpression,i.e.differences betweenexpressionvaluesatconsecutivetimepoints,are approx-imatedbyalinearcombinationofothergenes’expressionvalues. Expressionpatternsfortargetgenesarereplacedwithpatternsof changesinexpression[91–93]toinferinfluencecoefficients.Given thatbiologicalprocessesareassumedtobeinherentlynonlinear, linearOrdinaryDifferentialEquation(ODE)inferencealgorithms fortranscriptionalnetworksrelyontheassumptionthatthe sys-temoperatesclosetoastabilitypoint[93].Thesystemmaynotstay closetoastabilitypointinthecaseofstressinducedresponses, where a plant may transition from one stable steady state to another.NonlinearODEs,thoughpotentiallymorebiologically rel-evantbecausetheydonotrely onthesteadystateassumption, typically requirethe estimation of more coefficients associated withnonlinearterms[94].Coefficientestimationroutinesfor infer-encealgorithms searchtheparameterspacetofindcoefficients that yield solutions closest to measured expression values
[95,96].
Allof the described algorithms requireimplementation and validation in biological systems in order to assess theirutility. Anumberofvalidationtechniquesexist,dependingonthetype ofalgorithm[97–113].Thesevalidationtechniquesarevisualized withkeyreferencesinFig.4.ValidationforalgorithmsofTypes1
and2,whichpredictassociationsbetweenageneandaprocess oragene anda groupof genes,are limitedtoanalysisof Gene Ontology(GO)enrichmentorphenotypesinmutantsof transcrip-tionalregulators.Thesephenotypesrangewidelydependingonthe stressresponseinquestion,andcouldinvolveextensive experi-mentationtosearchforaphenotypeofinterest.Awiderrangeof techniquesexistforalgorithmsofTypes3–5,algorithmsthat pre-dictrelationshipsbetweentranscriptionfactorsandtargetgenes. Theserelationshipscanbetested indirectly throughexpression profiling,computationallythroughpromoteranalysis,ordirectly throughbindinginteractions.Giventhatno“goldstandard” vali-dationtechniqueexists[114],convincingsupportofteninvolves thecombinationofmultiplevalidationtechniques,suchas expres-sionanalysis and binding activityfor a regulator and target of interest.Similarly,complexpredictionssuchasthosederivedfrom
Type4andType5algorithmsrequireacombinationofstaticand dynamicvalidationtechniques–includingexpressionprofilingat multipletimepoints,preferablyalongwithdeterminationof bind-ingactivity.
4. Computationalapproaches
Computationalapproachesareusedwidelytogaininsightinto processesunderlyingplantresponse tostressconditions. These approacheshaveasimilarstructureintermsofthetypesof algo-rithms theyuseand differin thecombination of and order in whichthesealgorithmsareapplied.Inthefollowingexamples,we describehowalgorithmsofdifferenttypeshavebeencombinedin particularcomputationalapproachestoanswerresearchspecific questions.
4.1. Relevantgeneidentification
A large number of current computational approaches are focusedonidentifyinggenesthatplayakeyroleinaprocessof interest.The importanceofthese genesisthen typically tested throughmutantphenotypicanalysis.Maetal.[56]analyzedaset ofA.thalianaabioticstressresponsetranscriptomedatasetswith
6timepointstoidentifystressrelatedgenes.Thecomputational approachstartedbypartitioning eachstressdatasetinto “infor-mative” and “noninformative” genesusing differential network analysis(Type1algorithm).Theauthorsstatedthatdifferential net-workanalysisthatinvolvesmachinelearningandtrainingbasedon
aprioriinformationismoresensitivethandifferentialexpression analysis,whichisstatisticsoriented.TheGinicorrelationcoefficient wasthencalculatedforpairsof“informative”genestoestablish sig-nificantconnections(Type2algorithm).Stressrelatedgeneswere identifiedfromtheresultingnetworkbasedonthecombinationof 33topologyscoresobtainedfromthenetworkofsignificant con-nections(Type1algorithm).Theauthorsvalidatedtheiralgorithm byperformingaphenotypicscreenfor89candidatesidentifiedas saltstressrelated.Mutantsof2previouslyunreportedsalt stress-relatedgenesshowedphenotypes.
Dinnenyetal.[25]conductedDNAmicroarrayexperimentson
A.thalianarootresponsetoirondeficientmediawith7timepoints spanning72htoidentifycommonstressresponsebehavior pat-terns.Theauthorsapplieddifferentialexpressionanalysis[115]to identifygeneshavingatleasta1.5-foldchangeinexpressionwith afalsediscoveryratevaluelessthan10−4atasamplingtimepoint comparedtonotreatment(Type1algorithm).Theanalysisshowed thatthestrongesttranscriptionalresponseoccurredafter24hof treatment.Dinnenyetal.[25]thenappliedtheaffinity propaga-tionclusteringalgorithm[70]toformgroupsofsimilarlyexpressed genesandthusidentifygeneralpatternsofgeneexpression(Type 2algorithm).Longetal.[28]usedtheresultsofthisanalysisand screenedthroughmutantsof38identifiedgenescodingfor coex-pressedtranscriptionfactors.Thescreensledtoidentificationof importantironhomeostasisregulatorsPOPEYE(PYE)andBRUTUS (BTS).
Linetal.[30] investigatedtheeffectofphosphatestarvation onA.thalianarootgene signalingusingaDNAmicroarray time coursewith 3 time pointsto infer functional modules in early transcriptionalresponses.Theauthorsuseddifferentialexpression analysiswiththerequirementofa2-foldchangeinexpressionwith ap-valuecutoffof0.05toidentifystressrelatedgenes(Type1 algo-rithm).Additionalinformationfrom2671experimentaldatasets, 300ofwhicharerootspecific,wasusedtoselect187rootspecific genes(Type1algorithm).TheauthorsusedtheMulti-Array Corre-lationComputationUtility(MACCU)toolboxbasedonthresholding pairwise Pearson correlation coefficients toobtain 3 functional modulesofstressspecificgenes(Type2algorithm).Tovalidatethe results,Linetal.[30]conductedmutantscreenson31members ofaclusterwheremostofthegenesareknowntoparticipatein rootdevelopment.Only5testedlinesdidnotshowastatistically significantroothairlengthphenotype.
4.2. Genefunctionelucidation
Anothergroupofcomputationalapproachesaimtoassociate geneswithaspecificfunctionduringaprocessofinterest.The guilt-by-associationheuristic[43]isoftenusedtoassignafunctiontoan unknowngenebasedonknownfunctionsofco-regulatedgenes (GeneOntologyenrichment).Polanskietal.[48]analyzedsixA. thalianastressresponse transcriptomedatasetstoidentifygene modulesshowingevidenceforco-regulation.Thecomputational approachrevealed78modulesofco-regulatedgenes,71ofwhich wereoverrepresentedinGeneOntologycategoriesand51ofwhich wereenrichedin transcription factorbindingmotifs(compared to24and6of78randomlyassignedmodules,respectively).The approachusedinformationaboutwhichgenesweredifferentially expressedineachstressresponseasaninput(previously deter-minedinotherpublications usingType 1algorithms).For each genedifferentiallyexpressedunderatleast2conditions,the algo-rithmassembledasetofcorrelatedgenesforeachcondition(Type
Fig.4. Validationtechniquesforalgorithmtypeswithkeyreferences.Examplesshownarethosetypicallyseenincurrentcomputationalresearchapproaches,specifically forresearchprojectsinA.thaliana.
2algorithm).Aco-regulationrelationshipinapairofgeneswas establishedifthesegeneshadsharedasignificantnumberof corre-latedgenesacrossstressconditions(Type2algorithm).Theauthors usedGeneOntologyenrichment,promoteranalysis,andyeast one-hybridprotein–DNAinteractionstovalidatetheresultingmodules ofco-regulatedgenes.
MaandBohnert[116]integratedtimecourseandcellspecific transcriptomics data with gene promoter structures to iden-tifystressrelatedcis-elementsinA. thaliana.Thecomputational approach used in this work detectedknown stress related cis -elementsandidentifiedsecondarymotifs.Theauthorscombined abioticandbioticstress,hormoneand chemicaltreatmenttime coursesanddifferentlightconditionsamplestocreateone com-bined expression pattern of 145values pergene. Differentially expressedgeneswereidentified bycombiningtheresultsfrom fuzzyk-meansclustering[117]appliedtoallgeneprobesandthe ‘limma’statisticalprogram[118]whichidentifiedgenes differen-tiallyexpressedinatleastonecondition(Type1algorithm).Fuzzy
k-meansclusteringwasagainappliedtotheresultingsetto iden-tifystressrelatedclustersofgenes(Type2algorithm).Theauthors assignedfunctionstoclustersbasedonGOenrichment.Binding motif analysis using Plant Cis-acting Regulatory DNA Elements (PLACE)database[105]revealedmotifssignificantlyoverexpressed inthefunctionrelatedclusters.Furtheranalysisof22majorclusters resultedintheidentificationofnewDNAregulatorymotifs[119].
4.3. Generelationshipinference
Computationalapproachesthataimtounravelinfluential rela-tionshipsbetweenregulatorsandtheirtargetsarelesscommonbut areincreasinginfrequency.Windrametal.[22]applieda computa-tionalapproachtoidentifytranscriptionfactorfamiliesoperating atdifferentstagesofA.thalianapathogendefenseresponse.The authorsanalyzedtranscriptionalprofilesat24time pointswith 4 replicates per time point. The computational approach pre-dictedgeneregulatoryinteractions,confirmedexperimentallyor by bindingmotif enrichment.The analysisstarted with assess-ment of differentiallyexpressed genesbased ona combination ofMAANOVA(MicroArrayANalysisOf VAriance)[120], approx-imateFtests,GP2S(Gaussianprocess2sample)test[121],and Hotellingstatistic(T2)[122](Type1algorithm).Next,a
SplineClus-ter[123]algorithmseparateddifferentiallyexpressedgenesinto
clustersassociatedwithdifferentstagesofstressresponse(Type 2algorithm).TheclusterswerevalidatedbyGOenrichment anal-ysis.NonparametricmodificationofBayesiannetwork inference algorithm [124] wasapplied to clusterrepresentatives toinfer regulatoryconnectionsbetweenclusters(Type3algorithm).The authors validated the regulatory effect of one of the clusters throughexperimentswithaknockoutmutantlineforthe tran-scription factor TGA3. Experimental data showed altered gene expressioninpredictedTGA3targetclustersinthetga3-2mutant,
Fig.5. Transitionsbetweenalgorithmsofdifferenttypes.Typicalexperimentaltransitionsbetweenalgorithmsareindicatedwithbluearrowsandperspectivefuture transitions,lesscommonbutpossiblewithmorereliablesupportingalgorithms,areindicatedwithwhitearrows.
whereastargetsregulatedbynonTGA3clusterswerelessaffected. Theeffectofanothertranscriptionfactor,ANAC055,wasvalidated bybindingmotifenrichmentintargetclusters.
Redestigetal.[78]analyzedasetof18DNAmicroarraytime seriescorrespondingtoninedifferentabioticstresseswithseven timepointsobtainedfromrootandshootofA.thalianaseedlings withtheaimofassociatingstressresponsivetranscriptionfactors with theirtargets. The authors concluded that their computa-tional approach delivered a usable number of high-confidence targetgenes(12–59%ofidentifiedtruetargets)forstressrelated transcriptionfactors.Thecomputationalapproachidentifiedstress relatedtranscriptionfactorsbyselectingoneswithmaximum over-allresponseandmaximumchangeinresponsesatisfyingaspecific thresholdcriteria(Type1algorithm).Covariancevaluesbetween atranscriptionfactorandothergenesoverasetofdelayswere calculatedforasetofconditions(Type3algorithm).Highscores correspondedtoahighprobabilityofregulation.
Krouketal.[29]conductedDNAMicroarrayexperimentsonA. thaliananitrateresponsewithsixtimepointsspanning20minutes tocaptureageneregulatorynetworkunderlyingplantadaptation tonitrateprovision.Theinferredtemporalmodelofthereaction processbuiltfor20 clusterrepresentativesresultedin 70% cor-rect predictions of expression value direction changeafter the lasttimepoint inthetimecourse.Thecomputationalapproach startedwithANOVAtoidentifynitrogenregulatedgenes(Type 1algorithm).Next,MeVsoftware[125]wasusedtoseparatethe nitrogenregulatedgenesinto20clusters,eightofwhichappeared tohaveover-representedbiologicalfunctions(Type2algorithm). TheapplicationofLASSObasedalgorithmtocluster representa-tivesprovidedcoefficientsforasystemoflinearODEsdescribing thedynamicsofeachcluster(Type5algorithm).Predictionsonthe directionofchangeobtainedfromODEsweretestedbycomparing themwithexpressionvaluesfromatimepointthatwasnotused forinferencepurposes.
4.4. Summary
Ascanbesurmisedfromtheexamplesgiven,algorithmsfrom
Type1,Type2,andType3aremorecommonincurrent experimen-talapproachesappliedtoplants.Theproblemofdimensionality preventstheextensiveuseofType4andType5algorithmsfor individualgenes based onwhole genomedatasets due todata requirementsforsuchtype of inference[13].Thus, the dimen-sionoftheproblemistypicallyreducedbylimitingasetofgenes toonesknowntointeractorparticipate inthesame biological process.Recentnon-stressrelatedapproachesinA.thalianahave
employedsuchtechniques.Espinosaetal.[126]used experimen-tally obtainedknowledge aboutrelationshipsof 15 genes in A. thalianaflowerdevelopmentprocesstopredictdevelopment sce-nariosusingBooleannetworksapproach(Type4algorithm).Sankar
et al. [127] built a model to predict states of thecomponents
fromauxinandbrassinosteroidsignalingnetworksinA.thaliana
byapplyingBooleanlogicapproach(Type4algorithm)andthen transformed theresulting discrete network representation to a setofordinarydifferentialequations(Type5algorithm)toobtain quantitativepredictions.Cruz-Ramirezetal.[128]investigatedthe dynamicsofasymmetriccelldivisionwithintheA.thalianarootby analyzingasystemofnonlineardifferentialequationsfor7 inter-actingcomplexes(Type5output).Theanalysispredictedabistable behavioroftheprocess.Finally,Pokhilkoetal.[129]refinedthe interactionmodeldescribingcircadianrhythmsinA.thalianaby modelingtheprocesswithasystemofnonlinear ODEs(Type5
output).
Similaritiesinregulatoryprocesses onagenomiclevel allow fortheapplicationofcomputationalapproachesthatwere devel-opedfornon-plantspecies.Somecomputationalapproachesare availableinsoftware packages.Anextensiveuseofthese pack-ages showsthat even ifa technique wasdeveloped and tested for one species, it can be applied to a similar dataset from anotherspecies.Examplesoftheseapproachesarebrieflydescribed here. Vermeirssen et al. [130] combined the Learning Module Networksalgorithm[131]developedforyeast,ContextLikelihood ofRelatednessalgorithm[132]testedonEscherichiacoli,and Dou-bleTwo-wayt-testsalgorithmtestedonE.colitoidentifyoxidative stressregulatorytranscriptionfactorsinA.thaliana(Type3output). TheAlgorithmfortheReconstructionofGeneRegulatoryNetworks (ARACNE)[58]wasdevelopedtoinfertranscriptionalregulations inhumanBcells,butthenusedforotherapplicationsincluding theinferenceoftranscriptionalinteractionsunderlyingroot devel-opmentandphysiological processesinA.thaliana[133](Type3
output).Othersoftwarepackagesthatshowedtheabilitytorecover generegulatorynetworksfromtranscriptomicdatainclude CLR
[132],MRNET[134],C3NET[57],andARTIVA[135].TheDialogue
onReverseEngineeringAssessmentandMethods(DREAM)project attemptedto comparesuch GRNinferencemethods appliedto
E.coli,Staphylococcusaureus,Saccharomycescerevisiaeandinsilico
microarraydata[136].Theauthorsdiscoveredthatthesemethods havecomplementaryadvantagesandlimitationsunderdifferent contexts.Inthecaseofmulticellularorganisms,theperformance oftechniqueshassofarbeenmeasuredbasedongoalsachieved foraspecificapplication.Suchperformanceisdifficulttocompare betweenmethodssincegoalsandapplicationsareoftendiverse.
5. Conclusions
We presented a classification of computational algorithms basedonthetype of information theyaimtoinfer.This struc-turewasusedtodescribeapproachesintheliteraturethathave been used to gain insight into biological processes of interest basedontranscriptomicsdata.Examplesofexistingcomputational approachesappliedtoplantstresstranscriptionaldatasets demon-stratedapatternoftransitionbetweenalgorithmsofdifferenttypes (displayedgraphicallyinFig.5).Thisprogressiondemonstratesthat thequalityofpredictionsmadebyanalgorithminthescopeofa computationalapproachoftendependsonthequalityof predic-tionsmadebyaprecedingalgorithmaswellasonthequalityofthe originalbiologicaldata.Basedonavailablealgorithmsandexample implementations,wecanstatethateventhoughbothstressrelated geneidentificationandgroupingalgorithms(Type1andType2) arestill evolving,confidenceinType 2algorithm predictionsis sufficienttoallowforatransitiontocausalityinference(Type3).
Type3algorithmshavethepotentialtosupplyType4andType 5algorithmswithinformationaboutthestructureofgene regula-torynetworks.Thisinformationwillreducethenumberofpossible functionalrelationshipstoconsiderforthesetypesofalgorithms dramaticallyandthusallowfortheincreaseinscopeand predic-tivepower.Therefore,theperspectivetransitionsshowninFig.5
willlikelyappearmoreofteninfuturecomputationalapproaches asreliabilityofType3algorithmspredictionsincrease.
Acknowledgements
Thismaterialisbasedupon worksupportedbytheNational ScienceFoundationunderGrantNo.1247427andbytheNational ScienceFoundationGraduateResearchFellowshipunderGrantNo. 1252376.WethankRosangelaSozzaniandRobertFranksforcritical readingofthismanuscript.
References
[1]R.Mittler,E.Blumwald,Geneticengineeringformodernagriculture: chal-lengesandperspectives,Annu.Rev.PlantBiol.61(2010)443–462. [2]L.Vaahtera,M.Brosché,Morethanthesumofitsparts–howtoachievea
specifictranscriptionalresponsetoabioticstress,PlantSci.180(3)(2011) 421–430.
[3]W.Wang,B.Vinocur,A.Altman,Plantresponsestodrought,salinityand extremetemperatures:towardsgenetic engineeringforstresstolerance, Planta218(1)(2003)1–14.
[4]A.E.Valdés,Forcedadaptation:plantproteinstofightclimatechange,Front. PlantSci.5(2014)762.
[5]J.Riechmann,J.Heard,G.Martin,L.Reuber,J.Keddie,L.Adam,O.Pineda, O.Ratcliffe,R.Samaha,R.Creelman,etal.,Arabidopsistranscriptionfactors: genome-widecomparativeanalysisamongeukaryotes,Science290(5499) (2000)2105–2110.
[6]M.K.Udvardi,K.Kakar,M.Wandrey,O.Montanari,J.Murray,A.Andriankaja, J.-Y.Zhang,V.Benedito,J.M.Hofer,F.Chueng,etal.,Legumetranscription factors:globalregulatorsofplantdevelopmentandresponsetothe environ-ment,PlantPhysiol.144(2)(2007)538–549.
[7]R.Melzer,G.Theißen,MADSandmore:transcriptionfactorsthatshapethe plant,in:PlantTranscriptionFactors,Springer,2011,pp.3–18.
[8]G.R.Cramer,K.Urano,S.Delrot,M.Pezzotti,K.Shinozaki,Effectsofabiotic stressonplants:asystemsbiologyperspective,BMCPlantBiol.11(1)(2011) 163.
[9]S.Friedel,B.Usadel,N.VonWirén,N.Sreenivasulu,Reverseengineering:a keycomponentofsystemsbiologytounravelglobalabioticstresscross-talk, Front.PlantSci.3(2012)294.
[10]G.Krouk,J.Lingeman,A.M.Colon,G.Coruzzi,D.Shasha,etal.,Generegulatory networksinplants:learningcausalityfromtimeandperturbation,Genome Biol.14(6)(2013)123.
[11]C.Sima,J.Hua,S.Jung,Inferenceofgeneregulatorynetworksusing time-seriesdata:asurvey,Curr.Genomics10(6)(2009)416.
[12]K.Cho,S.Choo,S.Jung,J.Kim,H.Choi,J.Kim,Reverseengineeringofgene regulatorynetworks,Syst.Biol.IET1(3)(2007)149–163.
[13]M.Hecker,S.Lambeck,S.Toepfer,E.vanSomeren,R.Guthke,Gene regu-latorynetworkinference:dataintegrationindynamicmodels–areview, Biosystems96(1)(2009)86.
[14]G.Karlebach,R.Shamir,Modellingandanalysisofgeneregulatorynetworks, Nat.Rev.Mol.CellBiol.9(10)(2008)770–780.
[15]A.M.Middleton,E. Farcot,M.R.Owen, T.Vernoux, Modeling regulatory networkstounderstandplantdevelopment:smallisbeautiful,PlantCell24 (10)(2012)3876–3891.
[16]N.J.Atkinson,P.E.Urwin,Theinteractionofplantbioticandabioticstresses: fromgenestothefield,J.Exp.Bot.63(10)(2012)3523–3543.
[17]C.deSassi,J.M.Tylianakis,Climatechangedisproportionatelyincreases her-bivoreoverplantorparasitoidbiomass,PLOSONE7(7)(2012)e40557. [18]J.Kilian,D.Whitehead,J.Horak,D.Wanke,S.Weinl,O.Batistic,C.D’Angelo,E.
Bornberg-Bauer,J.Kudla,K.Harter,TheAtGenExpressglobalstressexpression dataset:protocols,evaluationandmodeldataanalysisofUV-Blight,drought andcoldstressresponses,PlantJ.50(2)(2007)347–363.
[19]L.López-Maury,S.Marguerat,J.Bähler,Tuninggeneexpressiontochanging environments:fromrapidresponsestoevolutionaryadaptation,Nat.Rev. Genet.9(8)(2008)583–593.
[20]R.F.Ditt,K.F.Kerr,P.deFigueiredo,J.Delrow,L.Comai,E.W.Nester,The
ArabidopsisthalianatranscriptomeinresponsetoAgrobacteriumtumefaciens, Mol.PlantMicrobeInteract.19(6)(2006)665–681.
[21]R.J.O’Connell,M.R.Thon,S.Hacquard,S.G.Amyotte,J.Kleemann,M.F.Torres, U.Damm,E.A.Buiate,L.Epstein,N.Alkan,etal.,Lifestyletransitionsinplant pathogeniccolletotrichumfungidecipheredbygenomeandtranscriptome analyses,Nat.Genet.44(9)(2012)1060–1065.
[22]O.Windram,P.Madhou,S.McHattie,C.Hill,R.Hickman,E.Cooke,D.J.Jenkins, C.A.Penfold,L.Baxter,E.Breeze,etal.,ArabidopsisdefenseagainstBotrytis cinerea:chronologyandregulationdecipheredbyhigh-resolutiontemporal transcriptomicanalysis,PlantCell24(9)(2012)3530–3557.
[23]B.-h.Lee,D.A.Henderson,J.-K.Zhu,TheArabidopsiscold-responsive transcrip-tomeanditsregulationbyICE1,PlantCell17(11)(2005)3155–3175. [24]A.S.Iyer-Pascuzzi,T.Jackson,H.Cui,J.J.Petricka,W.Busch,H.Tsukagoshi,P.N.
Benfey,Cellidentityregulatorslinkdevelopmentandstressresponsesinthe
Arabidopsisroot,Dev.Cell21(4)(2011)770–782.
[25]J.R.Dinneny,T.A.Long,J.Y.Wang,J.W.Jung,D.Mace,S.Pointer,C.Barron, S.M.Brady,J.Schiefelbein,P.N.Benfey,Cellidentitymediatestheresponseof
Arabidopsisrootstoabioticstress,Science320(5878)(2008)942–945. [26]S.González-Pérez,J.Gutiérrez,F.García-García,D.Osuna,J.Dopazo,Ó.
Lorenzo,Ó.Lorenzo,J.L.Revuelta,J.B.Arellano,Earlytranscriptionaldefense responsesinArabidopsiscellsuspensioncultureunderhigh-lightconditions, PlantPhysiol.156(3)(2011)1439–1456.
[27]T.J.Buckhout,T.J.Yang,W.Schmidt,Earlyiron-deficiency-induced transcrip-tionalchangesinArabidopsisrootsasrevealedbymicroarrayanalyses,BMC Genomics10(1)(2009)147.
[28]T.A.Long,H.Tsukagoshi,W.Busch,B.Lahner,D.E.Salt,P.N.Benfey,ThebHLH transcriptionfactorPOPEYEregulatesresponsetoirondeficiencyin Arabidop-sisroots,PlantCell22(7)(2010)2219–2236.
[29]G.Krouk,P.Mirowski,Y.LeCun,D.E.Shasha,G.M.Coruzzi,etal.,Predictive networkmodelingofthehigh-resolutiondynamicplanttranscriptomein responsetonitrate,GenomeBiol.11(12)(2010)R123.
[30]W.-D. Lin, Y.-Y. Liao, T.J. Yang, C.-Y. Pan, T.J. Buckhout, W. Schmidt, Coexpression-basedclusteringofArabidopsisrootgenespredictsfunctional modules in early phosphate deficiencysignaling, Plant Physiol. (2011) 110.
[31]L.Rizhsky,H.Liang,J.Shuman,V.Shulaev,S.Davletova,R.Mittler,When defensepathwayscollide.TheresponseofArabidopsistoacombinationof droughtandheatstress,PlantPhysiol.134(4)(2004)1683–1696. [32]S.Rasmussen,P.Barah,M.C.Suarez-Rodriguez,S.Bressendorff,P.Friis,P.
Costantino,A.M.Bones,H.B.Nielsen,J. Mundy,Transcriptomeresponses tocombinationsofstressesinArabidopsis,PlantPhysiol. 161(4)(2013) 1783–1794.
[33]C.M.Prasch,U.Sonnewald,Simultaneousapplicationofheat,drought,and virustoArabidopsisplantsrevealssignificantshiftsinsignalingnetworks, PlantPhysiol.162(4)(2013)1849–1866.
[34]N.Sewelam,Y.Oshima,N.Mitsuda,M.Ohme-Takagi,Asteptowards under-standingplantresponsestomultipleenvironmentalstresses:agenome-wide study,PlantCellEnviron.37(9)(2014)2024–2035.
[35]A.Hahn,J.Kilian,A.Mohrholz,F.Ladwig,F.Peschke,R.Dautel,K.Harter,K.W. Berendzen,D.Wanke,Plantcoreenvironmentalstressresponsegenesare systemicallycoordinatedduringabioticstresses,Int.J.Mol.Sci.14(4)(2013) 7617–7641.
[36]J.L.Riechmann,Transcriptionalregulation:agenomicoverview,The Ara-bidopsisBook16(1)(2002)1.
[37]X.Cui,G.A.Churchill,etal.,StatisticaltestsfordifferentialexpressionincDNA microarrayexperiments,GenomeBiol.4(4)(2003)210.
[38]G.K.Smyth,J.Michaud,H.S.Scott,Useofwithin-arrayreplicatespotsfor assessingdifferentialexpressioninmicroarrayexperiments,Bioinformatics 21(9)(2005)2067–2075.
[39]S.Anders,W.Huber,Differentialexpressionanalysisforsequencecountdata, GenomeBiol.11(10)(2010)R106.
[40]M.D.Robinson,D.J.McCarthy,G.K.Smyth,edgeR:abioconductorpackagefor differentialexpressionanalysisofdigitalgeneexpressiondata,Bioinformatics 26(1)(2010)139–140.
[41]K.Aoki,Y.Ogata,D.Shibata,Approachesforextractingpracticalinformation fromgeneco-expressionnetworksinplantbiology,PlantCellPhysiol.48(3) (2007)381–390.
[42]B.Zhang,S.Horvath,etal.,Ageneralframeworkforweightedgene co-expressionnetworkanalysis,Stat.Appl.Genet.Mol.Biol.4(1)(2005)1128.
[43]C.J.Wolfe,I.S.Kohane,A.J.Butte,Systematicsurveyrevealsgeneral appli-cabilityof“guilt-by-association”withingenecoexpressionnetworks,BMC Bioinform.6(1)(2005)227.
[44]B.Usadel,T.Obayashi,M.Mutwil,F.M.Giorgi,G.W.Bassel,M.Tanimoto,A. Chow,D.Steinhauser,S.Persson,N.J.Provart,Co-expressiontoolsforplant biology:opportunitiesforhypothesisgenerationandcaveats,PlantCell Envi-ron.32(12)(2009)1633–1651.
[45]I.Lee,B.Ambaru,P.Thakkar,E.M.Marcotte,S.Y.Rhee,Rationalassociationof geneswithtraitsusingagenome-scalegenenetworkforArabidopsisthaliana, Nat.Biotechnol.28(2)(2010)149–156.
[46]A. Gupta, C.D. Maranas, R. Albert, Elucidation of directionalityfor co-expressedgenes:predictingintra-operonterminationsites,Bioinformatics 22(2)(2006)209–214.
[47]J.Ehlting,V.Sauveplane,A.Olry,J.-F.Ginglinger,N.J.Provart,D. Werck-Reichhart,Anextensive(co-)expressionanalysistoolforthecytochrome P450superfamilyinArabidopsisthaliana,BMCPlantBiol.8(1)(2008)47. [48]K.Polanski,J.Rhodes,C.Hill,P.Zhang,D.J.Jenkins,S.J.Kiddle,A.Jironkin,
J.Beynon,V.Buchanan-Wollaston,S.Ott,etal.,Wigwams:identifyinggene modulesco-regulatedacrossmultiplebiologicalconditions,Bioinformatics 30(7)(2014)962–970.
[49]R.Balasubramaniyan,E.Hüllermeier,N.Weskamp,J.Kämper,Clusteringof geneexpressiondatausingalocalshape-basedsimilaritymeasure, Bioinfor-matics21(7)(2005)1069–1077.
[50]J.Nie,R.Stewart,H.Zhang,J.Thomson,F.Ruan,X.Cui,H.Wei,TF-Cluster:a pipelineforidentifyingfunctionallycoordinatedtranscriptionfactorsvia net-workdecompositionofthesharedcoexpressionconnectivitymatrix(SCCM), BMCSyst.Biol.5(1)(2011)53.
[51]X.Cui,T.Wang,H.-S.Chen,V.Busov,H.Wei,Tf-finder:asoftwarepackage foridentifyingtranscriptionfactorsinvolvedinbiologicalprocessesusing microarraydataandexistingknowledgebase,BMCBioinform.11(1)(2010) 425.
[52]H.Kishino,P.J.Waddell,Correspondenceanalysisofgenesandtissuetypes andfindinggeneticlinksfrommicroarraydata,GenomeInform.11(2000) 83–95.
[53]A.Wille,P.Zimmermann,E.Vranová,A.Fürholz,O.Laule,S.Bleuler,L.Hennig, A.Prelic,P.vonRohr,L.Thiele,etal.,SparsegraphicalGaussianmodelingofthe isoprenoidgenenetworkinArabidopsisthaliana,GenomeBiol.5(11)(2004) R92.
[54]J.Schäfer,K.Strimmer,Anempiricalbayesapproachtoinferringlarge-scale geneassociationnetworks,Bioinformatics21(6)(2005)754–764. [55]P.D’haeseleer,etal.,Howdoesgeneexpressionclusteringwork?Nat.
Bio-technol.23(12)(2005)1499–1502.
[56]C.Ma,M.Xin,K.A.Feldmann,X.Wang,Machinelearning-baseddifferential networkanalysis:astudyofstress-responsivetranscriptomesinArabidopsis, PlantCell26(2)(2014)520–537.
[57]G.Altay,F.Emmert-Streib,Inferringtheconservativecausalcoreofgene regulatorynetworks,BMCSyst.Biol.4(1)(2010)132.
[58]A.A.Margolin,I.Nemenman,K.Basso,C.Wiggins,G.Stolovitzky,R.D.Favera, A.Califano,ARACNE:analgorithmforthereconstructionofgeneregulatory networksinamammaliancellularcontext,BMCBioinform.7(Suppl1)(2006) S7.
[59]R.Steuer,J.Kurths,C.O.Daub,J.Weise,J.Selbig,Themutualinformation: detectingandevaluatingdependenciesbetweenvariables,Bioinformatics18 (suppl2)(2002)S231–S240.
[60]S.Kumari,J.Nie,H.-S.Chen,H.Ma,R.Stewart,X.Li,M.-Z.Lu,W.M.Taylor, H.Wei,Evaluationofgeneassociationmethodsforcoexpressionnetwork constructionandbiologicalknowledgediscovery,PLOSONE7(11)(2012) e50411.
[61]C.Ma,X.Wang,Applicationoftheginicorrelationcoefficienttoinfer regu-latoryrelationshipsintranscriptomeanalysis,PlantPhysiol.160(1)(2012) 192–203.
[62]L.Rueda,A.Bari,A.Ngom,Clusteringtime-seriesgeneexpressiondatawith unequaltimeintervals,in:TransactionsonComputationalSystemsBiology X,Springer,2008,pp.100–123.
[63]M.Triska,D.Grocutt,J.Southern,D.J.Murphy,T.Tatarinova,cisExpress:motif detectioninDNAsequences,Bioinformatics29(17)(2013)2203–2205. [64]S.Martin,Z.Zhang,A.Martino,J.Faulon,Booleandynamicsofgenetic
regu-latorynetworksinferredfrommicroarraytimeseriesdata,Bioinformatics23 (7)(2007)866–874.
[65]S.VanDongen,Aclusteralgorithmforgraphs,Rep.Inf.Syst.(10)(2000)1–40. [66]W.I.Mentzen,E.S.Wurtele,RegulonorganizationofArabidopsis,BMCPlant
Biol.8(1)(2008)99.
[67]Y.Zhang,H.Zha,C.-H.Chu,Atime-seriesbiclusteringalgorithmfor revea-lingco-regulatedgenes,in:InformationTechnology:CodingandComputing, 2005.ITCC2005,vol.1,IEEE,2005,pp.32–37.
[68]P. Tamayo,D. Slonim,J. Mesirov,Q. Zhu,S.Kitareewan,E.Dmitrovsky, E.S.Lander,T.R.Golub,Interpretingpatternsofgeneexpressionwith self-organizingmaps:methodsandapplicationtohematopoieticdifferentiation, Proc.Natl.Acad.Sci.U.S.A.96(6)(1999)2907–2912.
[69]M.B.Eisen,P.T.Spellman,P.O.Brown,D.Botstein,Clusteranalysisanddisplay ofgenome-wideexpressionpatterns,Proc.Natl.Acad.Sci.U.S.A.95(25) (1998)14863–14868.
[70]B.J.Frey,D.Dueck,Clusteringbypassingmessagesbetweendatapoints, Sci-ence315(2007)972–976.
[71]X.Li,Y.Ye,M.Ng,Q.Wu,MultiFacTV:moduledetectionfromhigher-order timeseriesbiologicaldata,BMCGenomics14(Suppl4)(2013)S2.
[72]J.-X.Liu,C.-H.Zheng,Y.Xu,Extractingplantscoregenesrespondingtoabiotic stressesbypenalizedmatrixdecomposition,Comput.Biol.Med.42(5)(2012) 582–589.
[73]T.Chen,V.Filkov,S.S.Skiena,Identifyinggeneregulatorynetworksfrom experimentaldata,in:ProceedingsoftheThirdAnnualInternational Con-ferenceonComputationalMolecularBiology,ACM,1999,pp.94–103. [74]A.T.Kwon,H.H.Hoos,R.Ng,Inferenceoftranscriptionalregulation
relation-shipsfromgeneexpressiondata,Bioinformatics19(8)(2003)905–912. [75]W.A.Schmitt,R.M.Raab,G.Stephanopoulos,Elucidationofgeneinteraction
networksthroughtime-laggedcorrelationanalysisoftranscriptionaldata, GenomeRes.14(8)(2004)1654–1663.
[76]W.Zhao,E.Serpedin,E.R.Dougherty,Inferringgeneregulatorynetworksfrom timeseriesdatausingtheminimumdescriptionlengthprinciple, Bioinfor-matics22(17)(2006)2129–2135.
[77]P.C.Ma,K.C.Chan,Inferringgeneregulatorynetworksfromexpressiondata bydiscoveringfuzzydependencyrelationships,IEEETrans.FuzzySyst.16(2) (2008)455–465.
[78]H.Redestig,D.Weicht,J.Selbig,M.A.Hannah,Transcriptionfactortarget pre-dictionusingmultipleshortexpressiontimeseriesfromArabidopsisthaliana, BMCBioinform.8(1)(2007)454.
[79]D.Heckerman,AtutorialonlearningwithBayesiannetworks,Innov.Bayesian Netw.(2008)33–82.
[80]N.Friedman,M.Linial,I.Nachman,D.Pe’er,UsingBayesiannetworksto ana-lyzeexpressiondata,J.Comput.Biol.7(3–4)(2000)601–620.
[81]K.Murphy,S.Mian,etal.,Modellinggeneexpressiondatausingdynamic Bayesiannetworks,Tech.rep.,TechnicalReport,ComputerScienceDivision, UniversityofCalifornia,Berkeley,CA,1999.
[82]N.Dojer,A.Gambin,A.Mizera,B.Wilczy ´nski,J.Tiuryn,Applyingdynamic Bayesiannetworkstoperturbedgeneexpressiondata,BMCBioinform.7(1) (2006)249.
[83]S.Liang,S.Fuhrman,R.Somogyi,etal.,REVEAL,ageneralreverseengineering algorithmforinferenceofgeneticnetworkarchitectures,in:Pacific Sympo-siumonBiocomputing,vol.3,1998,p.2.
[84]R.Albert,Booleanmodelingof geneticregulatorynetworks,in:Complex Networks,Springer,2004,pp.459–481.
[85]E.Dimitrova,L.García-Puente,F.Hinkelmann,A.Jarrah,R.Laubenbacher,B. Stigler,M.Stillman,P.Vera-Licona,ParameterestimationforBooleanmodels ofbiologicalnetworks,Theor.Comput.Sci.412(26)(2011)2816–2826. [86]R.Laubenbacher,B.Stigler,Acomputationalalgebraapproachtothereverse
engineeringofgeneregulatorynetworks,J. Theor.Biol. 229(4) (2004) 523–537.
[87]T.Akutsu,S.Miyano,S.Kuhara,etal.,Identificationofgeneticnetworksfroma smallnumberofgeneexpressionpatternsunderthebooleannetworkmodel, in:PacificSymposiumonBiocomputing,vol.4,WorldScientificMaui,Hawaii, 1999,pp.17–28.
[88]B.A.Rosa,J.Zhang,I.T.Major,W.Qin,J.Chen,Optimaltimepointsamplingin high-throughputgeneexpressionexperiments,Bioinformatics28(21)(2012) 2773–2781.
[89]G.Bernot,J.-P.Comet,A.Richard,M.Chaves,J.-L.Gouzé,F.Dayan, Model-ingandanalysisofgeneregulatorynetworks,in:ModelinginComputational BiologyandBiomedicine,Springer,2013,pp.47–80.
[90]R.Tibshirani,Regressionshrinkageandselectionviathelasso,J.R.Stat.Soc. B:Methodological(1996)267–288.
[91]R.Guthke,U.Möller,M.Hoffmann,F.Thies,S.Töpfer,Dynamicnetwork recon-structionfromgeneexpressiondataappliedtoimmuneresponseduring bacterialinfection,Bioinformatics21(8)(2005)1626–1634.
[92]M.Gustafsson,M.Hornquist,A.Lombardi,Large-scalereverseengineeringby thelasso,arXivpreprintq-bio/0403012.
[93]M.S.Yeung,J.Tegnér,J.J.Collins,Reverseengineeringgenenetworksusing singularvaluedecompositionandrobustregression,Proc.Natl.Acad.Sci.U. S.A.99(9)(2002)6163–6168.
[94]M.Gustafsson,M.Hörnquist,J.Lundström,J.Björkegren,J.Tegnér,Reverse engineeringofgenenetworkswithLASSOandnonlinearbasisfunctions,Ann. N.Y.Acad.Sci.1158(1)(2009)265–275.
[95]L.Palafox,N.Noman,H.Iba,Reverseengineeringofgeneregulatorynetworks usingdissipativeparticleswarmoptimization,IEEETrans.Evol.Comput.17 (4)(2013)577–587.
[96]M.Kabir,N.Noman,H.Iba,Reverseengineeringgeneregulatorynetworkfrom microarraydatausinglineartime-variantmodel,BMCBioinform.11(Suppl 1)(2010)S56.
[97]M.Ashburner,C.A.Ball,J.A.Blake,D.Botstein,H.Butler,J.M.Cherry,A.P.Davis, K.Dolinski,S.S.Dwight,J.T.Eppig,etal.,GeneOntology:toolfortheunification ofbiology,Nat.Genet.25(1)(2000)25–29.
[98]S.Maere,K.Heymans,M.Kuiper,BiNGO:aCytoscapeplugintoassess overrep-resentationofgeneontologycategoriesinbiologicalnetworks,Bioinformatics 21(16)(2005)3448–3449.
[99]J.M.Alonso,A.N.Stepanova,T.J.Leisse,C.J.Kim,H.Chen,P.Shinn,D.K. Stevenson,J.Zimmerman,P.Barajas,R.Cheuk,etal.,Genome-wide inser-tional mutagenesis of Arabidopsis thaliana, Science 301 (5633) (2003) 653–657.
[100] A.Sessions,E.Burke,G.Presting,G.Aux,J.McElver,D.Patton,B.Dietrich,P. Ho,J.Bacwaden,C.Ko,etal.,Ahigh-throughputArabidopsisreversegenetics system,PlantCell14(12)(2002)2985–2994.
[101] M.G.Rosso,Y.Li,N.Strizhov,B.Reiss,K.Dekker,B.Weisshaar,An Arabidop-sisthalianaT-DNAmutagenizedpopulation(GABI-Kat)forflankingsequence tag-basedreversegenetics,PlantMol.Biol.53(1–2)(2003)247–259.
[102] B.Ülker,E.Peiter,D.P.Dixon,C.Moffat,R.Capper,N.Bouché,R.Edwards,D. Sanders,H.Knight,M.R.Knight,Gettingthemostoutofpubliclyavailable T-DNAinsertionlines,PlantJ.56(4)(2008)665–677.
[103] P.Hilson,J.Allemeersch,T.Altmann,S.Aubourg,A.Avon,J.Beynon,R.P. Bhalerao,F.Bitton,M.Caboche,B.Cannoot,etal.,Versatilegene-specific sequencetagsforArabidopsisfunctionalgenomics:transcriptprofilingand reversegeneticsapplications,GenomeRes.14(10b)(2004)2176–2189. [104] A.Coego,E.Brizuela,P.Castillejo,S.Ruíz,C.Koncz,J.C.delPozo,M.Pineiro,
J.A.Jarillo,J.Paz-Ares,J.León,TheTRANSPLANTAcollectionofArabidopsis
lines:aresourceforfunctionalanalysisoftranscriptionfactorsbasedontheir conditionaloverexpression,PlantJ.77(6)(2014)944–953.
[105] K.Higo,Y.Ugawa,M.Iwamoto,T.Korenaga,Plantcis-actingregulatoryDNA elements(PLACE)database:1999,NucleicAcidsRes.27(1)(1999)297–300. [106] R.V.Davuluri,H.Sun,S.K.Palaniswamy,N.Matthews,C.Molina,M.Kurtz,E. Grotewold,AGRIS.Arabidopsisgeneregulatoryinformationserver,an infor-mationresourceofArabidopsiscis-regulatoryelementsandtranscription factors,BMCBioinform.4(1)(2003)25.
[107] S.K.Palaniswamy,S.James,H.Sun,R.S.Lamb,R.V.Davuluri,E.Grotewold, AGRISandAtRegNet.Aplatformtolinkcis-regulatoryelementsand transcrip-tionfactorsintoregulatorynetworks,PlantPhysiol.140(3)(2006)818–829. [108] T.R. O’Connor, C. Dyreson, J.J. Wyrick, Athena: a resource for rapid visualizationandsystematicanalysisofArabidopsispromotersequences, Bioinformatics21(24)(2005)4411–4413.
[109] S.M.Brady,N.J.Provart,Web-queryablelarge-scaledatasetsforhypothesis generationinplantbiology,PlantCell21(4)(2009)1034–1051.
[110] T.L.Bailey,M.Boden,F.A.Buske,M.Frith,C.E.Grant,L.Clementi,J.Ren,W.W. Li,W.S.Noble,MEMESUITE:toolsformotifdiscoveryandsearching,Nucleic AcidsRes.(2009)gkp335.
[111] M.K.Das,H.-K.Dai,AsurveyofDNAmotiffindingalgorithms,BMCBioinform. 8(Suppl.7)(2007)S21.
[112] K.W. Berendzen,K.Harter, D. Wanke,Analysisof plantregulatorydna sequencesbytransientprotoplastassaysandcomputeraidedsequence eval-uation,in:PlantSignalTransduction,Springer,2009,pp.311–335. [113] N.Wehner,L.Hartmann,A.Ehlert,S.Böttner,L.Onate-Sánchez,W.
Dröge-Laser, High-throughput protoplast transactivation (PTA) system for the analysisofArabidopsistranscriptionfactorfunction,PlantJ.68(3)(2011) 560–569.
[114] C.Olsen,K.Fleming,N.Prendergast,R.Rubio,F.Emmert-Streib,G.Bontempi, B.Haibe-Kains,J.Quackenbush,Inferenceandvalidationofpredictivegene networksfrombiomedicalliteratureandgeneexpressiondata,Genomics103 (5)(2014)329–336.
[115] T.-M.Chu,B.Weir,R.Wolfinger,Asystematicstatisticallinearmodeling approachtooligonucleotidearrayexperiments,Math.Biosci.176(1)(2002) 35–51.
[116] S.Ma,H.J.Bohnert,IntegrationofArabidopsisthalianastress-relatedtranscript profiles,promoterstructures,andcell-specificexpression,GenomeBiol.8(4) (2007)R49.
[117] A.P.Gasch,M.B.Eisen,Exploringtheconditionalcoregulationofyeastgene expressionthroughfuzzyk-meansclustering,GenomeBiol.3(11)(2002) 1–22.
[118] G.K.Smyth,Linearmodelsandempiricalbayesmethodsforassessing differ-entialexpressioninmicroarrayexperiments,Stat.Appl.Genet.Mol.Biol.3 (1)(2004)1–25.
[119] S.Ma,S.Bachan,M.Porto,H.J.Bohnert,M.Snyder,S.P.Dinesh-Kumar, Dis-coveryofstressresponsiveDNAregulatorymotifsinArabidopsis,PLOSONE 7(8)(2012)e43198.
[120]H.Wu,M.K.Kerr,X.Cui,G.A.Churchill,MAANOVA:asoftwarepackageforthe analysisofspottedcDNAmicroarrayexperiments,in:TheAnalysisofGene ExpressionData,Springer,2003,pp.313–341.
[121]O.Stegle,K.J.Denby,E.J.Cooke,D.L.Wild,Z.Ghahramani,K.M.Borgwardt,A robustbayesiantwo-sampletestfordetectingintervalsofdifferentialgene expressioninmicroarraytimeseries,J.Comput.Biol.17(3)(2010)355–367. [122]Y.C.Tai,T.P.Speed,etal.,AmultivariateempiricalBayesstatisticforreplicated
microarraytimecoursedata,Ann.Stat.34(5)(2006)2387–2412.
[123]N.A.Heard,C.C.Holmes,D.A.Stephens,D.J.Hand,G.Dimopoulos,Bayesian coclusteringofanophelesgeneexpressiontimeseries:studyofimmune defenseresponsetomultipleexperimentalchallenges,Proc.Natl.Acad.Sci. U.S.A.102(47)(2005)16939–16944.
[124]S.Klemm,etal.,CausalStructureIdentificationinNonlinearDynamical Sys-tems,Dept.ofEng.,Univ.ofCambridge,UnitedKingdom,2008(Master’s thesis).
[125]E.Howe,K.Holton,S.Nair,D.Schlauch,R.Sinha,J.Quackenbush,MeV: multi-experimentviewer,in:BiomedicalInformaticsforCancerResearch,Springer, 2010,pp.267–277.
[126]C.Espinosa-Soto,P.Padilla-Longoria,E.R.Alvarez-Buylla,Ageneregulatory networkmodelforcell-fatedeterminationduringArabidopsisthalianaflower developmentthatisrobustandrecoversexperimentalgeneexpression pro-files,PlantCell16(11)(2004)2923–2939.
[127]M.Sankar,K.S.Osmont,J.Rolcik,B.Gujas,D.Tarkowska,M.Strnad,I.Xenarios, C.S.Hardtke,Aqualitativecontinuousmodelofcellularauxinand brassinos-teroidsignalingandtheircrosstalk,Bioinformatics27(10)(2011)1404–1412. [128]A.Cruz-Ramírez,S.Díaz-Trivino,I.Blilou,V.A.Grieneisen,R.Sozzani,C. Zamioudis,P.Miskolczi,J.Nieuwland,R.Benjamins,P.Dhonukshe,etal.,A bistablecircuitinvolvingscarecrow-retinoblastomaintegratescuestoinform asymmetricstemcelldivision,Cell150(5)(2012)1002–1015.
[129]A.Pokhilko,A.P.Fernández,K.D.Edwards,M.M.Southern,K.J.Halliday,A.J. Millar,TheclockgenecircuitinArabidopsisincludesarepressilatorwith additionalfeedbackloops,Mol.Syst.Biol.8(1)(2012)574.
[130]V.Vermeirssen,I.DeClercq,T.VanParys,F.VanBreusegem,Y.VandePeer,
Arabidopsisensemblereverse-engineeredgeneregulatorynetworkdiscloses interconnectedtranscriptionfactorsinoxidativestress,PlantCell(2014), tpc-114.
[131]A.Joshi,R.DeSmet,K.Marchal,Y.VandePeer,T.Michoel,Modulenetworks revisited:computationalassessmentandprioritizationofmodelpredictions, Bioinformatics25(4)(2009)490–496.
[132]J.J.Faith,B.Hayete,J.T.Thaden,I.Mogno,J.Wierzbowski,G.Cottarel,S.Kasif, J.J.Collins,T.S.Gardner,Large-scalemappingandvalidationofEscherichiacoli
transcriptionalregulationfromacompendiumofexpressionprofiles,PLoS Biol.5(1)(2007)e8.
[133]R.A.Montes,G.Coello,K.L.González-Aguilera,N.Marsch-Martínez,S.de Folter,E.R.Alvarez-Buylla,ARACNe-basedinference,usingcurated microar-raydata,ofArabidopsisthalianaroottranscriptionalregulatorynetworks,BMC PlantBiol.14(1)(2014)97.
[134]P.E.Meyer,K.Kontos,F.Lafitte,G.Bontempi,Information-theoreticinference oflargetranscriptionalregulatorynetworks,EURASIPJ.Bioinform.Syst.Biol. 2007(2007)79879.
[135]S.Lebre,J.Becq,F.Devaux,M.P.Stumpf,G.Lelandais,Statisticalinferenceof thetime-varyingstructureofgene-regulationnetworks,BMCSyst.Biol.4(1) (2010)130.
[136]D.Marbach,J.C.Costello,R.Küffner,N.M.Vega,R.J.Prill,D.M.Camacho,K.R. Allison,M.Kellis,J.J.Collins,G.Stolovitzky,etal.,Wisdomofcrowdsforrobust genenetworkinference,Nat.Methods9(8)(2012)796–804.