Acta Cryst.(2001). E57, o145±o146 DOI: 101107/S1600536801000812 Bond, Shan and Jones C12H8N2
o145
organic papers
Acta Crystallographica Section E Structure Reports
Online
ISSN 1600-5368
4,7-Phenanthroline
Andrew D. Bond,* Ning Shan and William Jones
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, England
Correspondence e-mail: [email protected]
Key indicators Single-crystal X-ray study T= 180 K
Mean(C±C) = 0.007 AÊ Rfactor = 0.063 wRfactor = 0.161 Data-to-parameter ratio = 8.7
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2001 International Union of Crystallography Printed in Great Britain ± all rights reserved
The structure of 4,7-phenanthroline, C12H8N2, has been
determined at 180 K. The molecular unit possesses
pseudo-C2v point symmetry but does not possess crystallographic mirror symmetry. The molecules form stacks approximately along the b direction, with molecules in adjacent stacks forming an interplane angle ofca54.
Comment
As part of a continuing study of cocrystal formation between organic acids and N-containing organic bases, we have deter-mined the structure of 4,7-phenanthroline, (I), at 180 K. The molecular unit possesses pseudo-C2vpoint symmetry, but does not exhibit crystallographic mirror symmetry. Similar obser-vations have been made for the isomeric 1,10-phenanthroline (Nishigaki et al., 1978). In the crystal structure, 4,7-phenan-throline forms planar stacks approximately along the b
direction with molecules in adjacent stacks forming an inter-plane angle ofca 54 (Fig. 2); this contrasts with the
obser-vation of two approximately perpendicular layers in 1,10-phenanthroline. There is no conclusive evidence for direc-tional CÐH N contacts in 4,7-phenanthroline, with the shortest H N contacts, H1 N7i= 2.72, H8 N4ii= 3.00 and
H10 N7iii = 3.00 AÊ, exhibiting CÐH N angles of 153.6,
128.6 and 119.5, respectively [symmetry codes: (i) 3 2ÿx,
ÿ1 +y,1
2+z; (ii)12+x, 1ÿy, z; (iii)32ÿx, y,12+z].
Experimental
4,7±Phenanthroline was obtained from Aldrich and recrystallized from ethanol.
Crystal data
C12H8N2 Mr= 180.20 Orthorhombic,Pca21 a= 19.141 (4) AÊ
b= 3.8417 (4) AÊ
c= 11.564 (2) AÊ
V= 850.4 (3) AÊ3 Z= 4
Dx= 1.408 Mg mÿ3
MoKradiation Cell parameters from 6723
re¯ections
= 1.0±25.0
= 0.09 mmÿ1 T= 180 (2) K Plate, colourless 0.300.090.05 mm
Data collection
Nonius KappaCCD diffractometer Thin-slice!and'scans Absorption correction: multi-scan
(SORTAV; Blessing, 1995)
Tmin= 0.975,Tmax= 0.996
2186 measured re¯ections 1099 independent re¯ections
812 re¯ections withI> 2(I)
Rint= 0.080
max= 25.1 h=ÿ22!17
k=ÿ4!3
l=ÿ13!10
Re®nement
Re®nement onF2 R[F2> 2(F2)] = 0.063 wR(F2) = 0.161 S= 1.09 1099 re¯ections 127 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.078P)2] whereP= (Fo2+ 2Fc2)/3 (/)max= 0.002
max= 0.30 e AÊÿ3
min=ÿ0.27 e AÊÿ3
Absolute structure: Flack (1983) Flack parameter =ÿ3 (8)
The absolute structure was not determined. Friedel opposites merged prior to merging of data inPca21. H atoms were placed geometrically and allowed to ride during subsequent re®nement with an isotropic displacement parameter ®xed at 1.2 timesUisofor the C atom to which they are attached.
Data collection:COLLECT(Nonius, 1998); cell re®nement:HKL SCALEPACK(Otwinowski & Minor, 1997); data reduction:HKL
DENZO and SCALEPACK (Otwinowski & Minor, 1997);
program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to re®ne structure:SHELXL97 (Sheldrick, 1997); molecular graphics:XP(Sheldrick, 1993); software used to prepare material for publication:SHELXL97.
We thank the EPSRC for ®nancial assistance with purchase of the CCD diffractometers, and the Cambridge Overseas Trust and British Council for funding (NS).
References
Blessing, R. H. (1995).Acta Cryst.A51, 33±38. Flack, H. D. (1983).Acta Cryst.A39, 876±881.
Otwinowski Z. & Minor W. (1997).Methods Enzymol.276307±316. Nishigaki, S., Yoshioka, H. & Nakatsu, K. (1978).Acta Cryst.B34, 875±879. Nonius (1998).COLLECT. Nonius BV, Delft, The Netherlands.
Sheldrick, G. M. (1993).XP. University of GoÈttingen, Germany.
Sheldrick, G. M. (1997). SHELXL97 and SHELXS97. University of GoÈttingen, Germany.
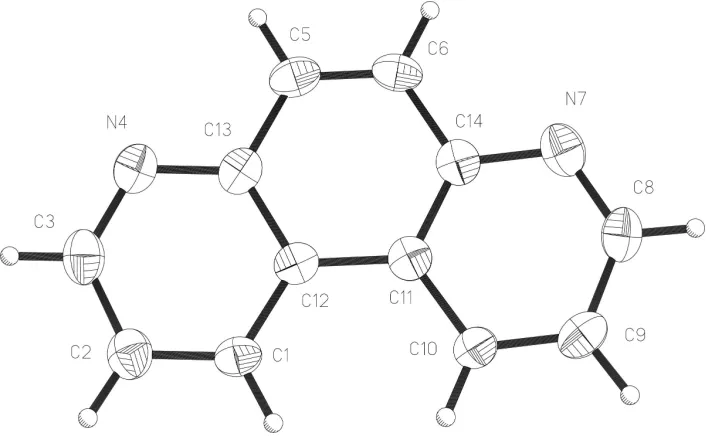
Figure 1
The molecular unit of the title compound showing displacement ellipsoids at the 50% probability level.
Figure 2
supporting information
sup-1
Acta Cryst. (2001). E57, o145–o146supporting information
Acta Cryst. (2001). E57, o145–o146 [doi:10.1107/S1600536801000812]
4,7-Phenanthroline
Andrew D. Bond, Ning Shan and William Jones
S1. Comment
As part of a continuing study of cocrystal formation between organic acids and N-containing organic bases, we have
determined the structure of 4,7-phenanthroline, (I), at 180 K. The molecular unit possesses pseudo-C2v point symmetry,
but does not exhibit crystallographic mirror symmetry. Similar observations have been made for the isomeric
1,10-phenanthroline (OPENAN; Nishigaki et al., 1978). In the crystal structure, 4,7-phenanthroline forms planar stacks
approximately along the b direction with molecules in adjacent stacks forming an interplane angle of ca 54° (Fig. 2); this
contrasts with the observation of two approximately perpendicular layers in 1,10-phenanthroline. There is no conclusive
evidence for directional C—H···N contacts in 4,7-phenanthroline, with the shortest H···N contacts, H1···N7i = 2.72,
H8···N4ii = 3.00 and H10···N7iii = 3.00 Å, exhibiting C—H···N angles of 153.6, 128.6 and 119.5°, respectively [symmetry
codes: (i) 3/2 - x, -1 + y, 1/2 + z; (ii) 1/2 + x, 1 - y, z; (iii) 3/2 - x, y, 1/2 + z].
S2. Experimental
4,7-Phenanthroline was obtained from Aldrich and recrystallized from ethanol.
S3. Refinement
The absolute structure was not determined. Friedel opposites merged prior to merging of data in Pca21. H atoms were
placed geometrically and allowed to ride during subsequent refinement with an isotropic displacement parameter fixed at
Figure 1
The molecular unit of the title compound showing displacement ellipsoids at the 50% probability level.
Figure 2
Projection onto (100) showing molecular stacks tilted with respect to each other with molecules in adjacent stacks
forming an interplane angle of ca 54°.
4,7-phenanthroline
Crystal data C12H8N2
Mr = 180.20
Orthorhombic, Pca21
a = 19.141 (4) Å b = 3.8417 (4) Å c = 11.564 (2) Å V = 850.4 (3) Å3
Z = 4 F(000) = 376
Dx = 1.408 Mg m−3
Melting point = 445–447 K Mo Kα radiation, λ = 0.71073 Å Cell parameters from 6723 reflections θ = 1.0–25.0°
µ = 0.09 mm−1
T = 180 K Plate, colourless 0.30 × 0.09 × 0.05 mm
Data collection Nonius KappaCCD
diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
Thin slice ω and φ scans
[image:4.610.128.483.330.434.2]supporting information
sup-3
Acta Cryst. (2001). E57, o145–o1462186 measured reflections 1099 independent reflections 812 reflections with I > 2σ(I) Rint = 0.080
θmax = 25.1°, θmin = 3.5°
h = −22→17 k = −4→3 l = −13→10
Refinement Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.063
wR(F2) = 0.161
S = 1.09 1099 reflections 127 parameters 1 restraint
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained w = 1/[σ2(F
o2) + (0.078P)2]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.002
Δρmax = 0.30 e Å−3
Δρmin = −0.27 e Å−3
Absolute structure: Flack (1983) Absolute structure parameter: −3 (8)
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
C1 0.6207 (3) 0.1033 (11) 0.5044 (4) 0.0310 (11)
H1 0.6586 0.0434 0.5535 0.037*
C2 0.5535 (3) 0.0311 (12) 0.5378 (5) 0.0375 (13)
H2 0.5440 −0.0760 0.6102 0.045*
C3 0.4995 (3) 0.1194 (12) 0.4625 (5) 0.0387 (13)
H3 0.4531 0.0682 0.4860 0.046*
N4 0.5083 (2) 0.2693 (10) 0.3606 (4) 0.0373 (11) C5 0.5853 (3) 0.5139 (12) 0.2191 (4) 0.0350 (13)
H5 0.5459 0.5658 0.1724 0.042*
C6 0.6492 (3) 0.5988 (12) 0.1814 (4) 0.0329 (12)
H6 0.6543 0.7110 0.1086 0.039*
N7 0.7727 (2) 0.6174 (10) 0.2027 (4) 0.0390 (12) C8 0.8289 (3) 0.5474 (13) 0.2656 (5) 0.0390 (14)
H8 0.8732 0.6078 0.2346 0.047*
C9 0.8271 (3) 0.3908 (11) 0.3745 (5) 0.0382 (13)
H9 0.8692 0.3517 0.4162 0.046*
C10 0.7644 (2) 0.2944 (11) 0.4206 (4) 0.0312 (11)
H10 0.7622 0.1868 0.4945 0.037*
C12 0.6336 (2) 0.2649 (12) 0.3981 (4) 0.0268 (11) C13 0.5751 (2) 0.3463 (11) 0.3286 (4) 0.0307 (12) C14 0.7098 (3) 0.5227 (12) 0.2492 (4) 0.0316 (12)
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
C1 0.035 (3) 0.030 (3) 0.028 (3) 0.003 (2) −0.005 (2) −0.002 (2) C2 0.038 (3) 0.035 (3) 0.040 (3) 0.005 (2) 0.006 (3) −0.004 (2) C3 0.033 (3) 0.035 (3) 0.048 (3) −0.006 (2) 0.007 (3) −0.006 (2) N4 0.032 (3) 0.038 (2) 0.041 (2) 0.0010 (18) 0.000 (2) −0.006 (2) C5 0.040 (3) 0.035 (3) 0.030 (3) 0.004 (2) −0.011 (3) −0.005 (2) C6 0.046 (3) 0.029 (3) 0.024 (3) 0.003 (2) −0.003 (2) 0.000 (2) N7 0.044 (3) 0.038 (2) 0.035 (3) −0.0064 (18) 0.009 (2) −0.0063 (19) C8 0.030 (3) 0.043 (3) 0.044 (3) −0.003 (2) 0.006 (3) −0.007 (2) C9 0.035 (3) 0.040 (3) 0.039 (3) 0.002 (2) −0.009 (3) −0.006 (3) C10 0.030 (3) 0.033 (2) 0.031 (3) 0.002 (2) −0.002 (2) −0.001 (2) C11 0.033 (3) 0.024 (2) 0.032 (3) 0.0033 (19) 0.002 (2) −0.005 (2) C12 0.032 (3) 0.021 (2) 0.027 (2) 0.0007 (18) −0.004 (2) −0.0080 (18) C13 0.031 (3) 0.028 (3) 0.034 (3) 0.0007 (19) −0.002 (2) −0.010 (2) C14 0.035 (3) 0.028 (3) 0.032 (3) 0.001 (2) 0.003 (2) −0.006 (2)
Geometric parameters (Å, º)
C1—C2 1.371 (7) C6—H6 0.9500
C1—C12 1.398 (7) N7—C8 1.327 (6)
C1—H1 0.9500 N7—C14 1.367 (6)
C2—C3 1.394 (7) C8—C9 1.396 (7)
C2—H2 0.9500 C8—H8 0.9500
C3—N4 1.322 (7) C9—C10 1.364 (7)
C3—H3 0.9500 C9—H9 0.9500
N4—C13 1.364 (6) C10—C11 1.410 (7)
C5—C6 1.338 (7) C10—H10 0.9500
C5—C13 1.434 (7) C11—C14 1.403 (6)
C5—H5 0.9500 C11—C12 1.455 (6)
C6—C14 1.431 (7) C12—C13 1.414 (6)
C2—C1—C12 120.3 (5) C9—C8—H8 117.9
C2—C1—H1 119.9 C10—C9—C8 119.4 (5)
C12—C1—H1 119.9 C10—C9—H9 120.3
C1—C2—C3 118.0 (6) C8—C9—H9 120.3
C1—C2—H2 121.0 C9—C10—C11 118.9 (5)
C3—C2—H2 121.0 C9—C10—H10 120.6
N4—C3—C2 124.6 (5) C11—C10—H10 120.6
N4—C3—H3 117.7 C14—C11—C10 117.7 (4)
C2—C3—H3 117.7 C14—C11—C12 119.2 (4)
C3—N4—C13 117.1 (5) C10—C11—C12 123.2 (4)
supporting information
sup-5
Acta Cryst. (2001). E57, o145–o146C6—C5—H5 119.3 C1—C12—C11 124.0 (4)
C13—C5—H5 119.3 C13—C12—C11 118.8 (4)
C5—C6—C14 120.8 (5) N4—C13—C12 122.8 (4)
C5—C6—H6 119.6 N4—C13—C5 117.7 (4)
C14—C6—H6 119.6 C12—C13—C5 119.6 (4)
C8—N7—C14 116.4 (4) N7—C14—C11 123.4 (4)
N7—C8—C9 124.2 (5) N7—C14—C6 116.4 (5)
N7—C8—H8 117.9 C11—C14—C6 120.2 (4)