0022-538X/88/031008-08$02.00/0
Copyright ©O 1988, American Society for Microbiology
Development of Avian Sarcoma and Leukosis Virus-Based
Vector-Packaging Cell Lines
ANDREW W. STOKER* ANDMINA J. BISSELL
Laboratory of Cell Biology, Division of Biology and Medicine, Lawrence Berkeley Laboratory, University of California, Berkeley, California 94720
Received 16September 1987/Accepted 13 November 1987
We haveconstructed anavian leukosis virus derivative witha5' deletionextending from within the tRNA
primerbinding site toa Saclsite in the leader region. Our aimwas toremovecis-acting replicative and/or
encapsidationsequencesand tousethisderivative,RAV-1*-, todevelop vector-packaging cell lines. We show
that RAV-14-canbestably expressed in the quail cell line QT6 and chicken embryo fibroblasts and that it is completely replication deficient in both cell types. Moreover, wehave demonstrated that QT6-derived lines expressing
RAV-1*-
canefficiently package four structurally different replication-defective v-srcexpressionvectorsinto infectious virus, withverylowor undetectablehelper virus release. TheseRAV-l*--expressing cell linescomprise the first prototype avian sarcomaand leukosis virus-based vector-packaging system. The construction ofour vectors has also shown usthatasequence present withingag, thought to facilitate virus
packaging, is notnecessaryfor efficient vectorexpression and high virus production. We show that quantitation and characterization ofreplication-defective viruses can be achieved with a sensitive immunocytochemical procedure, presentinganalternative to internal selectable vector markers.
Retroviral vectors have proved to be highly efficient for introducing clonedgenes into culturedcells and organisms. Recently developed retroviral vector packaging cell lines provide infectious, replication-defective (rd)viruses without helper virus contamination, thus avoiding potential problems withviral interference and helpervirus-induced disease(22, 24, 41). The mostextensively used system, the murine i2 cell line, has facilitated the transduction of genes into hemopoetic, neuronal, and embryonal carcinoma cells for studies ofhemopoetic reconstitution and for chromosomal marking (7, 11, 14, 29, 32, 37, 40, 42). Such work has enhanced theunderstandingofdifferentiation, celllineages, and thebehavior ofretroviral vectorsin vivo.
Our studies have centred upon the interaction of Rous
sarcoma virus (RSV) and the oncogene v-src, with both hatched andembryonicchicken tissues. We have shown that RSV-inducedtumorigenesis is both inhibitedin ovo(9)and
dependent upon wounding in hatched birds. (10). These unforseencomplexitiesofRSV-induceddisease raisea
num-ber ofquestions that could be addressed appropriately by usingrdv-src expression vectors. Althoughthere has been recent interest in its use, a packaging system designed specifically for avian sarcoma and leukosis virus (ASLV)
vectors is not presently available. The feasibility of
con-structingsuchasystem'with avian leukosis virus has been in some doubt because of the poorly characterized avian
leu-kosis virus RNA encapsidation region and the possibility thatatleastpartof theregion lies 3' tothesplice donor site withingag(31). The leader (L) region, however, is believed
tocontainatleastapartof thecis-actingRNAencapsidation
sequences(4i), sincetwonaturally occurringRSVpackaging mutants,TK15(27)andSe21Qlb (34),bothcontaindeletions in L(fora reviewsee reference6). Arecent study by Katz etal. also describedaspecific sequencewithin the Lregion necessary for RNA encapsidation in RSV (17). We show here thatremoval ofarelated sequencewithin the Lregion ofacloned avianleukosis virushelperis sufficienttoabolish
*Correspondingauthor.
virus replication in cis. Using this crippled helper virus, designated RAV-1-, we have establishedprototype pack-agingcelllines for ASLV-based vectors.
With sensitive immunocytochemical techniques (36) we
show that transfected pRAV-1h- directs the expression of viralproteins but doesnot supportreplicatingvirus
produc-tion from either chicken embryo fibroblasts (CEF) or the
quailline QT6 (25). Stable, RAV-14--expressing QT6lines have remained helper virus free since their development. These same cells were shown to package three different ASLV-based rd vectors into infectious virions in transient
assays, without significant helper virus titers. Packaging cells stably releasing vector-derived virus provide greater than i05 viruses per ml, with very little or no helper virus
release. We have thusdemonstrated both the feasibility of designing ASLV-based packaging cell lines and the
con-struction ofspecific vectorsforusein these cells. MATERIALS ANDMETHODS
Cell culture.QT6isachemicallyderivedtumorcell line of
the Japanese quail, free ofexogenous ASLV (kindly
pro-videdbyP.Vogt, University of Southern California, School ofMedicine, Los Angeles) (25). CEF were prepared from day 10 white leghorn embryos (specific pathogen free) as
described previously (3). All cells were routinely grownin medium 199(GIBCO Laboratories) supplemented with 10%
tryptose phosphate broth, 4% newborn calf serum, 1% chickenserum, and 100 p.gofgentamicinperml.
Manipulationof cloned viral DNA. All recombinant DNA manipulations wereperformed by standardtechniques (21). Proviral DNA inplasmid formis referred to with prefix p,
whereas the viruses expressed havenoprefix.
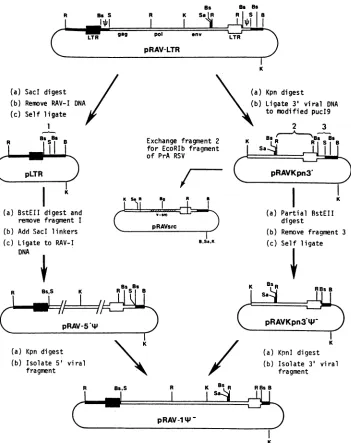
pRAV-1*-
(Fig.1and2).The clonedhelpervirusgenomepRAV-LTR was'kindly given by B. Vennstrom (Differenti-ationProgramme, EuropeanMolecularBiology Laboratory, Heidelberg, Federal Republic of Germany). This plasmid containsacloned, permuted Rous-associated virus(RAV-1)
inserted asa Sacl fragment 3' toan avian erythroblastosis 1008
on November 10, 2019 by guest
http://jvi.asm.org/
R as S
L~
L*
C
LTR- 9a9Bs Bs Bs
R K SasR R|S B
I
I
1
Lp
I
pol env
L--pRAV-LTR LTR
pRAV-LTR
K
(a) (b)
(a) Sacl digest
(b) Remove RAV-I DNA /
(c) Self ligate
I
Kpn digest
Ligate 3' viral DNA to modified pucI9
|K
(a) BstEII digest and
remove fragment I
(b) Add SacI linkers (c) Ligate to RAV-I
DNA
-Exchange fragment 2 for EcoRIb fragment of PrA RSV
K So R 9g R a
pRAVsrc
I
0,S&,K
IN 2 3
Bs B--s Bs
Ba R B Ba
Sa
pRAVKpn3'
K
(a) Partial BstEII digest
(b) Remove fragment 3
(c) Self ligate
R Bs,S aRs Bs B
.14 1-
//I-pRAV-5
IWJ
(a) Kpn digest
(b) Isolate 5' viral fragment
Q
/
R K RBaB
RRA1K Ss R B
.1
I
s^l-Q
I
pRAV-1WI
-(a) KpnI digest
(b) Isolate 3' viral fragment
K
FIG. 1. Schematic representation of
pRAV-14-
construction. Boxed regionsrepresent proviral DNA, RAV-1 (O) andAEV10(U).Single linesrepresentbacterialplasmid DNA. DuplicatedRNAencapsidationsequences(Oi)
areindicatedin pRAV-LTR. Relevant enzyme cleavage sitesareindicated: B,BamHI(sites within RAV-1 DNA are notshown);Bg,BgII;Bs, BstEII; K,KpnI;R, EcoRI;5,Sacl;Sa,Sall.L
RAV-LTR
RAy-lW-U5 I
I
_.,,,,,,g
-Dr7.aa
BstEll SacI BamHI
r-pr76gsg f
[image:2.612.132.483.63.506.2]sd
FIG. 2. Diagram representing 5' regions of pRAV-LTR and pRAV-li- proviruses: AEV10 DNA (_), RAV-1 DNA (mi and =!). TheU5 region of theAEV10 LTR is shown, with thetRNA
PBS, the noncoding leader region (L), and the startof the pr76xag coding region. sd, Splice donor site. The 5' region deleted from pRAV-LTRtoformpRAV-14- is indicated.
virus (AEV10) long terminal repeat (LTR) in pSV2gpt (26, 39). Due to the permuted structure of this provirus, two similar deletions were introduced to remove putative 4, sequences.Thedeletionin the 5'AEV10 L regionwasmade first by cleaving pRAV-LTR with SacI to remove RAV-1 sequences. The resulting plasmid pLTR was digested with BstEII to removethe L-regionsequence; SacIlinkers were added, and the RAV-1 DNA was ligated back to create pRAV-5'iP, containing the correct 5' deletion. Deletion of 4 sequence 3' of the RAV-1 LTR was initiated by cleav-ing pRAV-LTR with KpnI and ligating the 3' viral DNA fragment to amodified pUC19 plasmid (lackingEcoRIand SacI restriction sites). This plasmid, pRAVKpn3', was di-gested partially with BstEII and self-ligated to create
pRAVKpn3'*-,
which now contained the 3' deletion(ap-proximately 100basepairs
[bpl
largerthan the 5' deletion).R as's
I
ad
I
J4
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.56.296.563.709.2]pAll LTR
_r
H
9ag9 Apol W-arc LTR
6
a c
M0S
PASrci LTR 4 v-arc * LTR
pASrc1l
B/S
pASrcneol
U3 RU5
RESV _|_RAV1
EcoRI
1 Kb
L--SH IB/BgI
pASrcneo6
LR-ac
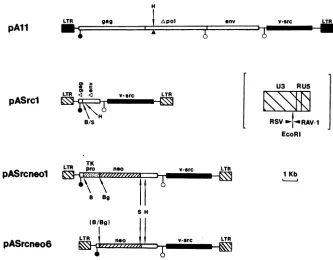
FIG. 3. Replication-defectivev-srcexpressionvectors.pAll isapol-B77RSV;pASrclandpASrcneolarederived fromRAV-1and PrA RSVDNAand havechimericLTRs(inset contains theLTRsofpASrclandpASrcneol,showingboundaries ofRSVand RAV-1sequences). Agag, Apol, and Aenvaretruncatedgenes. Symbols:positionof deletedHindlllfragmentinpAll(A) andsplicedonor(0)andacceptor
(0)sites. Unique cloning sites invectorspASrclandpASrcneolareindicated. B,BamHI;S,Sall;H,Hindlll;TK pro,thymidinekinase gene promoter; neo,neomycin resistancegene.
To construct pRAV-1I-, 5' and 3' proviral halves were ligated together as KpnI fragments from pRAV-5'4i and pRAVKpn3O'%-, respectively.
pAll (Fig. 3).pAll proviralDNAwaskindly givenbyD. Gillespie and J. Wyke (The Beatson Institute, Glasgow, United Kingdom). The pAll provirus is aBratislava strain (B77) of RSV cloned from an infected rat cell line and reconstructed with a 130-bp deletion between twoHindlll sites withinpol(19). Toestablish long-term
packaging
lines expressing thisvector,10 ,ugofpAllwascotransfected with 1 ,ug of pY3 containing the hygromycin resistance gene (hphr) (4)onto106Q2bn cells.hphrpopulationswereisolated andscreened forvirus release.pASrcl (Fig. 3). A cloned EcoRI b fragment (containing v-src) from wild-type PrA RSV (kindly supplied by V. Fincham and J. Wyke [12]) was exchanged for the EcoRI fragment in pRAVKpn3' containing 3' env to 5' LTR se-quences. The resulting plasmid pRAVsrcl was used to constructthecomplete proviruspresentinpASrcl. Unique, internalBamHI, Sall,andHindlIl cloning sites arepresent inthis vector,as aresplicesites for thev-srcmRNA(RAV-1 donor, PrAacceptor).
pASrcneol (Fig. 3).ProviruspASrcneolwasderived from pASrcl. A BamHI-SalI fragment containing the neo gene and TK promoter from pIPB1 (a plasmid containing the neomycinresistance (neo) geneof TN5 (16) preceded by the herpes simplex virus thymidine kinase gene promoter (23), kindly given by R. Sweet (College ofPhysicians and Sur-geons, Columbia University, New York) was inserted into
the
BamHIISalI
cloning sitein pASrcl in thesametranscrip-tionalorientationas v-src.
pASrcneo6 (Fig. 3). Provirus pASrcneo6 was derived from pASrcneol.Thethymidinekinase promoterwasremovedas
aBamHI-BglII fragment, and the plasmidwas self-ligated. The neo geneisexpresseddirectly fromthe viralLTR.
pASrcneo7. Provirus pASrcneo7 is similarto pASrcneo6, exceptthat the RSV EcoRI bfragment is derivedfrom B77 RSV and notPrARSV.
DNA transfection anddrug selection. Calcium phosphate-mediated DNA transfection was performed essentially as describedbyGraham and Van der Eb(15).Precipitateswere left on cells for 4 to 6 h, and the cells were then glycerol shocked(completemediumplus15%glycerolfor 100s).To establish stableQT6linesexpressing
pRAV-1*-,
the follow-ing DNAs weretransfected (per 106cells): 1 ,ug of BamHI cutpIPBj;
10 ,ug of Sall-cleavedpRAV-lq-;
10 ,ug of calf thymus DNA carrier. These cells were split 1 day after transfection and placed under G418 selection (200 to 400 jig/ml), and resistant colonies were isolated 2 to 3 weeks later. A second series ofRAV-1--expressing QT6
lines weredevelopedinasimilarmannerwith thehphrexpression plasmid PY3 (4)asthecotransfected marker.Plasmids containingv-srcexpression vectors were trans-fecteduncut(10 ,ug)with 10 ,ugofcalfthymusDNAcarrier per 106 cells.
Virus collection and characterization withexpressionfocus unit (EFU) assay. Viruseswere collected from cells in fresh medium for 2.5 h. These supernatants were cleared of cell debris by centrifugation (5 min, 600 x gto 1,000 x g) and either used directlytoinfect cellsorstored frozenat-70°C before use. After transfection of v-src expression vector DNA into packaging cells, virus was collected 18 to 24 h posttransfection asdescribed above.
An immunocytochemical assay has been specifically de-veloped forquantitationof bothreplicatingand rdinfectious viruses (36). Briefly, CEF were infected with virus and
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.142.475.65.325.2]TABLE 1. pRAV-LTR-andpRAV-1%--derived virus titers releasedfrom transfected and infected CEF andquailcell lines
Maximal virus Cell type Virusexpressed (titer(EFU/ml)
CEF NONE <1
CEF RAV-LTRa 1.0 x 106
CEF RAV-lh-a <1
QT6 RAV-LTRb 1.3 x 105
QT6
RAV-lq-F
line Qlan <1QT6 RAV-1lt-line Q2an <1
QT6 RAV-ltl- lineQ2bn <1
aViral DNA wasintroduced by transfection, and virus titers were assayed
8days later after one cell passage.
bRAV LTRwasintroducedby infection, and virus titers were measured
after 12 days.
IViral DNA was introduced by transfection, and virus titers were assayed
at numerousintervals up to the present.
overlaid withagar 14 to 16 hlater. The agar was removed 5 to 10 days later, andthecells werefixed in2% paraformal-dehyde. A three-layer immunodetection system was em-ployed: layerone was an antiviral serum(either
anti-pl9g'g,
[image:4.612.338.541.381.675.2]two different monoclonal antibodies kindly provided by T. Pawson [MountSinaiHospital Research Institute, Toronto, Ontario, Canada] and D. Boettiger [University of Pennsyl-vania, School of Medicine, Philadelphia], or anti-pp60vsr( [JB327] kindly supplied by J. Brugge [State University of New York at Stony Brook, School of Medicine, Stony Brook]); layer two was a biotinylated anti-mouse F(ab')2 (Amersham Corp.); layer three was a streptavidin-conju-gated alkaline phosphatase complex (Enzo Biochem or Zymed).Aftersequential binding of these threereagents, the color reaction was developed to reveal foci of antigen-expressing cells (virus titers are expressed in EFU). For characterization of nonreplicating virus, the agar overlay step was omitted, and cells were stained 3 to 5 days postinfection. Thisassayishighly sensitive, detectingasfew as oneinfectioneventper testplate. Inaddition, ASrcneol-andASrcneo6-derived viruseswere quantitated bycounting the number ofQT6 neomycin-resistant (neor) colonies re-maining after virus infection andG418 selection (200 to 400
jig/ml).
RESULTS
Introduction of L-region deletion in cloned helper virus genome.
Analysis
ofnaturally occurring packaging-deficient RSV mutants suggested that critical RNA packaging se-quenceslie intheviral Lregion. We used suitablerestriction sites located in a cloned helper virus genome pRAV-LTR (containingcombined sequences fromRAV-1 andAEV10), to removepart ofL;adeletion ofapproximately150bpwas introducedbetween theBstEIIsite within theprimer-binding site(PBS)abutting the5' LTRandthe downstream Saclsite in L (Fig. 1and2) (see MaterialsandMethods). Inaddition to deleting a part of L, this removed 10 of theoriginal
18 nucleotides from the PBS. Because the Lregion
isdupli-cated in the downstream permuted RAV-1 sequences, this region and approximately 100 bp of 3' adjoining sequence were also removed to avoid recombination. The final pro-viral construct was named
pRAV-1V-.
RAV-1*-
is replication defective. To test whether the deletionmadeinpRAV-1*-
had removedsequences critical for virusreplication,wetransfectedcircularpRAV-ltf-
and pRAV-LTR DNAs into CEF and monitored viral proteinexpression
byimmunocytochemistry
and virus release.Within8 days of transfection with pRAV-LTR, virus spread had occurredtomostcellsasjudged by expression of p199a9 (data not shown). High titers of infectious virus were re-leased from these cultured cells(Table 1). CEF transfected with
pRAV-1lq-,
however, did not show this pattern. Rather, a minor population of CEF (<0.01%) showedp199a9 expression, demonstrating stable transfection of RAV-1-in a small numberof cells without any indication of virus spreadorinfectious virusrelease (Table 1; seeFig. 5f).To further characterize the behavior of pRAV-1l-, we transfected thecloned provirus intoQT6cells. As with quail cells in general, this tumor cell line has no endogenous viruses closely related to ASLV (13), although distantly related sequences have recently been described (5). The absence of endogenous viruses in QT6 is advantageous if recombination of introduced viral sequences (here
pRAV-1*-)
withcellulart4
sequences istobeavoided. The pRAV-1- DNAwascotransfected into QT6 cells withaselectable neorplasmid,and neorcolonieswereisolated, amplified,and immunocytochemically stained for p199a9 expression. Three positive lines, Qlan, Q2an, and Q2bn, were chosen for detailed study.Usingthe EFU virusassay (see Materials and Methods), we tested whether these lines released replicating helper virus and found none (Table 1). Moreover the lines have been nonproducers for more than 9 months and are thus stable. QT6 cells infected with the pRAV-LTR-derived helper virus produced high titers ofreplicating virus(Table
1).
Intact proviral DNA has been detected in each line by
TRANSfECTED
18-24 HR LATER COLLECT VIRUS IN SUPERNATANT
ASrcneol
I
INFECT 0T6 CELLS WITH VIRUS
I
G418 SELECTION
t
COUNT G418r COLONIES
All, ASrcl, ASrcneol
INFECT CEF WITH VIRUS
I
3-4 DAYS
(2) (1)
COLLECT
SUPERNATANT
INFECT CEF
3
3 OAYS FIX ANDIMMUNOSTAIN
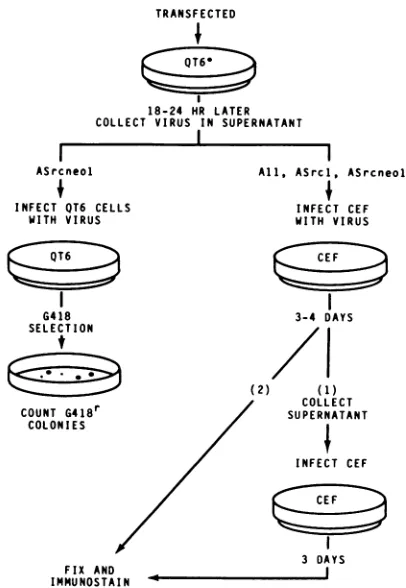
FIG. 4. Regimen for testing rd virus packaging in RAV-lF--expressing QT6 cell lines. QT6* indicates QT6-derived packaging
cells.Supernatantswerecollected fromCEF(step 1)beforefixation ofthesamecells(step 2). Methodsfortransfection,viruscollection, andimmunostainingaredescribed inMaterials and Methods.
on November 10, 2019 by guest
http://jvi.asm.org/
a
c
b
d
af
f
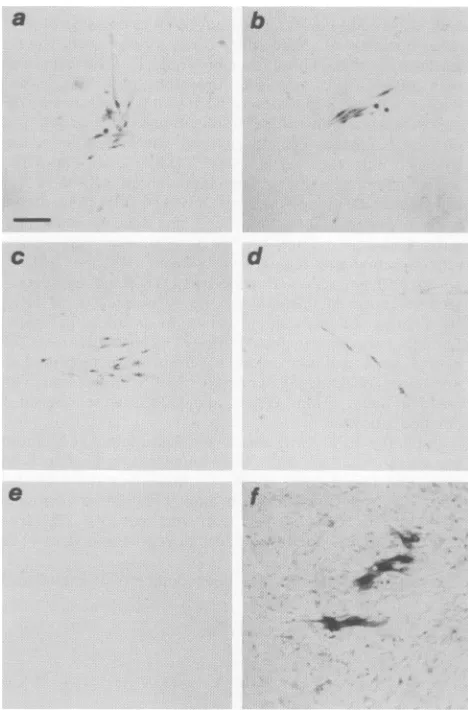
FIG. 5. Alkaline phosphatase-immunostained foci of virus-in-fected CEFexpressing pp6-src (dark cells)in anuninfectedCEF
background. Panels: representative foci of cells 3 to4 daysafter
infection with virus derived frompAll (aandb), pASrcl (c), and pASrcneol (d), uninfected CEF 4 days after inoculation with
supernatant from pAll-transfected QT6 cells, stained with anti-pp6Ov-src (negative control, e); CEFexpressingpRAV-lp- 8days after transfection andasinglepassage, stained withanti-p199'9 (f). Bar,0.1mm.
Southern analysis of restriction endonuclease-digested cel-lular genomic DNA(data not shown). This rules outgross
proviral rearrangements as the reason for the absence of virus production by these lines. The lack of
RAV-1*-replication in QT6 cells corroborates the CEF transfection data discussed above, confirming that the deletion in RAV-1l- eliminated its ability to releasereplicating virus, whereas viral p199ag expressionwas maintained.RAV-14--expressingcellscanpackage replication-defective
vectors. (i) Packaging ofpol- rd RSV. To test whether the mutationinRAV-lW-wascisacting,wetested whetherthe expressing QT6 cell lines couldpackage rd viral RNA into infectious viruses without accompanying helper virus. The first rdvectorwe testedwaspAll, apol- Bratislava strain
(B77) RSV provirus (19). Uncut pAll DNA (10 ,g) was
transfected into the lines Qlan, Q2an, andQ2bn, and tran-sient virusreleaseinto culturesupernatantswastested 18to 24 hlater (Fig. 4; Fig. 5a and b). Traditional focus assays were not performed because we have shown that our
im-munocytochemical assay is more accurate (36; A. Stoker,
unpublished
observations). pAll-derived
viruswasreleased from linesQlanandQ2bn, butnone wasdetected from line Q2anorQT6 cells(Table
2).Thefact thatQ2an
cells didnot release virussuggested
that theprovirus(es)
in these cells were debilitated through mutations or rearrangements not detectableby
Southernanalysis,
and this linewastherefore notstudiedfurther.To examine whether
replicating
viruswasconcomitantly
released withrd
virus,
supernatantsfrom the infected CEF were collected before cell fixation and used to infect fresh CEF(Fig. 4).
These cells werelater fixed andanalyzed
forp199a9
EFU.Using
RAV-LTRhelper
virusweconfirmed thesensitivity
of thisprocedure: single plates
of CEF infected with1 to2 EFU ofreplicating helper
virus released detect-ableprogeny virus(up
to 10EFU/ml)
into the supernatants 3days
after infection.Intesting
thepAll-derived virus,
two ofnineindependent experiments
revealed recombinant in-fectious virus in supernatants from infected CEF.However,
virus titerswere verylow(12 and1
EFU/ml, respectively),
and the virus was
transforming
asjudged by positive
pp60v-src staining
and the transformedmorphology
of in-fected CEF. These recombinant viruses appear to have arisenpredominantly by
recovery of the pol sequenceby
All.
(ii)
Packaging
of vectors with no viral structural genes. From the above datawe could conclude that the cell linesprovided
pol functions andexpressed
gagproteins.
We wished to ascertain whetherpRAV-l1--expressing
cells wouldprovide
all viral structuralproteins
in trans, andalso whetherby
incorporating larger
deletions into vectors the recombination withRAV-1b-
would be reduced. Two smallerv-src vectors wereconstructed which lackedall viral structuralgenes.pASrcl
retainscomplete
5'*
sequences inL,
the 3'polypurine
tract(35),
viralsplice signals
for v-srcmRNA,
and thev-srcgeneofPrARSV.
Only
truncatedgag andenvsequences remain of the viral structural genes. Transient virus releaseassayswere
performed
asdescribed aboveforpAll.
pASrcl-derived
virus was released from both cell linestested,
attiterscomparable
to those found withpAll
(Table 2).
pASrcneol
expresses bothv-srcand thedominant select-able neo gene(under
the internal control ofthe HSV TK genepromoter)
(Fig. 3).
The titer ofpASrcneol-derived
virus can be estimated from the number of neor colonies induced in infectedQT6 cells;
transientexpression
ofpASrcneol
inQ2bn
cells thus gave maximal virus titers of 5.1 x 103 EFU/ml. ASrcneol virus was alsopackaged by
lineQlan,
butatlowerefficiency
thanQ2bn
(Table
2).
CEF infectedwith this rd viruswereimmunostained forpp6Ovsrc
and foci with similar characteristics to those inducedby
ASrcl were observed(Fig.
5c andd).
The virus titers estimatedfromneorcolony
formationwerebroadly
compa-rabletothepp6Ov-src EFU, indicating
thatbothquantitation
procedures
have similar accuracy.Replicating helper
virus was not detected whenpASrcl
was tested in transient assays.
Similarly,
noreplicating
helper
was found whenpASrcneol
wastested,
with oneexception: helper
virus wasfound afterone transfection ofQ2bn cells,
butonly
at the threshold of our detectiontechnique
(1 to 2EFU/ml).
Helper
release was notstable,
however,
since a later virus collection failed to detect its presence.Long-termvirus-producinglines. The virus
analyses
using
thetransientassay demonstrated the
packaging
behavior ofRAV-1b--expressing
celllines,
but thetitersobtainedwere J.VIROL..o
.
or
:.w
..
...10
K;Fw-t
7
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.67.301.69.424.2]TABLE 2. Transient vector-derived virus release from quail cell lines
Celltype Titer (EFU/ml) with the following rd vectorintroduceda: colonies/ml'
transfected pAll pASrcl pASrcneol
QT6 <1 (<1) <1 (<1) <1(<1) <1
<1 (<1) <1 (<1) <1(<1) <1
Qlan 3.0 x 102(<1) 5.5 x 102 (<1) 1.8 x 102(<1) 1.1 x 102
4.0 x 102(<1) 6.6 x 102(<1) 1.4 x 102 (<1) 1.4 x 102
7.4 x 102(<1) 2.0 x 102(<1) 1.2 x 102 (<1) 3.1 x 102
1.5 x 103(<1)
Q2bn 9.2 x 102(<1) 9.6 x 102(<1) 3.8 x 102(<1) 3.5 x 102
1.0 X 103(12) 1.0 x 103(<1) 3.4 x 103(1-2) 5.0 x 103
1.5 x 103(<1) 2.8 x 103(<1) 6.0 x 102 (<1) 5.1 x 103
1.8 x 103(<1) 3.5x103(1 )
Q2an <1 (NT)
<1(NT)
a Titer of virusreleased measuredwithpp6Ov-srcimmunostaining inCEF.Figures within parenthesesrepresenttiters ofreplicatingvirus releasedfrom infected CEF, measured in pl9gagEFU permilliliter. NT, Nottested.
bpASrcneol-derived viruswasanalyzed.
not satisfactory foruse in in vivo studies. In an attempt to
obtain higher titers, stable virus-releasing clones of cells
were isolated. Two approaches were used. In the first
approach we cotransfected pAll and an hph selectable
plasmid into line Q2bn and isolated stable hphr cells. These cells released pAll-derived virus at titers up to 30-fold
greaterthan the comparable transient titers (Table 3). Rep-licating viruses (including nontransforming helper virus)
were also detected at low levels with this large vector.
Although certain populations released as few as 0.01%
replicating viruses, we have not been able to subclone any
which were completely helper free, indicating that a
rela-tively high recombination rate was occurring between this
vectorand RAV-14- (either beforeor after virus release).
Our secondapproach wasto establishpackaging popula-tions by direct selection for transfected neo vectors. For this, wefirstestablishedasecond series ofpackaging lines,
using hph cotransfection with pRAV-1l-, which behaved essentially like Qlan and Q2bn described above in transient virus releaseassays (datanotshown). One such line, Q4dh,
wastransfected with pASrcneo6 and selected for neor (neo
and hph are independently selectable [4]). pASrcneo6 ex-presses neo directly from the viral LTR (Fig. 3), since we
wished to select for LTR activity and not an independent
internal promoter (pASrcneo6 gave titers comparable to
those ofpASrcneolin transientassaysin lineQ2bn). neor populations cloned in this way stably released ASrcneo6 virusattitersgreaterthan 105 EFU/ml (Table 3). Virus stocks were screened for replicating viruses as
de-scribed for Asrneol in Fig. 3. Virus screens up to7 weeks after clone isolation did not not reveal any helper virus.
Tests after 9 weeks did detecthelper virus, onlyatthe lowest threshold ofourdetection(Table 3). Other stable lines have
been isolated with vectorpASrcneo7, which is structurally similar to pASrcneo6; again, maximal rd virus titers were
greaterthan 105 EFU/ml. The majority of these lines have remainedhelper free since isolation (>10 weeks) (Table 3).
DISCUSSION
Our goals in this work were twofold: (i) to design and
express, inappropriate cells, acrippled ASLV-based helper
virus which would complement rd vectors in trans and yet withhold its own ability to replicate, and (ii) to design specific rd vectors for use in these packaging cells. In accomplishingthesegoals,wehave used novel immunocyto-chemical techniques to screen for replicating virus and infectious rd viruses.
The defective helper virus pRAV-14- was successfully expressed aftertransfection into QT6 cells, and these lines did not release replicating helper virus. The helper-free stability of the cells, an important consideration in their long-term use, may arise from the absence ofendogenous viruses available for recombination. We have also shown that stable expression ofpRAV-1t- occurs in CEF after transfection, without detectable virus replication (Fig. 5f; Table 1). These latter data are at odds with thosepresented by Cooper and Okenquist, who suggested that stable inte-gration of transfected rd viral DNA in CEF requires first virus replication and then reinfection (8). Our data with
[image:6.612.318.560.529.679.2]pRAV-l1i-
argueagainstthisinterpretationastheexclusiveTABLE 3. Virus titers from stablevirus-releasing packaging cells
Cell Vector Virus- Virustiter Replicating
line expressed cell clonesa (EFU/ml) virus/ml
Q2bn pAll 1 2 x
104b
1-52 3 x 104 1-5
Q4dh pASrcneo6 1 2 x 105c 1-2
2 2 x 105 1-3
3 2x105 1-3
4 3 x 104 NTd
5 3 x 104 NT
6 6 x 103 NT
Q4dh pASrcneo7 1 2 x 105 <1
2 3 x 105 1-4
3 2x104 <1
aPopulationsisolated from colonies after transfection ofpackagingline and
drug selection.
bVirustitermeasuredbyp199agEFU.
cVirus titer measuredbyneorcolonyformation.
dNT,Nottested.
on November 10, 2019 by guest
http://jvi.asm.org/
route of stable expression; by immunocytochemistry we detect stable pRAV-l+- expression in CEF populations 8 days posttransfection. This form of expression after trans-fection into CEF is clearly very low, however, and differen-tial sensitivity of assay techniques may explain the differ-ences between ourdata and those ofCooper andOkenquist. Until recently the characterization of the 5' RNA packag-ing signals in ASLV genomes had not been extensive. The construction ofpRAV-li- was therefore foundedupon data from two naturally occurring RSV packaging mutants (27, 34).We have shownthat the deletion in pRAV-1l-, extend-ing from within the PBS region to a Sacl site in L, is sufficient to prevent the release of replicating virus from avian cells and that this defect is cis acting. While pRAV-lt-was being tested in our laboratory, Katz et al. presented adetailed analysis of theijregion in RSV, concluding that a critical30-bppackaging sequenceliesupstream ofthesingle SacI site in L (17). This 30-bp sequence is absent in pRAV-1-,and ourdata thus corroborate those of Katz and co-workers. When compared with TK15, however, the re-moval of L sequences does not satisfactorily explain the total lack ofpRAV-l+- replication. The mutant TK15 con-tains adeletion extending from the 3' junction of the PBSto near the gag AUG start codon (28). Although this region is about 100 bp larger than that deleted in
pRAV-1*-,
low-titer infectious virus is released fromTK15-expressingcells (18). Acritical difference between TK15 andRAV-1l-islikely to bethe removal of PBS sequences in the latter. A study by Pugasch and Stacey (30) showed that removal of the entire PBS from avian leukosis virus eliminated virus replication, presumably through blocking minus-strand DNA priming (for a review, see reference 38). The pRAV-l1i- genome retains only 8 of the original 18 bases of the PBS (additional base matches may occur with the newly juxtaposed sequence), and this may also be insufficient for specific tRNA binding. Although we have no biochemical data, we believe primarily that the deletion in L severely reducesor abolishes the specific packagability ofRAV-1- RNA, and that the PBS deletion prevents reverse transcription of any nonspecifically packaged viral RNA.We have shown that pRAV-l+- linescanpackagea pol-B77 RSV and two specifically designed rd v-src vectors in transient assays. Significant recombination between vector pAll and
RAV-1*-
was shown to occur in these assays, and the evolution of replicating viruses in stable All-releasing populationsconfirmed this finding. WithvectorspASrcl and pASrcneol, replicating viruses were not detected in tran-sient assays exceptfor the single casediscussedabove. The stable lines releasing packaged vectors ASrcneo6 and ASrcneo7 also demonstrated that high titers of rd viruscould beisolated frompRAV-lip--expressing
cells, with very low orundetectablerelease of replicating virus. Thus removalof unnecessary coding sequences in the pAS series of vectors significantly reduces recombination frequencies in compari-son to pAll.Although the removal of internal sequences also generates space for nonviral inserts, maximizing the size of the dele-tion must be balanced against the retention of necessary cis-acting sequences. Recent studies of both avian and murine retroviruses indicate that sequences in the 5' gag region may augment virus expression and/or genomic RNA encapsidation and also influence the viral disease spectrum (1, 2, 33). RSV hasbeen shown tocontainasequencein gag with transcriptional enhancing activity and also a sequence flanking the BamHI site at position 532 (PrC RSV base equivalent) which may enhance encapsidation (32). Our pAS
vectors,however, donotcontain these regions. Removal of the gag enhancer segment appears not to hinder vector expression as judged by the ability of the vector-derived viruses to morphologically transform CEF (A. Stoker, un-published observations). Nevertheless, we are presently examining thisquestionin moredetail. Loss of theputative encapsidation region does not significantly affect the virus titers derived from our vectors in comparison with either pAll (in both transient and long-term virusrelease;Table3), or the titer of RAV-LTR released from QT6 (Table 1). Therefore RNApackagingremains efficient after loss of this region, afindingalso notedbyothers(20, 28).
Wehave shown withimmunocytochemistrythataccurate quantitation of vector-derived viruses is possible without using internal selectable markers. Quail cellsarealso resis-tant toinfection by a number of commonASLV subgroups (25),and therefore the useofthe neor
colony-forming
assay for such viruses would not be applicable. If immunocyto-chemical detection isafeasiblealternative,
selectablevector genes may be either removed or replaced by sequences of biological interest in size-constrainedvectors.In conclusion, we have constructed an ASLV-based helper virus derivativewithacis-acting mutationpreventing the release ofreplicating virus. Stable quail cell lines ex-pressing pRAV-l+- are helper virus nonproducers but are able to package rd ASLV-based vectors into infectious virus. Using cell clones selected tostablyexpressrdvectors, we have obtained high-titer rd virus with very low or undetectable helper contamination, suitable foruseinvivo. Theavailability of this cell systemwill furtherour examina-tion ofthe complex oncogene-host interactions in hatched andembryonic avian tissues. Moreover, suchasystem will facilitate experiments in vivo
addressing
broaderquestions
ofviralexpression, target
specificity,
and celllineage
in the developing avian embryo.ACKNOWLEDGMENTS
We thank Jill Hatier for preparation of CEF and the persons mentioned in thetextforprovisionofantibodies andplasmids. We thankL.-H.Chen,M. Glotzer,A. R.Howlett, andR.Schwartz for helpfulcommentsconcerningthemanuscript.
This work wasfunded inpartby aEuropean MolecularBiology Organization fellowship toA.W.S. (ALTF 271-1985), and by the Office of Health and Environmental Research, Department of En-ergy,undercontract DE-AC-03-76SF00098.
LITERATURECITED
1. Arrigo, S.,M.Yun, and K. Beemon. 1987.Cis-actingregulatory elements within gag genes of avian retroviruses. Mol. Cell. Biol. 7:388-397.
2. Bender, M. A.,T. D. Palmer, R. E.Gelinas, andA. D. Miller. 1987. Evidence that the packaging signal of molony murine leukemia virus extends into the gag region. J. Virol. 61:1639-1646.
3. Bissell, M. J., D. Farson, and A. S. C. Tung. 1977.Cell shape
and hexose transport in normal and virus-transformed cellsin culture. J. Supramol. Struct.6:1-12.
4. Blochlinger, K., and H. Diggelmann.1984. HygromycinB phos-photransferase as aselectable marker for DNAtransfer exper-iments with higher eucaryotic cells. Mol. Cell. Biol. 4:2929-2931.
5. Chambers, J. A., A. Cywinski, P.-J. Chen, and J.M. Taylor. 1986. CharacterizationofRous sarcomavirus-relatedsequences in theJapanese quail.J. Virol. 59:354-362.
6. Coffin,J. 1982. Structure of the retroviralgenome, p. 261-368. In R. Weiss, N. Teich, H. Varmus, and J. Coffin (ed.), RNA tumor viruses, vol. 1. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
on November 10, 2019 by guest
http://jvi.asm.org/
7. Cone, R. D., and R. C. Mulligan. 1984. High-efficiency gene transfer into mammalian cells: generation of helper-free recom-binant retrovirus with broad mammalian host range. Proc. Natl. Acad.Sci. USA 81:6349-6353.
8. Cooper, G. M., and S. Okenquist. 1978.Mechanism of transfec-tion of chicken embryo fibroblasts by Rous sarcoma virus. J. Virol. 28:45-52.
9. Dolberg, D. S., and M. J. Bissell. 1984. Inability of Rous sarcomavirus to cause sarcomas in the avian embryo. Nature (London) 309:552-556.
10. Dolberg, D. S.,R. Hollingsworth, M. Hertle, and M. J. Bissell. 1985.Wounding and its role in RSV-mediated tumor formation. Science 230:676-678.
11. Eglitis, M. A., P. Kantoff, E.Gilboa, and W. F. Anderson. 1985. Gene expression in mice after high efficiency retroviral-medi-ated genetransfer. Science 230:1395-1398.
12. Fincham, V. J., and J. A. Wyke. 1986. Localization of temper-ature-sensitive transformation mutations and back mutations in the Rous sarcomavirus src gene. J. Virol. 58:694-699. 13. Frisby,D. P., R. A.Weiss, M. Roussel,and D. Stehelin. 1979.
The distribution of endogenous retrovirus sequences in the DNA of galliform birds does not coincide with avian phyloge-netic relationships. Cell 17:623-634.
14. Gilboa, E., M. A. Eglitis, P. W. Kantoff, and W. F. Anderson. 1986. Transfer and expression of cloned genes using retroviral vectors. Biotechniques 4:504-512.
15. Graham,F.L., andA.J.Van der Eb.1973.A newtechnique for theassayofinfectivity of human adenovirus 5 DNA. Virology 52:456-467.
16. Jorgensen, R. A., S.J. Rothstein, and W. S. Reznikoff. 1979. Restriction enzyme cleavage map ofTnS and location of a region encoding neomycin resistance. Mol. Gen. Genet. 177:65-72.
17. Katz,R.A., R. W.Terry,and A. M.Skalka. 1986.Aconserved cis-acting sequence in the 5' leader of aviansarcoma virus is required for packaging.J. Virol.59:163-167.
18. Kawai, S., and T. Koyama. 1984. Characterization of a Rous sarcoma virus mutantdefective in packaging itsowngenomic RNA:biologicalproperties ofmutantTK15 andmutant-induced transformants. J. Virol. 51:147-153.
19. Levantis, P.,D. A. F.Gillespie,K.Hart,M.J. Bissell, and J.A. Wyke. 1986. Control ofexpression ofan integrated Rous sar-coma provirus in rat cells: role of 5' genomic duplications reveals unexpectedpatternsof genetranscription and its regu-lation. J.Virol. 57:907-916.
20. Lipsick, J. S., C.E.Ibanez,and M. A. Baluda.1986.Expression of molecular clones ofv-myb in avian and mammalian cells independently of transformation. J. Virol. 59:267-275. 21. Maniatis, T.,E. F. Fritsch, and J. Sambrook. 1981. Molecular
cloning: alaboratory manual. Cold Spring Harbor Laboratory, ColdSpringHarbor, N.Y.
22. Mann,R.,R.C.Mulligan,and D. Baltimore.1983.Construction ofaretroviruspackaging mutant and itsuse toproduce helper-free defective retrovirus. Cell 33:153-159.
23. McNight, S., E. R. Gavis, R. Kingsbury, and R. Axel. 1981. Analysis of transcriptional regulatory signals of the HSV thy-midinekinase gene: identification ofanupstream controlregion. Cell 25:385-398.
24. Miller, A. D.,andC. Buttimore. 1986. Redesign of retrovirus packaging cell lines to avoid recombination leadingto helper
virusproduction. Mol. Cell. Biol. 6:2895-2902.
25. Moscovici,C.,M.G. Moscovici,H.Jiminez,M. M. C.Lai, M. J. Hayman, and P. K. Vogt. 1977. Continuous tissue culture cell lines derived from chemically inducedtumorsof Japanesequail. Cell11:95-103.
26. Mulligan, R. C., and P. Berg. 1980. Expression ofabacterial gene inmammalian cells. Science 209:1422-1427.
27. Nishizawa, M., T. Koyama, and S. Kawai. 1985. Unusual features of the leader sequence of Rous sarcoma packaging mutant TK15. J. Virol.55:881-885.
28. Norton,P. A., and J. Coffin.1985. BacterialP-galactosidaseas amarker of Rous sarcomavirusgene expression and replica-tion. Mol. Cell. Biol. 5:281-290.
29. Price, J.,D.Turner, and C. Cepko. 1987.Lineage analysis inthe vertebratenervous system by retrovirus-mediated gene trans-fer. Proc.Natl.Acad. Sci. USA84:156-160.
30. Pugatsch, T.,and D. W.Stacey. 1982.Analysis by microinjec-tion of the biological effects of site-directed mutagenesis in cloned avian leukosis viral DNAs. J. Virol. 43:503-510. 31. Pugasch, T., and D. W. Stacey. 1983. Identification of a
se-quence likely to be required for avian retroviral packaging. Virology 128:505-511.
32. Robertson, E., A. Bradley, M. Kuehn, and M. Evans. 1986. Germ-line transmission of genes introduced into cultured pluri-potential cells by retroviral infection. Nature (London) 323:445-448.
33. Robinson, H. L., S. S. Reinsch, and P. R. Shank. 1986. Se-quences near the 5' long terminal repeat of avian leukosis virusesdetermine the abilitytoinduceosteopetrosis. J. Virol. 59:45-49.
34. Shank, P. R., and M. Linial. 1980. Avian oncovirus mutant (SE21Q1b) deficient in genomic RNA: characterization ofa
deletion in theprovirus. J. Virol. 36:450-456.
35. Sorge, J.,andS. H.Hughes. 1982. Polypurine tract adjacentto the U3 region of the Roussarcoma virus genomeprovides a cis-acting function. J. Virol. 43:482-488.
36. Stoker, A.W., and M.J. Bissell. 1987. Quantitative immuno-cytochemical assay for infectious avian retroviruses. J. Gen. Virol. 68:2481-2485.
37. Turner,D.L.,and C. L.Cepko. 1987.Acommonprogenitor for neurons and glia persists in rat retina late in development. Nature(London)328:131-136.
38. Varmus, H., and R. Swanstrom. 1982. Replication of retrovi-ruses, p. 369-512. InR. Weiss, N. Teich, H. Varmus,andJ. Coffin (ed.), RNA tumorviruses, vol. 1. Cold Spring Harbor Laboratory, Cold Spring Harbor,N.Y.
39. Vennstrom, B., L. Fanshier, C. Moscovici, andJ. M. Bishop. 1980. Molecular cloning of the avian erythroblastosis virus genome and recovery of oncogenic virus by transfection of chicken cells. J. Virol. 36:575-585.
40. Wagner,E.F.,M.Vanek,and B.Vennstrom. 1985. Transfer of genes into embryonal carcinoma cells by retrovirus infection: efficient expression from an internal promoter. EMBO J. 4:663-666.
41. Watanabe, S.,and H. M. Temin. 1983.Construction ofahelper cell line foravian reticuloendotheliosis virus cloning vectors. Mol. Cell. Biol.3:2241-2249.
42. Williams, D. A., S. H. Orkin, and R. C. Mulligan. 1986. Retrovirus-mediated transfer of human adenosine deaminase gene sequencesinto cells in culture and intohematopoieticcells invivo. Proc. Natl. Acad.Sci. USA83:2566-2570.