CATALYSIS
A T h esis P resented B y Jam es M adden
In Partial F u lfillm ent O f The R equirem ents For T he A w ard O f T he D egree O f D octor
O f P h ilo so p h y
Christopher In g old L aboratories,
U n iv ersity C o lle g e L ond on ,
2 0 G ordon Street,
W C IH O A J
All rights reserved
INFORMATION TO ALL USERS
The quality of this reproduction is dependent upon the quality of the copy submitted.
In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uest.
ProQuest U642958
Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author.
All rights reserved.
This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC.
ProQuest LLC
789 East Eisenhower Parkway P.O. Box 1346
The thesis describes an investigation into the use o f functionalised polym ers and surfactant m olecules to influence the rate, regiochem istry and stereochem istry o f som e carbon-carbon bond form ing reactions in aqueous and organic m edia.
T o this end, a series or polym er-supported and non-polym er-supported surfactants (e.g. A and B ) were syn thesised. For comparative purposes these differ from each other in tw o w ays: i) the type/length o f the alkyl chain and ii) the type o f head group used, i.e. large/sm all, hom ochiral/non-chiral, anionic/cationic.
= polmer support
Com pounds related to these have been reported in the literature and used to induce high enantioselectivity and rate enhancement in the addition o f diethyl zinc to aldehydes.'--^ The com pounds described in the thesis - both polym supported and non-polym er-supported - contain a neutral/charged moiety and can be used in aqueous or organic media. N on-polym er-supported surfactants generate m icelles in water and reverse m icelles in organic media.
The critical m icelle concentration (CM C) o f each non-polym er-supported surfactant in H2O and hexane has been determined and the surfactants are then used at the CMC in
reaction system s.
Som e D iels-A lder cycloaddition reactions are described using aqueous su spension s o f the polym er-supported surfactants and aqueous solutions o f the non-polym er-supported surfactants. Different yields and product selectivities have been observed compared to the use o f water, or conventional organic solvents.
A c k n o w l e d g e m e n t s ... 7
A b b r e v i a t i o n s ... 8
1. I n t r o d u c t io n ... 9
1.1 Polym er Supported Chem istry... 9
1.1.1 The Polym er B a c k b o n e ...9
1.1.2 The L in k er... 11
1.1.3 Functional Group L oading... 14
1.1.4 B ead S i z e ...14
1.1.5 M olecules Attached to R e s in s ...15
1.1.6 R esin-B ound Catalysts and R eagents... 18
1.1.7 Spectrometric Techniques for Characterisation o f the S olid Support... 19
1.2 Aqueous M edia and The D iels-A lder Reaction... 21
1.3 S u rfactan ts... 24
1.3.1 Determination o f C M C ... 25
1.3.2 The Structure o f M ic e lle s ...27
1.3.3 Properties o f Surfactants... 28
1.4 Surfactants in Non-Polar M e d ia ... 33
1.4.1 Determination o f Reverse Critical M icelle C oncentration...33
1.5 D ialkylzinc Additions to Aldehydes 1.6 M ichael R ea ctio n s...37
1.7 A im s o f the Project...40
2 . R e s u lts & D is c u s s io n ... 41
2.1 The Synthesis o f C 8-12 M ethylene Spacer Surfactants and R e sin s... 41
2.1.1 (O -B enzyloxyalkylam ines... 42
2.1.2 (o-Polym er-Bound Alkylam ine Surfactants... 44
2.2 Determination o f Critical M icelle Concentrations in W ater... 49
2.2.1 The D y e Solubilisation M e th o d ... 49
2.2.2 The Conductivity M eth od ... 51
2.3.3 R esin Absorption o f M ethyl O range... 53
2.3 Determination o f Reverse Critical M icelle Concentrations in Organic S o lv e n ts... 54
2.4 Initial Investigations into the Effects o f Polym er-Bound & N on-Polym er-Bound Surfactants... 56
2.4.1 The D iels-A lder C y c lisa tio n ... 56
2.4.1.3 The Reaction B etw een N V K and C pH ... 64
2.4 .1 .4 The Reaction B etw een A lkyl (2-benzoyI)-acrylate and C p H ... 66
2.5 The Synthesis o f Surfactants Containing a C 16 M ethylene S p a c e r 70 2 .6 The Synthesis o f Polym er-Bound and N on-Polym er-B ound Surfactants Containing O xygen S p a c e r s...76
2.6.1 PEG S urfactants... 76
2 .6 .2 PEG R esin s... 78
2.6.3 The Synthesis o f Achiral Surfactants Containing One O xygen Linker... 82
2.6.4 The Synthesis o f Chiral Surfactants Containing One O xygen Linker... 84
2.6.5 Preparation o f Polym er-Bound Tentagel Surfactants... 87
2.7 Further Investigations into the E ffects o f Polym er-Bound & N on-Polym er-Bound Surfactants... 91
2.7.1 The D iels-A lder C y c lisa tio n ...91
2.7.1.1 The Reaction B etw een M V K and C p H ... 91
2 .7 .1 .2 The Reaction B etw een N V K and C pH ... 95
2.7.1.3 The Reaction Between M enthyl acrylate and C p H ... 96
2.7 .1 .4 The Reaction B etw een 2,6-D M B Q and CpH ... 98
2.7.1.5 The Reaction Betw een Benzoquinones and m yrcene...99
2 .7 .1 .6 The Reaction B etw een B enzoquinones & 2,3-dimethylbuta-1,3 -d ie n e ... 100
2.7.2 The Reduction o f Prochiral K etones...101
2.7.3 Dialkyl Zinc Addition To A ld e h y d e s... 103
2.7.4 M ichael R eactions... 112
2.7.4.1 The Reaction Betw een m ethylcyclohexan-1,3-dione and E V K ...112
2.7.4.2 The Reaction B etw een D E M and Cyclopentenone...114
2.7.4.3 The Reaction Betw een M ethylethylacetoacetate and E V K ...118
2 .7 .4 .4 The Reaction B etw een 2 -A lkylindan-1 -ones and E V K ...119
2.8 S um m ary... 123
3 . E x p e r im e n t a l... 126
3.1 The Preparation o f Polym er-Bound and N on-Polym er-Bound Surfactants...126
3.1.1 The Preparation o f Surfactants containing m ethylene spacer groups... 126
3.1.2 The Preparation o f R esins containing m ethylene spacer g r o u p s... 143
3.1.3 The Preparation o f Surfactants Containing Short Chain Polyethylene G lycol (PEG) L inkers...151
3.1.4 The Preparation o f R esins Containing Short Chain PEG L in k e r s...154
3.1.5 The Preparation o f R esins Containing Long Chain Polyethylene G lycol (PEG) Linkers (Tentagel R esin s)... 158
3.1.6 The Preparation o f Surfactants with a C l 6 Spacer...164
3.1.7 The Preparation o f Surfactants Containing One O xygen Linker...175
3.2 The Preparation o f Precursor M olecules for U se with Surfactants & R e s in s ... 185
3.2.1 The Preparation o f D ie n o p h ile s...185
3.2.2 The Preparation o f A ld eh y d es...191
3.2.3 The Preparation o f 2 -A lk yl-l-In d an on es...191
3.3 C-C Bond Form ing Reactions in The Presence o f Polym er-Bound and N on-Polym er-Bound Surfactants... 194
3.3.1 The D iels-A lder R e a c tio n ...194
3.3.1.1 The Reaction Betw een M V K & C p H ...194
3 .3 .1 .2 The Reaction B etw een M V K & Isoprene...200
3.3.1.3 The Reaction B etw een N V K and C pH ...202
3.3.1.4 The Reaction Betw een M enthyl Acrylate and C p H ... 205
3.3.1.5 The Reaction B etw een A lkyl-2-benzoyl-acrylates and C p H ... 207
3 .3 .1 .6 The Reaction B etw een 2,6-D M B Q and C pH ...209
3.3.1.7 The Reaction Betw een 2,6-D M B Q and M y rcen e... 211
3.3.1.8 The Reaction Betw een Q uinones and 2,3-Dim ethylbuta-1,3-diene... 212
3 .3.4 M ichael Additions...231 3.3.4.1 The Addition o f 2-M
ethylcyclohexane-1,3-dione to E V K ...231
3.3.4.2 The Addition o f 2-M ethylethylacetoacetate to
Ethyl Vinyl K etone... 237 3.3.4.3 The Addition o f Diethyl Malonate to
Cyclopentenone... 239 3.3.4.4 The Addition o f 2-M ethyl-1-indanone to Ethyl
Vinyl Ketone (E V K )...245 3.3.4.5 The Addition o f 2-n-H exyl-1-indanone to Ethyl
Vinyl Ketone (E V K )...248
M y thanks go firstly to H elen for everything.
My parents have also been wonderful throughout, and I don’t mean just the last three years. I hope this book full o f incomprehensible chem ical jargon sat on their book shelf is not a disappointing culmination o f their spiritual and financial support.
Thank you also to Dr. N eil Harris for guidance at U C L and particularly at Dagenham,
thanks also to Rhone-Poulenc Rorer for the expensive chemicals and for periodically topping up my bank account.
Thanks to Darren, Ruk, Raf, Y ing Yang, N eha and everyone in labs 4 6 9 and 435 w ho have helped keep m e sane. The technical staff o f the department are gratefully acknowledged and last, but by no means least, thank you Janene, for all your encouragement.
A b s - absorption
A c - acetyl
A T R - attenuated total reflectance
A r - aryl
B n - benzyl B u - (C H2)3C H3
- C(CH3)3
cat - catalyst or catalytic
C bz - carb ob en zyloxy C l - ch em ica l ion isation
C M C - critical m ic e lle concentration
C pH - cyclopentadiene
C T A B - cetyltrim eth ylam m on iu m brom ide
C T A C l - cetyltrim eth ylam m on iu m chloride D A B C O - l,4 -d ia za b icy clo 2 .2 .2 ]o cta n e
D C C /D C C I - d icycloh exylcarb od iim id e D C M - dichlorom ethane
dCpH - dicyclopentadiene d.e. - diastereom eric e x cess
D E A D - diethylazodicarboxylate D E M - diethyl m alonate
D IP E A - A^,A/-diisopropyl-jV-ethylamine D M B Q - d im eth lb en zo-1,4-quinone
D M F - dim ethylform am ide
D M S O - dim ethyl su lfo x id e D N P H - dinitrophenylhydrazine
E D C - ethyl diazocarboxylate e.e. - enantiom eric e x c e ss E l - electron ion isation
E S I - electrospray ionisation
+ v e /-v e - p o sitiv e/n eg a tiv e E t - C H2C H3
E V K - eth yl v in y l k eton e
F A B - fast atom bom bardm ent G C - gas chrom atography
H PL C - high pressure liquid chrom atography
H O B T - 1-hydroxybenzotriazole
IR - infra red
L A H - lithium alum inium hydride E D A - lithium diisop rop ylam id e
L G - lea v in g group M e - C H3
M s/m e sy l - m ethane su lfo n y l M S - m ass spectrom etry
M V K - m ethyl v in y l k eton e N M R - nuclear m agnetic resonance
N V K - nonyl vin yl k eton e
PC C - pyridinium ch lorochrom ate P E G - p o ly eth y len e g ly c o l
Ph - phenyl Pr - (C H2)2C H3
T r - C H (C H3 ) 2
p y - pyridine
p y .S0 3 - pyridine sulfur trioxide co m p lex
R C M C - reverse critical m ic e lle concentration
R M M - relative m olecu lar m ass
S A M P ( 5 ) l A m in o 2 -(m ethoxym eth y l)pyrrol idi ne S D S - sodium d od ecyl sulfate
S E M - scanning electron m icroscop y SP O C - solid p h ase organic chem istry SP PS - solid p hase p eptide syn th esis
S P S - solid p hase syn th esis
T B A B - tetrabutylam m onium brom ide
T B D M S - tertiarybutyldim ethylsilyl T E A - triethylam ine
T E M P O - 2 ,2 ,6 ,6 -tetra m eth y lp ip erid in y lo x y
tert - tertiary
T EA - trifluoroacetic acid TH E - tetrahydrofuran
T IP S - triio sp ro p y lsily l
tic - thin layer chrom atography T M S - trim eth y lsily l
T O C S Y - total correlation sp ectroscop y
T s/to sy l - p a ra tolu en esu lfon yl U V - ultra v io le t
1.1 Polymer Supported Chemistry
With the arrival o f S olid Phase Synthesis (SP S) over 30 years ago"^ came a new era across the spectrum o f first peptide, then synthetic organic and more recently inorganic chem istry.5 Early work on solid phase peptide synthesis (SPPS) show ed, to an initially
doubting scientific comm unity, the advantages o f polym er supported strategies i.e. high chemical yields, facile reaction work-up, ease o f product isolation/by-product removal and the recyclability o f the robust polymers. A s investigations were carried out into the uses o f solid supports, applications were found in not only peptide synthesis but oligonucleotide synthesis, and the synthesis o f small and medium size organic m olecules.
Developm ents in the synthesis and applications o f solid supports have meant that it is now possib le to 'fine tune' a resin or polymer's properties to a particular m ethodology. These properties derive from:
The backbone o f the resin and its flexibility.
The type o f group that links the substrate to the backbone known as the linker. This can dictate not only the chemical properties o f the resin but the physical properties as well. The density o f functional sites on the resin (usually expressed in m m ol/g). This is termed as their loading.
The size o f the polym er beads, often referred to as the m esh.
1.1.1 The P o lym er B ackbone
Merrifield's resin was a mixture o f 1,4-divinylbenzene and vinylbenzene in a ratio that
established upon polymerisation beads with a semi-rigid polymer backbone. Figure 1.1.1.1 gives a schematic representation o f the polymer. Merrifield functionalised his vinyl benzene groups post-polym erisation, how ever contemporary methods introduce the functional groups from the outset, as discussed below .
to 5 m l/g. The amount o f sw ellin g observed depends on the degree o f solvation, itself a function o f the solvent added. If too much divinylbenzene is present, the polym er will be highly cross-lin k ed and therefore clo sely packed^. T hese covalent constraints result in poor accessibility for the solvent m olecules and affects the ability o f the polym er to sw ell. This results in steric interactions and will hinder the approach o f substrate m olecules w hile at the
divinyl benzene cross linkers
polym er backbone
R = H an C H X l
CHjCI
figure 1.1.1.1
Polym er bead
ca. 4 x 1 0 ' m
sam e time increase the chance o f cross-coupling reactions (see 1.1.3 b elo w ). If there is insufficient divinylbenzene present, the polym er will lack structure and strength resulting in poor handling properties and the tendency to fragment. The polym er may even d issolve com pletely in solvents such as form ami de.
supported chem istry, including the use o f paper, polythene/polystyrene m ixes, cotton w ool and glass beads, it is still resins based on polystyrene that are m ost com m only u sed.
1.1.2 The Linker
Inclusion o f a chlorom ethyl group in the resin provides a m eans o f covalently bonding chem ical substrates on to the polymer. M errifield achieved this by activating the phenyl ring with a nitro or bromine group follow ed by reaction with the triethyl amm onium salt o f the first amino acid in the peptide chain‘d as shown in figure 1.1.2.1
N O ) O
, ^ ^ N . fT ' C b z
O Cbz
-EtjNH^ pi
figure 1.1.2.1
Solid phase peptide syn th esis (S P P S ) can thus be achieved by an iterative procedure involvin g the deprotection o f the amine and reaction with the next A^-protected amino acid with a suitable cou p lin g agent (such as D C C P ).
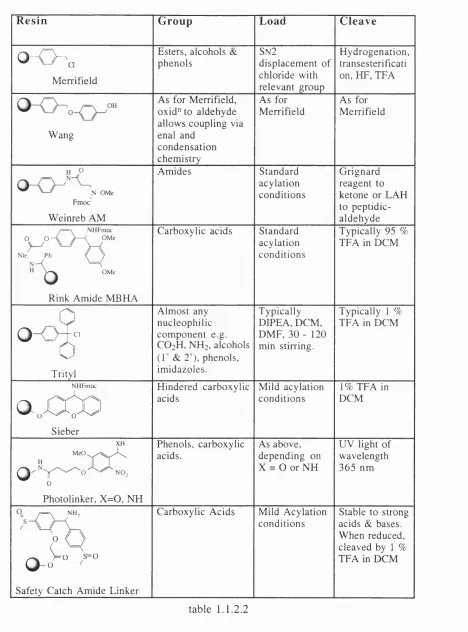
The Merrifield resin lends itself to substrate attachment via the W illiam son ether sy n th e sis’®, ester formation as outlined a b o v e” and attachment o f phenols via a nucleophilic d isp lacem en t’^, how ever the cleavage o f the substrate after chemical
m odification often requires harsh conditions such as hydrogenation or
hydrofluorination (to cleave benzyl ethers), or the use o f sodium hydroxide or m ethoxide for cleavage o f the ester. Furthermore, the type o f functional group available for attachment to the solid support is frequently a m oiety other than a hydroxy, a carboxylic acid or a phenol. Indeed, a large number o f exam ples to be found in the literature pertain to acid and/or base sensitive m olecules that require connection through functionalities such as am ino, im ino, thiol, guanidino and carboxamido®. For these reasons it has been necessary to develop linkers amenable to facile connection and disconnection procedures, under m ild conditions.
R e s in G ro u p L o a d C le a v e
0 “ 0 - Q
M e r r ifie ld
E ste r s, a lc o h o ls & p h e n o ls
SN 2
d is p la c e m e n t o f c h lo r id e w ith r e le v a n t g r o u p
H y d r o g e n a t io n , tr a n s e ste r ific a ti o n , H P , T E A
W a n g
A s fo r M e r r ifie ld , o x id " to a ld e h y d e a llo w s c o u p lin g v ia en a l and
c o n d e n s a t io n c h e m is t r y
A s fo r M e r r if ie ld
A s fo r M e r r ifie ld
O M .
F m o c
W e in r e b A M
A m id e s S ta n d a r d
a c y la t io n c o n d it io n s
G r ig n a r d r e a g e n t to k e to n e o r L A H to p e p tid ic -a ld e h y d e
/ = \ NHFmoc
0 O —{ )---- < OMe
Me Ph ( 7
H OMe
R in k A m id e M B H A
C a r b o x y lic a c id s S ta n d a r d a c y la t io n c o n d it io n s
T y p ic a lly 9 5 % T E A in D C M
0
o - o T
-Ô
T r ity l
A lm o s t a n y n u c le o p h i li c c o m p o n e n t e .g . C O2H , N H ] , a lc o h o ls
(1° & 2 °), p h e n o ls , im id a z o le s .
T y p ic a l ly D IP E A , D C M , D M F , 3 0 - 1 2 0 m in stir r in g .
T y p ic a lly 1 %
T E A in D C M
NHFmoc
a.cCO
S ie b e r
H in d e r e d c a r b o x y lic a c id s
M ild a c y la t io n c o n d it io n s
1% T E A in D C M
XH
H
^
I
P h o to lin k e r , X = 0 , N H
P h e n o ls , c a r b o x y lic a c id s .
A s a b o v e , d e p e n d in g o n X = 0 or N H
U V lig h t o f w a v e le n g th 3 6 5 n m
/ = \ N H ,
o
^2)
0 - o ^ °
S a fe t y C a tc h A m id e L in k e r
C a r b o x y lic A c id s M ild A c y la t io n c o n d it io n s
S ta b le to stro n g a c id s & bases.
W h e n r e d u c e d , c le a v e d b y 1 % T E A in D C M
table 1.1 .2.2
ca. 30 0 - 4 0 0 Â). This has tw o main effects; the first is to distance the hydrophobic hydrocarbon backbone from the attached substrate to be manipulated, the second is to change the physical properties o f the resin so that it is less hydrophobic and more compatible with the use o f more polar solvents. Early work into SPPS was hampered by low chemical yields and slow reaction kinetics resulting from peptide folding, an attempt by the supported peptide to minimise hydrophobic interactions. To overcome this problem, dimethylacrylamide has been supported on polystyrene and silica gel and
PEGA supports (bis 2-acrylamidoprop-1 -ylpolyethyleneglycol, 2-acrylam idoprop-l-yl2-am inoprop-l-yl] polyethylene glycol dimethyl acrylamide co-polym er, which is com posed o f the olefins shown in figure 1 .1 .2 .3 ) have been d ev elo p ed ’^, although the handling properties o f the dimethylacrylamide supports are not as good as w as initially envisaged.
O M e
M e O
M e
N T he terminal am in e is
H 7 _ , availab le for linker attachm ent
M e
figure 1.1.2.3
In SPS, it is som etim es necessary to carry out a synthetic step in a solvent that does not swell the resin {e.g. M eO H , H2O). In such reactions, diffusion o f the reagent into the
1.1.3 Functional G roup Loading
Merrifield determined the quantity o f chloride groups present in his 2 % cross-linked resin after functionalisation to be 1.89 mmolg-^ which corresponds to the
chlorom éthylation o f 22 % o f the aromatic rings o f the polym er. A s mentioned above, functionalisation o f the polymer leads to a higher distribution o f the chloromethyl groups am ong the ortho and meta sites and this may result in poor accessibility o f the functional sites to reactants. Present methods o f co-polym erisation using divinylbenzene, vinylbenzene and 4-chlorom ethylvinylbenzene result not only in lower
backbone cross-linking, but exclusive para chloromethyl substituents, and a good degree o f hom ogeneity o f the chloromethyl sites throughout the polym er. Overall, this
leads to low er steric crowding when the substrates are subsequently loaded as well as a reduction in the likelihood o f site-site interreaction or cross-coupling. For example, when loading an imidazole on to a chlorotrityl derived resin, one nitrogen is im m obilised on to the resin, leaving the other nitrogen free for synthetic manipulation. H ow ever, if another trityl chloride group is adjacent, then the likelihood o f cross coupling is increased.
For the m ost part, resins are synthesised from divinylbenzene (1 or 2 %) vinylbenzene and the relevant p-functionalised vinylbenzene in such a ratio as to m axim ise the quantity o f substrate per gram whilst m inim ising site-site interactions. Consequently, loadings are typically between 0.25 - 2.0 mmolg-^ depending on the molecular mass o f the linker. In the case o f PEG linkers, the loading w ill be relatively low due to the high molecular m ass o f the PEG chain, whereas in the case o f Merrifield resins, loadings can be higher.
1 .1 .4 B e a d Size
The size o f the polym er bead dictates the diffusion coefficient and handling properties
1) Calculate the surface area on one gram o f beads from the average size o f the beads,
assum ing bead density is ca. 1 g/ml.
2) The cross-sectional area o f a saturated C H2 chain, from studies o f m onolayers on
water, is 18 Â^. Therefore, functional groups on the surface o f the beads probably occupy at least 50 Â^.
3) Establish how may functional groups there are per mmol and how many mmol/g beads.
4) H ence estimate the percentage o f groups on the surface. There are many
approximations, but the key m essage is the same; only a very small percentage are on the exterior.
1.1 .5 M olecu les A tta ch ed to Resins
The range o f polym er supported chemistry is continually grow ing, as exem plified by the number o f publications each y ea r^ i 6.17,18,19,20
Although reference has been made above to the use o f solid supports in the fields o f oligonucleotide, peptide and inorganic chem istry, it is the domain o f synthetic organic chemistry that has seen the greatest expansion o f polymer-compatible m ethodology over the past 20 years. Peptide chemistry can now be largely automated thanks to a range o f chemistries that have been perfected that are capable o f selectively deprotecting and coupling amino acids on the solid support. The application o f inorganic chemistry to the use o f polymeric supports is limited by the chemical modification and poisoning o f the polym er backbone by the transition metal species involved.
T o adapt a single phase hom ogenous organic manipulation to a two-phase heterogenous organic reaction at the solvent phase/solid phase interface has proven to be complicated, although som e solution phase chemistries have been found to be directly transferable to reaction on the solid support. H ow ever, once an organic reaction has been adapted for
the solid phase, it has several advantages over its solution phase counterpart, these include:
• A s mentioned above, ex cess reagent can be em ployed to drive the reaction equilibrium such that high chemical yields may be observed.
In cases where there is more than one group o f a m olecule that can react (e .g . d io ls), attachment o f the m olecule at the appropriate terminus effectively protects that group, resulting in higher ch em oselectivity towards the desired product.
Pseudo-dilution phenom ena can be exploited, e.g. favouring intramolecular cyclisation over intermolecular cyclisation, or the Dieckm ann condensation (see figure 1.1.6.1 b elo w ). When using labelled pim elic acid tw o products are generated from the base induced cyclisation. H ow ever, product B can be rem oved by w ash ing and isolated w hereas product A can subsequently be cleaved and isolated^!.
figure 1.1.6.1
• Substances hazardous to health can be handled more safely than with the non bound analogues {e.g. p-bound tosyl azide).-°
• And lastly, it is amenable to automation.
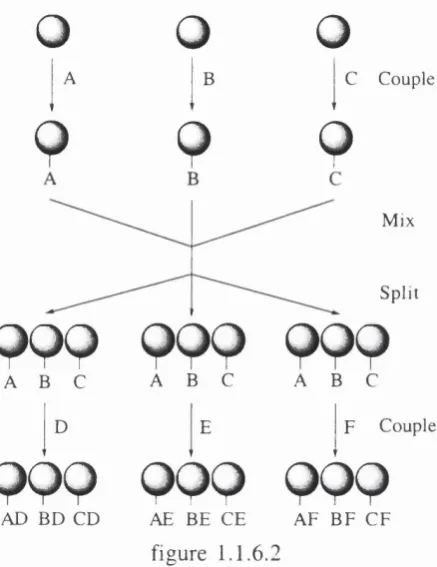
Polym er supported chemistry has found one o f its greatest applications in the pharmaceutical industry where the scientific developm ent o f biological techniques for screening new com pounds has forced lead discovery departments to look for means o f accelerating their production o f drug candidates. This has resulted in the developm ent o f tw o new form s o f solid phase organic chemistry (SPO C); Combinatorial Chemistry and Parallel synthesis.
o
0
1
^0
1
^1 ^
9
9
9
C C ouple
M ix
S p lit
999 999 999
A B C A B C A B Cj
DI
Ej
F C ouplep p p p p p 9 P P
A D B D C D AE BE CE A F B F C F
figure 1 .1.6.2
Early screening techniques relied on the cleavage and analysis o f com pound libraries as m ixtures. The limitations o f this method are twofold: firstly, the com pounds are a mixture and therefore the characterisation o f individual m olecules is som etim es not p ossib le without further purification steps. S econ d ly, if a biologically active com pound is present in a mixture with an anti-active com pound, or antagonist, then a potential biological ‘hit’ may be m issed. Current m ethods allow the beads to be separated and random p osition s in the library are sequentially defined^^ on or o ff the bead using techniques described in section 1.1.7 b elow .
1.1.6 R esin-B o u n d C a ta ly s ts a n d Reagents.
W hilst many review articles have covered polym er bound substrates with a particular em phasis on the combinatorial chem istries and parallel syn th eses on these im m obilised m olecules, polym er-bound catalysts and reagents are also extrem ely important. The advantages to be found in this area reflect those m entioned above:
• The reagent/catalyst can be filtered o ff and, if necessary, regenerated. • The pure product can be isolated from the solvent phase.
• Reagents hazardous to health can be more easily and safely handled on the polym eric support.
An exam ple o f a polym er bound catalyst is N oyori's Ruthenium Binap catalyst which has been used to selectiv ely reduce y-ketoesters (figure 1.1.7.1) in > 98 % enantiomeric e x c ess.
Pli Cl
figure 1.1.7.1
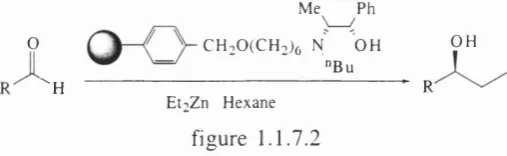
Frechet- and Soai^-^ have reported the use o f polym er bound catalysts w hich have been em ployed in the asym m etric addition o f diethyl zinc to aldehydes (figure 1.1.7.2).
Me^ ^Ph
O CH. QCCHoic N OH OH
"Bu
"H " R
E t iZ n Hex an e
figure 1.1.7.2
One o f the main disadvantages o f SPS is the m onitoring o f extent o f reaction as well as characterising interm ediates in m ulti-step solid phase syntheses. Initial methods were o f the cleave-and-characterise type which is costly both in terms o f tim e and material.
N uclear m agnetic resonance spectroscopy has been em ployed with resin slurries in 50 % CDCl^/DMSOd^) taking advantage o f the parity in T] and T j relaxation times between the pendant substrate backbone and the polym er backbone. H o w ev er, acquisition times are in the order o f hours which rules out its use in the rapid m onitoring o f reactions. A dditionally, the spectra tend to suffer from line broadening as the individual environm ents vary from substrate to substrate. The use o f ’^C-enriched building b locks greatly im proves the ability to obtain high resolution spectra, though this method is costly . Kurth and coworkers^^ have fo llo w ed the reaction show n in figure 1.1.8.1 by infra red. H ow ever this exam ple is the exception rather than the rule o w in g to the distinct infra red stretches ob served for each o f the intermediates.
C H X l --- CHO
-/ = \ OT
—
-N O ,
/ = \ OTMS
N O ,
figure 1.1.8.1
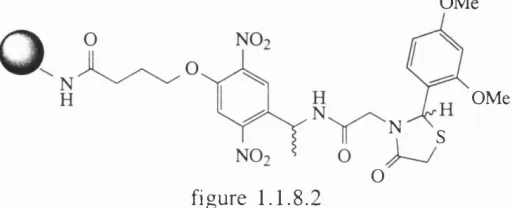
’H N uclear m agnetic resonance spectroscopy is now a valid tool for on-bead analysis. U sin g 2-D N M R and T O C SY , Anderson and coworkers-^ were able to totally assign the structure o f supported F m o c-L ysin e-B oc. U sin g a 6 0 0 M H z Varian spectrometer, A ffym ax workers-^ com pletely assigned the com plex m olecule sh ow n b elow in figure
1. 1 . 8 . 2 by use o f a m agic angle n a n o p r o b e ,a probe originally d esigned for use on sam ples that may otherw ise have unacceptably broad line shapes due to susceptibility discontin u ities, such as w hen using very sm all sam ples o f liquid (< 4 0 p i).
OMe
OMe N
S
figure 1 .1.8.2
Matrix assisted laser desorption ionisation tim e-of-flight (MALDI-TOF) mass
s p e c t r o m e t r y 2 8 . 2 9 has also been used which enables beads to be cleaved within the
spectrometer without previous cleavage reactions. This technique can be used to characterise com pound mixtures produced from combinatorial techniques, although more com plex combinatorial mixtures require methods such as H PLC-M S or affinity capillary electrophoresis.^^
The availability o f 'desktop' m ass spectrometers, which can analyse sam ples in a matter o f minutes, now means that reactions can effectively be monitored either by cleavage o f
1.2 Aqueous Media and The Diels-Alder Reaction
The use o f water as a reaction solvent for many organic reactions had not been fully realised until 1980 when B reslow and coworkers^^ em phasised the remarkable rate
acceleration o f the Diels-Alder reaction o f methyl vinyl ketone (but-3-en-2-one) with cyclopentadiene (see figures 1.2.1 and 1.2.2 (below )) in water compared to organic solvents. Since that initial observation, the bulk o f research into aqueous media has focused on the D iels-A lder reaction, from ab initio molecular m odelling calculations^^ to its applications in natural product s y n t h e s i s . A s a result, this section will deal primarily with aqueous media as they apply to the D iels-Alder reaction with less em phasis on other aqueous carbon-carbon bond forming reactions.
o
o
-Endo product E xo product
figure 1.2.1
Breslow rationalised^"^ that the sparingly soluble cyclopentadiene (10 mM in H2O) was
forced to aggregate in solution with the methyl vinyl ketone to reduce the hydrophobic interactions with water, and given the negative activation volume o f the transition state (ca. - 30 m lm ol'O, he concluded that the hydrophobic effect w as responsible for the rate acceleration. This theory is lent credibility by the effects o f additives such as lithium chloride (figure 1.2.2) which increases the degree o f hydrogen bonding in solution. This increases the net polarity o f the solution, making it more lipophobic, which results in an increase in rate o f reaction for the D iels-Alder addition o f methyl vinyl ketone and cyclopentadiene.
S o lv e n t R elative R ate E lectrostatic C onstant, E Endo
H2O 100 78 2 1 .4
M eO H 1.71 3 2 .7 9 .5 5
Isooctane 0 .1 3 5 2 .3 3 .5 3
4 .8 6 M L iC l (aq) 245 -
-figure 1.2.2
increasing the overall lipophilicity o f the solution. Other additives such as cyclodextrins and surfactants have different effects on rates and selectivities that will be discussed below .
The hydrophobic effect alone is not sufficient to explain the observed rate
enhancement.^'^ The high cohesive energy density o f water (cohesive pressure) compared to other solvents must be taken into account. This important effect is a result o f H -bonding which reduces the surface contact between solvent and solvated m olecules. This effectively reduces the cavity size o f the solvate which can enforce molecular aggregation. The overall process described above has been termed the enforced hydrophobic interaction 38 and is also amplified by salting out agents such as lithium chloride and sugars. In addition to the enforced hydrophobic interaction, the solvent may have a stabilising effect on the transition state o f the reaction, resulting in a rate enhancement. Berson^^ illustrated in 1961 the correlation between rate, selectivity
and solvent polarity. Therefore, despite the fact that the D iels-Alder reaction is considered to go through a non-polar [4+2] pericyclic transition state, it is not inconceivable that water stabilises a partially polar transition state,^^-^’ especially given that it can be accelerated in aprotic organic s o l v e n t s , ' ^ ^ aqueous solvents and by strong
Lew is acids."^3
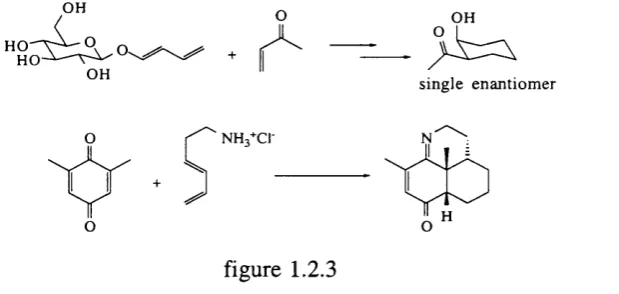
Although biphasic reaction mixtures o f water and reactants have show n rate accelerations^'^ and enhanced selectivities, the grafting o f a hydrophilic m oiety such as a sugar to the diene or dienophile (figure 1.2.3) also accelerates the reaction. This is due partly to the solubilisation o f the reactant and partly to the augmentation o f the hydrophobic effect"^.
O H
ho^ V ^ 9
H O
O H
O H
sin g le enantiom er
NHg+CI-figure 1.2.3
presence o f additives also effects the endo/exo selectivity (section 1.3.3).
Although the Diels-Alder reaction has been the main subject o f discussion in this section, other pericyclic reactions have also been shown to proceed with increased rates o f reaction and enhanced selectivities in water compared to organic solvents. For example, the hetero D iels-A lder cyclisation and the Claisen rearrangement both, like the Diels-Alder cyclisation, involve apolar reactants and both have negative reaction
activation volum es, thus they can benefit from the same hydrophobic effects as with the Diels-Alder reaction. Gajewski'^ and Grieco'^'^ also observed that the attachment o f a hydrophilic moiety to the relevant precursor results in rate enhancements when the reaction is performed in aqueous media figure 1.2.4.
O
R02C(H2C)6
6 0 °C
R02C(H2C)6; 0
R solvent relative rate
Na
Na
M e
H2O
M eOH
Benzene
106
4 .6
1
figure 1.2.4
Other reactions compatible with water include the M ichael addition^^. Aldolisation"^^, the Bay lis Hillman reaction^®, Mannich reaction^’ and a broad range o f organometallic
1.3 Surfactants
A s highlighted previously, the use o f aqueous media can effect the rate and selectivity o f reactions. H ow ever, these benefits are frequently unobtainable ow ing to the poor solubility o f substrates in water. Surfactants^^, have special properties in aqueous solution, one o f which is the ability to solubilise fatty m olecules.
Surfactants are water soluble m olecules that are amphiphilic, i.e. they contain three distinct moieties; the apolar alkyl chain (although the use o f a polyethylene glycol has been reported^"^), the polar head group (which can be cationic, anionic or apolar) and the counterion (although som e surfactants are neutral and have no counterion). They form oriented m onolayers at phase-phase interfaces (hence their name is derived from su r fa c e a c tiv e a g e n ts) and their properties include m icellisation, detergency, foaming
and émulsification^^. With their ability to form multi-lamellar structures they have certain biomimetic properties, and as such have certain biological properties such as catalysis^^ and drug transportation.^^
Their ability to m icellise in aqueous solution is a balance o f hydrophobic and hydrophilic properties. Hydrophobic Van Der Waals interactions results in an enthalpically favourable aggregation term, overcom ing the large negative entropie term as surfactant m olecules are ordered and structured, as expressed in equation 1.
R T In Xw = - AH -H TAS eq. 1
Xw is the solubility o f the hydrophobic m oieties, AH is the change in enthalpy and AS
the change in entropy. H ow ever, Frank and Evans^^ have shown that upon addition o f the alkyl chains, the macromolecular aqueous structure is disturbed and reorganises into assem blies with strong attractive interactions which are mainly responsible for the
hydrophobic bonding. The charge repulsion o f the polar head groups counteracts the aggregation processes, how ever this has been found to be m inim ised by varied degrees o f intrusion o f the polar head group into the micelle core.^^ The combination o f these different parameters dictates the size o f the micelle as w ell as the number o f surfactant m olecules that it consists of, or aggregation number n. For ionic surfactants n is typically 10-100 whereas for non-ionic surfactants n m ay be greater than 1000.
concentration (C M C ). It has been observed^- that surfactant solution s above the CMC have different physico-chem ical properties to those b elow the CM C. These properties include conductivity, osm otic pressure, solubilisation, magnetic resonance and self diffusion. Any o f these properties can be exp loited not only to determine the C M C, but also in practical chem ical applications.
figure 1.3.1, The stages o f m icellisation
1 3 . 1 D etenn ination o f CM C:
A popular method o f CMC determination is dye solubilisation.^^-^ This reflects the difference in stabilisational capability between the bulk polar aqueous phase and the apolar hydrocarbon interior o f the m icelle. A m olecule such as methyl orange (figure 1.3.1.1 below ) has a delocalised n electron system , tw o canonicals o f which are
sh o w n .
N
O N a O N a
N+'
figure 1.3.1.1
In apolar media, the left hand canonical will be preferred as it has only one negative and one positive charge whereas the right hand canonical with tw o positive and two negative charges will be preferred in polar media such as water. Therefore, below the CMC, the doubly charged canonical will prevail and at and above the CM C, the singly charged species will prevail. This change can be monitored by U V /vis spectroscopy and by the eye, for the two species absorb in the orange/yellow region o f the visible spectrum. The disadvantage o f this method is that by their presence, the dye molecules intrinsically affect the aggregation o f the surfactant m olecules and hence the value o f the CM C. Figure 1.3.1.2 show s tw o further examples o f solvatochromie com pounds. Reichardt's dye, A , is negatively solvochrom ic and has been used to determine the polarity o f solvent mixtures, in the same manner as the positive solvochrom ic dye, B.
A , R = B , R = Et
figure 1.3.1.2
Many studies have been undertaken into the size and shape o f m icelles and H artley's model<^5 (figure 1.3.2.1), show n b elow , is generally accepted.
Schematic representation of a micelle
G ou y-C h ap m an layer,
th ic k n e ss < 1 0 0 Â A
P o la r h ead group (A n io n ic , c a tio n ic , n o n -io n ic or a m p h o litic)
C ou n ter ion
Surfactant molecule:
^ ^ ^ /\/V '\zx/\/\y\zx/\z'
H ydrocarbon chain
A Q U E O U S E X T E R IO R
S tem layer,
th ic k e ss fe w Â
M ic ella r core (h yd rocarb on interior),
th ic k n e ss 1 0 -2 8 Â
figure 1.3.2.1
The G ouy-Chapm an layer contains the d iffu se counterions (3 0 -5 0 % o f total counterions) and according to the Hartley m od el, extends approximately to twice the volum e o f the Stem layer and core. The Stem layer con sists o f the ionic head groups and the tightly bound remainder o f the counterions and is just a few  thick. The core is split into tw o regions; the outer core which includes the first four methylene groups and, as a result o f repulsion between head groups, may contain a few water m olecules w hile the inner core is the rest o f the alkyl chain packed in clo sely together in a hydrophobic organic phase held together by Van Der W aals forces. The core is generally accepted as being o f radius less than or equal to the length o f the fully extended alkyl chain. The region including the head groups and the first few m ethylenes is som etim es called the palisade layer or mantle.
can affect the dynam ic equilibrium between the surfactant, the m icelle, the rod, or other structure. The effect depends on the nature o f the additive.
figure 1.3.2.2
The Hartley model indicates that the methylene groups in the alkyl chain are trans, though this is not feasible as the crowding in the core o f the m icelle w ould be too great. More probably, the alkyl chains fold in on them selves to varying extents as proposed by D ill and Flory*^<^. ’^C Chem ical shift experim ents do indeed indicate that the terminal CD-carbons are in various different environm ents.
Studies by Ulnius^^ and cow orkers into the nature o f the interior o f the m icelles have show n that cetyl trimethylammonium bromide (C T A B ) has the same core molecular motion as both the pure surfactant and benzene in their liquid crystalline phases. He therefore con clu ded, given that m icelles and liquid crystals start to form at approximately the same temperature, that the interior o f a m icelle is liquid like in character.
1.3.3 P r o p e r tie s o f Surfactants
electrostatic interactions. Substrates and reagents are brought into clo se proximity in the m icelle due to these, and the individual effect depends on the nature o f the com pound. More polar m olecules will be held in close proximity to the Stem layer, w hile apolar m olecules w ill be held in the hydrocarbon core. M olecules consisting o f a lipophilic
m oiety and a hydrophilic m oiety will be located in the m icelle with the polar moiety near the Stem layer and the apolar moiety in the hydrocarbon core. These proximity and orientational effects are the com er stones o f micellar effects and are further illustrated below .
The medium effect arises from cage, m icroviscosity, and polarity effects and the result is that m icelles can hold reactive species together for a longer period o f time than in the bulk solution. For exam ple, B a m i^ and coworkers observed that cyanine dyes were over tw o orders o f magnitude more stable to continuous irradiation from a
high-pressure mercury lamp in solutions o f sodium dodecyl sulfate (S D S ) and especially dioctadecyldim ethylam monium chloride (D O D A C ) than in an aqueous solution.
o o
R =0(C H2)3CH 3 o
M edium Y ield (%)
H2O 0
S D S (0 .0 5 M ) 5 0
figure 1.3.3.1
Photochemical reactions in particular are catalysed in micellar media. Radicals generated
in aqueous solution in the T^ excited state are normally quickly quenched by intersystem crossing (ISC) to the S ’ state. H ow ever, in micellar m edia, the sequestered radical pair can retain its geminate character for sufficient periods to access the singlet surface,^^’^^ leading to the formation o f closed shell products, as typified in the use o f m icellar m edia in the dimérisation o f the coumarin derivative above (figure 1.3.3.1).7o
o f C o (N H3)$CI^+ in SD S. A lso, Singh and coworkers’^^ have reported the rate
acceleration o f the reaction between benzoquinones, a family o f substrates with limited
solubility, and cyclopentadiene. In one exam ple, (figure 1 .3 .3 .2 )
methylnaphthoquinone failed to react in water but in an aqueous solution o f cetyltrimethylammonium bromide (C T A B ), the reaction proceeded in 8 6 % chemical
yield.
C T A B 3 h, 3 0 °C
figure 1.3.3.2
Al-Lohedan has reported"^^ that the hydrolysis o f long chain glycinate esters is accelerated in cetyltrimethylammonium hydroxide (figure 1.3.3.3) solution compared to solutions o f C TA C l/N aO H and just NaOH. This was m ost likely due to the orientation o f the polar ester moiety in the stem layer which lies in close proximity to the reactive hydroxide counterions:
figure 1.3.3.3
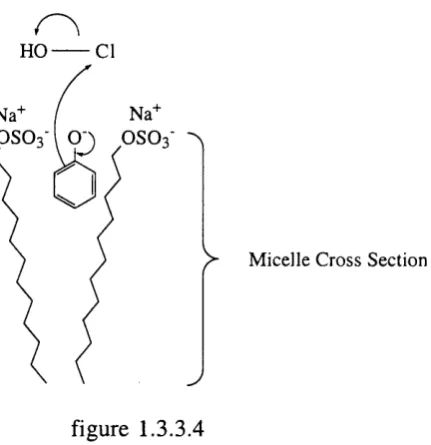
Rate accelerations in micellar media are not always observed, indeed the change in rates o f reaction with surfactants can often be contradictory.^"^ In addition, ow ing to the lipophilic nature o f the micellar interior, rates can be low er (as they w ould be in a lipohilic solvent) than in water. H owever, the rate o f reaction can also be enhanced due to the internal pressure in a micelle being higher than that o f the corresponding hydrocarbon solvent. Furthermore, the micellar medium can im pose stereo- and regioselective constraints on to reacting substrates.
Na+ I Na+
O S O3
I
O S O3 ^)> M ic e lle C ross S ectio n
J
figure 1.3.3.4
Preorientation in the m icelle can be fixed by attaching one o f more o f the reactants to the surfactant m olecule. Jaeger and coworkers'^'^ have attached a diene to a cationic head group and observed regioselectivity in the D iels-A lder reaction o f surfactant dienes with aryl vinyl ketones (figure 1.3.3.5). In examples where both dienophile and diene have ionic head groups, such as in figure 1.3.3.5 below , greater regioselectivity can be obtained compared to when performing the reaction in water.^5
NHC02(CH2)6N^Me3Br' +
Ar
Ar = Q H 5, NHC02(CH2)6N*Me3Br
C«H,
CgH] ? 0
CgHnOzC
S. S—
(CH2)4N^Me3Bp-0 (CH2)6N^Me3Br‘
C02(CH2)6N^Me3Br'
figure 1.3.3.5
Shinitzky and Haimovitz^^ have reported the existence o f chiral surfaces in (5)-am ino acid derived surfactants. Circular dichroism spectra o f m icelles o f the surfactant suggest that they arrange them selves repetitively across the micelle surface resulting in supramolecular chirality o f the aggregate. Previous work within our research group'^'^ has also observed enantioselectivity (see table 1 .3 .3 .6 ) when reacting cyclopentadiene
figure 1 .3 .3. 6
O
OC9H1 9
O H
N+cr ^16^33
O
0 C9H] 9
M edium
H2O
Chiral Surfactant
Y ield (%)
72
75
EndolExo
1.7
2.2
e.e. (%)(/?)
0
1.4 Surfactants in Non-Polar Media
When surfactants are dissolved in apolar m edia, they can form what is termed reverse micelles where the polar head groups form a core, minim ising lipophobic interactions with the apolar solvent and the alkyl chains. The use o f polyethylene glycol (PEG) instead o f alkyl chains has also been reported'^^-'^^ where the PEG points out into the apolar solvent, m axim ising lipophilic interactions. Reverse m icelles are capable o f
catalysing reactions by solubilising polar substrates and concentrating them in the polar head group region. Thus reaction selectivities can be achieved as a result o f preorientational and concentration effects analogous to those explained above.
As with their aqueous counterparts, these reverse micellar solutions exhibit unique changes in their physico-chem ical properties such as light scattering, turbidity, conductivity^^ and solubilisation above a certain concentration. These reverse micelles are capable o f solvating water in the vicinity o f the head groups, creating 'water p ools' in otherwise hydrophobic immiscible organic solvents. This property can be exploited when ascertaining a reverse critical m icelle concentration (RCMC).^^
1.4.1 Determ ination o f R everse C ritical M icelle Concentration
A review o f the literature has indicated that the solubilisation o f either water or dye molecules to be the most comm on methods used in RCMC d e t e r m i n a t i o n s . ^ ^
T y p ic a lly ,w h e n water is added to a sub-RCM C solution o f surfactant, the water will either form an em ulsion or a separate phase. A bove the RCM C, the solution w ill remain clear and dissolve added water (up to a certain point). The disadvantage o f this method is that the quantity o f water present intrinsically affects the RCM C and the solution is rendered incompatible with water sensitive reactions.
2 h
Reverse ^ M icelle X -Section
V
figure 1.4.1.1; Schematic representation o f reverse m icelle
As with their aqueous counterparts, the RCMC and subsequent reverse micellar properties vary as a function o f length o f alkyl chain, nature and size o f the head group and counterion, the solvent and the presence o f additives or solubilisâtes.^’
Ei sen berg and coworkers have reported"^^ a RCMC for polym erised aggregates o f polystyrene/polysodium acrylate and polystyrene/polyacrylic acid, or block copolymer m icelles. These aggregates have uses in the areas o f detergency, oil recovery, drug delivery and catalysis. F e n d l e r ^ ^ and coworkers observed rate acceleration in the
mutarotation o f 2,3,4,6-tetram eth yl-a-D -glucose (figure 1.4.1.2) by a factor o f 600 in
reverse m icelles o f dodecylamm onium butyrate in cyclohexene.
CH2OCH3
HjCO-—
H
3
C
0
I
OH
1.5 Dialkylzinc Additions to Aldehydes:
Dialkyl or diaryl zinc com pounds are weakly nucleophilic. In the presence o f carbonyl
com pounds they w ill not react to form the corresponding alcohol without the presence o f a catalyst. Protic catalysts such as amines and alcohols have been show n to catalyse the addition o f the organometallic reagent via the loss o f RH, and subsequent formation o f amides or alkoxides respectively. In this manner, the L ew is acidity o f the metal is increased and the carbonyl can then bind to the metal atom with concomitant increase in its electrophilic character. Intra- or intermolecular attack o f R to the carbonyl is thus facilitated, and in the presence o f a homochiral protic catalyst can result in the formation
o f secondary alcohols o f high enantiomeric purity, as outlined in figure 1.3.1 below .
H X
R Z nX
R O ZnX
R OZnR
figure 1.3.1
This method has several advantages over other organometallic reactions such as organolithiums and Grignards where the rate o f reaction is not com pletely controlled by binding o f the carbonyl to the catalyst and the reaction environment is therefore not always chirally pure. A s a result, the addition o f diaryl, or more com m only dialkyl zinc to ketones, or more readily to aldehydes, is a good model reaction for studying the effects o f homochiral catalysts. Several chirally pure hom ogenous and heterogenous catalysts have been investigated^’^^-^'^-^^’^^’^® and attention has focu sed on the use o f P~
amino alcohols.^’ This family o f com pounds catalyse the reaction m ost effectively by deprotonation o f the hydroxyl group (although free OH groups are almost universally reported, metal salts have been used^^) and binding o f the zinc-alkyl group to the
Zn
Zn
/
figure 1.3.2
The degree o f enantioselectivity is governed by the chirality particularly at the a-carbon although other factors such as the bulk o f the (3-substituents and N-substituents are also
important.^’ W hile m ost m odels such as that show n in figure 1.3.2 require an amine with a free lone pair o f electrons, Soai reported^ a cationic salt o f (1 5 , 2/?)-ephedrine (figure 1 .3 .3 ) which, in its solid state in apolar solvents such as hexane, efficiently catalysed the diethyl zinc reaction in up to 74 % e.e.
Me Ph
figure 1.3.3
A s outlined in section 1.1.7, the amino alcohol catalyst when attached to polymeric
1.6 Michael Reactions
Compounds containing electron withdrawing groups (such as ketones, esters, nitriles, nitrosos, sulfones etc.) can add conjugately, usually in the presence o f a base, to alkene or alkyne m olecules containing electron withdrawing groups. This is the Michael reaction (figure 1.4.1) and has traditionally been carried out in protic polar solvents which have the effect o f stabilising the initial enolate anion and the transitional enolate anion.
M eO M eO
X H
M eO M eO
figure 1.4.1
In a Michael reaction where R ’ and are different from R^ and R^, then four diastereoisomers can be formed. Diastereoselective processes have been reported, as well as enantioselective ones.^^ Enantioselective Michael reactions can be achieved in three main ways; firstly by the attachment o f a chiral handle to the nucleophile and/or electrophile, secondly by use o f chiral enamines instead o f enolates and thirdly by use o f a chiral catalyst.
Attachment o f a chirally pure auxiliary often requires expensive materials, additional connection steps as well as cleavage o f the auxiliary. The example given in figure 1 .4 .2
reported by Enders^"^ em ploys a pyrrolidine m oiety (SAM P) that afforded e.e.'s greater than or equal to 95 %. The SA M P is cleaved and recycled after the Michael reaction, although the cleavage is achieved using som ewhat harsh ozonolysis conditions.
M eO M eO
S A M P
C0 2Me
COiMe
M e [ , \
C0 2Me
C O oM e
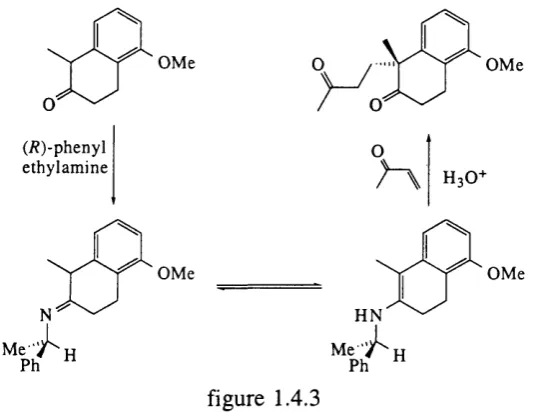
Chirally pure amines such as (/?)-(-)-phenylethylamine have been reacted with ketones to form an intermediate imine which can exist as the enamine. They are therefore capable o f 1,4 attack on a double bond as shown by d'A ngelo and coworkers^^ and are illustrated in figure 1.4.3
O M e
(/?)-p h en yl eth y la m in e
O M e
O
O M e
figure 1.4.3
O M e
H N
Ph
H owever, as above, this method requires the connection, cleavage and recovery o f the (expensive) chiral amine.
Catalysis o f the Michael addition is often feasible as the substrate to catalyst ratios are high.96 The catalyst can be removed at the purification step and re-used, or indeed if attached to a polymeric support, as outlined later in this section, work-up and recycling are made even more facile. Yura and coworkers^'^ obtained enantiomeric excesses o f up to 93 % using chirally pure pyrrolidinyl diamines and tin (U) enolates.
Manickam^^ also achieved high yields and stereoselectivities with the optically pure (R)-styrene oxide/benzylam ine derived catalyst as shown in figure 1.4.4.
EtO
O O
OEt
_ Bn _
I o
OH OH / 2 / A i - L i
figure 1.4.4
copper catalyst in the addition o f methylindan- 1 -one-2-carboxylate to methyl vinyl
ketone (figure 1.4.5). The product was obtained quantitatively in either R (54 % e .e .) or S (50 % e.e.) configuration by using the corresponding R or S configured catalyst respectively.
O O
+
O M e
Et
cat. ^ ^
O
figure 1.4.5
catalyst =
O
1.7 Aims of the Project
It w ould seem that although the sphere o f SPS is extensive, and still expanding
(particularly in the area o f combinatorial chemistry), the sphere o f polymer supported catalysis is comparatively unexplored. The number o f domains o f m ethodology available that remain unprobed are considerable. For exam ple, the Diels-Alder cyclisation displays enhanced regioselectivity, stereoselectivity and rate o f reaction in aqueous micellar media (section 1.2), how ever, no research pertaining to this reaction
using recoverable surfactants have been published. Therefore, for exam ple, the attachment o f surfactants to polymeric supports follow ed by the investigation o f the cycloaddition in aqueous or organic suspensions o f these polym ers w ould be o f interest.
Therefore the aims o f this project from the outset were to synthesise a range o f
polymer-bound and non-polymer-bound surfactants and to investigate their
CHAPTER 2. RRSUT.TS & DTSCUSSTON
As outlined above in section 1.7, the aim o f this research was to prepare novel surfactants for use in aqueous and organic media as catalysts for asymmetric
carbon-carbon bond forming reactions. W e were also interested in the preparation and application o f several novel polymer-supported surfactants, in order to assess their properties and potential in comparison with the non-polymer-supported surfactant analogues.
2,1 The Synthesis of Cs-i2 Methylene Spacer Surfactants
and Resins
A s m entioned above, Soai 3,io,io3 ^nd Fréchet^ have prepared optically active ephedrine
catalysts attached to crosslinked polystryrene polymeric support w hich were used in diethyl zinc additions to afford products o f high enantiomeric purity in high chemical yield. N otably, Soai reported that optimal results were obtained with catalysts containing six methylene spacers between the catalytic m oiety and the polymer backbone.
Our initial aim was therefore to synthesise a range o f supported surfactants making use o f Soai's attachment procedures^. The first range o f head groups selected was N -trimethylamine and A-triethylamine to compare the relative effects o f the bulkiness o f the head group. The non-chiral supported surfactants w ould then be used to test their catalytic effect on the Diels-Alder cyclisation compared to commercially available analogous surfactants such as cetyltrimethylammonium bromide (C T A B ). It was also decided to prepare a chirally pure supported surfactant. The head group em ployed by
Soai was chosen using his synthetic m ethodology since ephedrine is readily available and ideal as it possesses tw o chiral centres.
Solution analogues o f these materials were also prepared to test the synthetic route and to monitor the comparable behaviour o f these materials. Different chain lengths o f Cg, Cio and were used to indicate the required chain length for micellisation o f these