0022-538X/89/010250-09$02.00/0
Copyright © 1989, American Society for Microbiology
The Gene
Encoding the
glll
Envelope Protein of Pseudorabies
Virus Vaccine Strain Bartha Contains
a
Mutation
Affecting Protein Localization
A. K. ROBBINS, J. P. RYAN,t M. E. WHEALY, AND L. W. ENQUIST* Central Research & Development Department, E. I. du Pont de Nemours & Co.,Inc.,
Experimental
Station, E3281B31,
Wilmington,
Delaware 19898 Received 13July 1988/Accepted 9 September 1988Pseudorabies virus(PRV)vaccine strain Bartha hasa diminishedcapacity tocausedisease andharborsa
varietyof mutationsaffecting virulence. It has beenreportedthatPRVBarthaproducesvirions withreduced amountsof the major envelope glycoproteingIH. Onehypothesiswasthatthisphenotypewasdue toreduced
expression of thegIIIgene. Inthis report,wedemonstrate that the reduced amount ofgIllinvirionswasnot mediated at the level of transcription, but rather reflected a defect in protein localization. We describe experimentswithgenereplacementtechnologytoprovethat theexpressiondefectwascloselylinked to thegIII gene itself. Using pulse-chase experiments, we found a defect similar to that observed for certain signal sequence mutations of PRV Becker gIII. The Bartha gIII protein was translated, but was inefficiently
introduced into the membrane proteinexportpathway. Consequently, onlyafraction of theprimaryBartha gIll translation product was glycosylated and matured. The remaining fraction stayed presumably in the cytoplasm,where itneverbecameglycosylatedorinserted into cellor virus membranes. The resultwasthat
Bartha-infected cells producedvirions with reduced amounts ofgIIIin theirenvelopes.Comparisonof theDNA sequenceof thepromoterand amino-terminalcoding regionsof Becker and BarthagIllgenesrevealedasingle
base pair difference inBartha, changing codon 14 of the signalsequencefrom aleucine (CTC)toa proline
(CCC) codon.We suggest thatthesignalsequencemutation isresponsiblefor theapparent reducedexpression
phenotype of this attenuated strain. This mutation represents, toourknowledge, the firstreportednatural signalsequencemutationinaherpesvirusglycoprotein.
Pseudorabies virus(PRV)isan alphaherpesviruscausing
anaturalinfection in swine similartothatofherpes simplex virus in humans (4, 8). Because the disease in swine is of
economic importance, there has been considerable effortto
control thisproblem through vaccinationprograms. Accord-ingly, there hasbeen an increased efforttodevelop
attenu-atedlive vaccine strains. TheseattenuatedPRVstrainshave
been studied in detailbyanumberof laboratoriesto under-standthegeneralmechanisms ofherpesvirus virulence (1-3,
5, 7, 10, 11, 13, 14, 20, 21).
PRV strain Bartha (PRV-Ba) is a well-knownattenuated
vaccine strain (1) and is thebasis for the Duvaxyn vaccine sold in Europe (21). Although this strain grows well in
culture, itharbors a number of mutations, some of which
have been shown to contribute to its avirulent phenotype. Forexample,PRV-Bacontainsadeletionintheuniqueshort
region removing coding sequences for glycoproteinsgI and
gp63 (11, 14). Mettenleiteretal. (13) proved that glycopro-teingI played amajorrole in virulence but that itdid soin
conjunctionwithatleast one otherundefined viral function. These workers also demonstrated that the absence of gI
affects virus release from certaincelltypes (12).Lomniczi et al. (10) noted that theBamHI4fragment of PRV-Ba
encod-ing fourgenesinvolved in capsid assembly contained defects
contributingtothe lack of virulence.
Thesubject of this report is the characterization of another
defect in PRV-Ba, the reduced amount of glycoproteingIll found in the virusenvelope (2, 20). To explain this
observa-*Correspondingauthor.
tPresent address: Department of Microbiology, The Health ScienceCenter, Universityof Tennessee, Memphis, TN 38163.
tion, Ben-Poratetal. (2) havesuggested that PRV-Ba gIII is expressed in lesseramounts. We describeexperimentsusing genereplacementtechnologytoprovefirst that the
expres-siondefect iscloselylinkedtotheglllgeneitself and second thatitisnotmediatedatthelevel oftranscription,but rather
reflectsadefect inproteinlocalization, mostlikelyduetoa signal sequence mutation. Given that the gIll protein plays animportantrolein virusadsorption (20)and release(18,20, 22), aswell asbeing a major immunogen (2, 9), it is likely that the defective localization phenotype is yet another
deficiency contributing totheavirulence of PRV-Ba. MATERIALS AND METHODS
Cells, virus, and DNA. The growth and properties of the Becker strainofPRV(PRV-Be) and theporcine kidneycell line PK15 have beenpreviously described(16). PRV-Ba and PRV strain Ka (PRV-Ka) were generous gifts from T. Ben-Porat.
Cloning of the PstI fragments containing the gIll gene from PRV-Be, PRV-Ba, and PRV-Ka intoEscherichia coli pBR322plasmidswasdoneasdescribed previously(17). The structure ofeachplasmid was verified by restriction
endo-nuclease analysis.
Construction ofisogenic setofPRV-Be strains containing
gIlI gene ofPRV-Ba and PRV-Ka.Recombinantviruseswere obtainedby the calcium phosphate cotransfection and gene replacement techniques described previously (17, 18). Fig-ure1outlines the gene replacement strategy. Briefly, E. coli
plasmids containingthe4.3-kilobasepair (kbp) PstI fragment
harboringthe
gIll
genewerecleavedwith PstI and cotrans-fected with PRV10 DNA. PRV10 is a virus lacking a func-tional gIll gene (17). Itcontains adeletion ofa 1,480-base-250on November 10, 2019 by guest
http://jvi.asm.org/
gillmRNA
Pstl Xhol Xhol Sacl Sacl Xhol
I
(ClonedPRV-gIIl Gene) ` /
Pstl Xhol Xhol Xhol Psti
-/
I_I_I___
PRV10: aglll Deletion
FIG. 1. Construction of isogenic derivatives of PRV-Be carrying theglll gene of PRV-Ba and PRV-Ka. The topline indicates the cloned4.3-kbp PstI fragment from PRV-Be, PRV-Ba, orPRV-Ka.
Relevant restrictionenzymecleavage sites areindicated. The glll
gene ofPRV-Ka contains an additional Sacl cleavage site. The
shaded boxdelineates theglll-codingsequences.Thearrowabove thebox indicates the approximate startsite, direction of transcrip-tion, andstopsite of the gIll mRNA. The bottom line indicates the homologous regiononthe glll nullmutantvirus, PRV10 (17). The
gll mutation is a deletion of the indicated XhoI fragment. The
potentialcrossoversrequired forgenereplacement and
reconstruc-tion ofanactiveglllgene areindicated by largeXs.Themethod for
genereplacement is described in Materials and Methods.
pair (bp) XhoI fragment removing 230 bp of the upstream glll promoter region and 87% of the gIll coding sequence.
PRV10 produces no detectable glll protein and forms a
nonreactive orwhite plaque in the black-plaque assaywith
glll-specific antibodies followed byaperoxidase-linked
sec-ond antibody (17). In the same assay, Ml antibody gives reactiveorblack plaques for the parentalPRV-Be, PRV-Ba,
and PRV-Ka. After cotransfection, virus plaques were
screened in the black-plaque assay for production of the
parentalglll protein. With monoclonal antibody Ml, black plaques werefound atfrequencies of 5% or more from all three cotransfection lysates, and one such black-plaque
recombinant from each was picked for further analysis.
Viruseswere namedas follows: PRV20(glllgene of
PRV-Be), PRV21(gIIIgeneof PRV-Ka), and PRV22(glllgeneof
PRV-Ba). In these three recombinant viruses, we can be confidentthatatleastthe1,480-bpXhoIfragment lackingin PRV10 has been replaced by DNAprovided by the cloned PstI fragment. It is highly likely that more DNA upstream and downstream of the XhoI fragment also has been
re-placed.
DNA and RNA analysis. Viral DNA preparation and Southern blot analysis of viral DNA with a gIII-specific
32P-labeled probe were performed as described previously
(15, 17). ForDNA sequencing, a 1.1-kbp XhoI-KpnI
frag-ment containing the putative promoter and 5'-coding
se-quence of glll was cloned into M13mpl8. The dideoxy
method (19) was used to sequence the glll promoter and signal sequence coding region with asynthetic
oligonucleo-tideprimerthathybridized upstreamof thegene (6). Theisolation of totalcytoplasmicRNAfrom infected cells andsubsequentNorthern(RNA)and slot-blotanalysiswere
conducted aspreviously described (6, 17).
The invitrotranslation of totalcytoplasmicRNAobtained fromPRV-infectedcells has been describedpreviously (17). Translatedextractswereresolvedbysodiumdodecylsulfate (SDS)-polyacrylamide gel electrophoresis or used for the immunoprecipitation ofglll-specific proteins with 282 anti-serum(18).
Fractionation and immunoprecipitation of PRV glycopro-teins. Forglucosamine labelingofglycoproteins,PK15cells
were infected at a multiplicity of infection of 10 with the
indicated viruses and grown throughout a 16-h infection in mediumcontaining 100,uCi of [3H]glucosamine per ml. For
cysteine labeling of proteins, a similar protocol was used exceptthat 100,uCi of [35]cysteine per ml was present from 6 to 16 h postinfection. Infected cell, virion, and medium fractionation ofPRV-infected PK15 cells was performed as previously described (18). The preparation of infected cell extractsand the immunoprecipitation procedure have been described previously (18). As indicated in the text, some samples were denaturedby boiling priortoadding antibody
to improve 282 serumreactivity.
Antibody reagents. The antibodies used in these studies included mouse monoclonal antibodies Ml and M16 (reac-tiveonlyagainstthe nativeglycosylatedforms ofgIll; these
antibodies do not react with in vitro-translated gIll or intracellular nonglycosylated gIll [6, 9]), goat polyvalent
anti-gIll antiserum 282(reactivewith both native and dena-tured forms of glycosylated or nonglycosylated gIll [18]), and goatpolyvalentanti-gIIantiserum 284 and mouse
mono-clonalantibodyM3(bothreactive onlyagainstthe gIIfamily ofglycoproteins [9, 18]).
Pulse-chaseanalysis ofgIII protein. The pulse-chase pro-cedure used has beendescribedpreviously (18; J. P. Ryan, M. R.Whealy, A. K.Robbins,C. L. Keeler, Jr.,andL. W.
Enquist, UCLA Symp. Mol. Cell. Biol., in press). Briefly,
PK15cellswereinfectedatamultiplicity ofinfection of10, andat 4 hpostinfection, cells were pulse-labeled for2 min with 100 ,Ci of
[35S]cystine
per ml. The radiolabel wasremoved, and the cells were incubated in the presence of
excess nonradioactive cysteine and methionine for various times. At the desired chase times, media (containing re-leasedvirus) andinfected cellfractions wereharvested and thegIIIspecieswasimmunoprecipitated with 282serum. In these experiments, the extracts were denatured prior to
immunoprecipitationwith 1.0%SDS and 10mM
dithiothrei-tol.
Black-plaque assay. The black-plaque assay has been
described previously(6).
RESULTS
Rationale.Wesoughttodetermine thecauseofthe appar-entlow
gIll
expression by attenuated vaccine strain PRV-Ba. Wereasonedthatsuchanexpressiondefect could either be linked to the gIII gene itselforbe caused byone ofthe manyknown defects ofPRV-Ba. We therefore deleted the gIII gene andits promoterfromPRV-Be,astraincontainingawell-characterizedandfully expressedgIII gene(6, 15-18,
22; Ryanetal.,inpress) andreplacedit withthe
gIl-coding
region of PRV-Ba. As controls, we also replaced PRV-Be gIII gene with the parental PRV-Be
gIll
gene and thegIll
gene of the widely used PRV-Ka strain. If the reduced expression phenotype was linkedto the gIII gene, itwouldmanifestitselfin therecombinant virus.
If thedefectwaslinkedto
gIll,
it could be duetoreduced transcription or RNAaccumulation,
reduced translation efficiency,poorimmunoreactivity owing
toalterations inthegIll
proteinitself,or someposttranslationaldefectincluding proteolysis or improper protein localization. Using theseisogenic strains, we
performed
a setofexperiments
exam-iningthesteady-state levelsofgIll-specific
RNA,theability
of this RNAtobetranslated invitro,
thesteady-state
level,
location, and immunoreactivity of
gIll
proteins,
and thepulse-chasekinetics of the gIII
primary
translationproducts.
Constructionofan isogenicsetof PRV-BestrainswithgIIIgenes from PRV-Ka and PRV-Ba. The gene replacement
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.55.293.76.171.2]strategyis describedinFig. 1and inMaterialsand Methods. PRV-Be, PRV-Ka, and PRV-Ba all expressed detectable amounts ofgIllon the surface of infected cells since these
viruses formed black plaques with glll-specific Ml mono-clonal antibody. We used this observation to screen for
isogenic viruses with the desiredgIII genereplacementafter
cotransfection ofPRV10(17) with cloned fragments
encod-ing theglllgenes ofPRV-Be, PRV-Ba, PRV-Ka. Thehigh frequency ofgene replacement
(>5%)
reflected the stronggrowth advantage of viruses expressing gIII over viruses
lacking a functional gIII gene (22). The isogenic set of viruses derived was PRV20 (gIII geneofPRV-Be), PRV21
(gIllgene ofPRV-Ka), and PRV22 (gIll gene ofPRV-Ba). The viral DNA was digested with a variety of restriction
enzymesand analyzed by Southern blothybridizationwith
the
32P-labeled
4.3-kbpPstIfragment ofPRV-Be. Withtheexception ofSacI, no restriction fragment polymorphisms
were detected (data not shown). BothgIII-Be (PRV-Be or
PRV20) and gIII-Ba (PRV-Ba or PRV22) contained two
apparently identicalSacl sites within thebody ofthe gene,
while gIII-Ka (PRV-Ka or PRV21) contained an additional
Sacl site. Fragments from all viruses hybridizing to the PRV-Be gIIIprobe appeared with the same intensity,
indi-catingno major sequence divergence. There were no
obvi-ousdeletionsorrearrangementsofthe4.3-kbpPstIfragment
amongtheviruses.
Analysis of total cytoplasmicRNAfrom PRV-infected cells. To determine whether transcription of the gIII gene was
altered in PRV-Ba or PRV22, we studied the quantity and
quality of
glll-specific
RNA by using Northern blot andslot-blottechniques. Total cytoplasmicRNAwasextracted fromPK15cellsinfected withtheindicated viruses. Estima-tionof the quantity of
glll-specific
RNAwas accomplished with a slot-blot format. The purified RNA (10 ,ug) wasserially
dilutedtwofold, denatured,andtransferredto nitro-cellulose membranes inaslot-blotapparatusasdescribedinMaterials and Methods. The quality of
glll-specific
RNA was determined by Northern blotting. For these studies, 5 ,ug of each RNA sample was analyzed as described inMaterialsandMethods.Theradioactiveprobeusedfor both
blots was an NcoI-BamHI fragment containing only gIII gene sequencesfrom PRV-Be.
Autoradiograms ofthe slotblots
(Fig.
2) are shown. Theslot-blot analysis indicatedthat the amount ofsteady-state gIII-specific RNA was nominally the same for all viruses tested. The experimenthas been repeatedusing RNA
pre-pared at different times after infection with similar results (datanotshown). The Northern blotanalysis indicatedthat anidentically sized species of gIII-specificRNAcomigrating with the 1.55-kilobase PRV-Be RNA was found for all
viruses (datanot shown).
In vitro translation of total RNA extracted from infected PK15cells.The sameRNAanalyzed by slot-blot and North-ern analysis was used for in vitro translation experiments (Fig. 3). RNA from uninfected cells was included as a control. The polypeptides translated in vitro were labeled
with [35S]methionine. Either in vitro-translated samples werefractionateddirectly on a 10% polyacrylamide-SDS gel
(Fig.3, left panel), or gIII-specific translation products were
immunoprecipitated with polyvalent 282 serum and then
fractionatedon a10% polyacrylamide-SDS gel (Fig. 3, right
panel). Translationproducts from uninfected cell RNA are in
lanesmarked C.
By examining the intensity of bands in the
nonimmuno-precipitatedsamples (seven lanes on the left), it is clear that all samples received approximately the same amount of
tt K,,i R i 20 2 1 22
mom_, m .M~ mum
_ __
_.
-. - 4p1
FIG. 2. Slot-blotanalysis ofglll-specific RNA. Total cytoplas-mic RNA was extracted from PRV-Be-, PRV-Ka-, PRV-Ba-, PRV20-, PRV21-, or PRV22-infected PK15 cells as described in Materials and Methods. A10-p,gsample of eachRNAwasserially dilutedtwofold, denatured, and subsequently transferred to nitro-cellulosein aslot-blot apparatus. The probe used was a 1,400-bp NcoI-BamHIfragment from pALM3 (17)thathybridizedonlytothe body of the glll gene. Lanes: Be, PRV-Be; Ka, PRV-Ka; Ba, PRV-Ba; 20, PRV20; 21, PRV21;22,PRV22.
RNA, althoughthe pattern forPRV-Ba isdistinctlydifferent fromthat forPRV-Be orPRV-Ka. Fortheisogenic strains, PRV20, PRV21, and PRV22, the patternswere identicalto thatfound forPRV-Be,asexpected.Itisnoteworthythata novelprotein product tightly linked to gIII appeared to be alteredinbothPRV-KaandPRV-Ba.This is best seen in the left panel comparing the last three isogenic samples. The
profilesareidentical except for theindicated band(shownby adot) migrating slightly fasterfor PRV21 and PRV22, but notPRV20. The identity of this proteinis unknown.
Itis clear thatfor all viruses,282 antiserum immunopre-cipitated proteins of approximately 58-kilodaltons, charac-teristic of thepredicted 479-amino-acid gIII primary trans-lationproduct (15, 17). However,forPRV-Ka, PRV-Ba,and theisogenicstrainsPRV21 andPRV22,theprimary transla-tionproduct migrated slightlyfaster than that from PRV-Be orPRV20. This may reflect differencesin amino acid com-position or may indicate that the genes contain small in-frame deletions. Comparedwith the amountof PRV-BegIII
immunoprecipitated by282 serum,bothPRV-Ba and PRV22 RNA yielded about two- to threefold-reduced amounts of immunoreactive gIII. Given that theamountsofgIII-specific
RNA in these preparations were essentially identical (Fig. 2), this result suggests a lower translation efficiency ofthe PRV-Ba gIII message in the rabbitreticulocyte system.
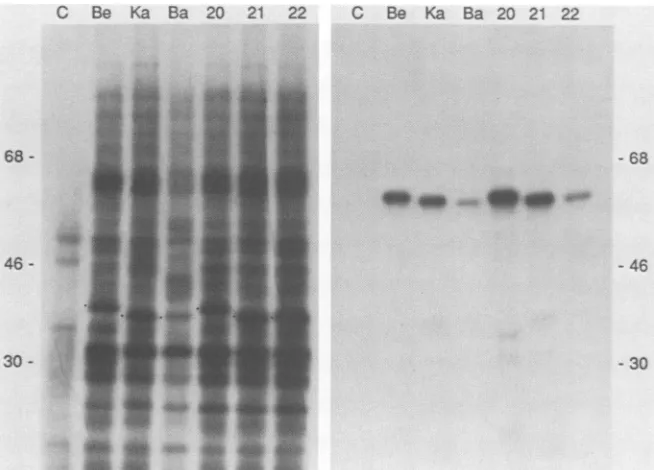
Immunoprecipitation of glucosamine-labeled PRV-specific
proteins fromvirus particles. PK15 cells were infectedwith
PRV-Be, PRV-Ka, PRV-Ba, PRV20, PRV21, orPRV22 as described in Materials and Methods. [3H]glucosamine was added at the time of infection to label the glycoproteins.
After 16 h, medium containingextracellular virus particles
was collected and the virus was purified by centrifugation
and analyzed as described in Materials and Methods.
Re-maining infected cellswere used in thesubsequent
experi-ment. Thepurifiedvirusparticles wereanalyzed directlyor
lysed and reactedwith gIII- orgll-specific antibodies. The
immunoprecipitates were collected and fractionated on a
10% polyacrylamide-SDS gel. The3H-labeled polypeptides detectedby fluorography aredisplayedin Fig. 4.
ThegIII defectof PRV-Ba is demonstrated inFig.4Awith the Ml monoclonal antibody. Reduced amounts of the mature, 92-kDa form ofgIll were observed in PRV-Ba as
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.337.547.76.217.2]C Be Ka Ba 20 21 22 C Be Ka Ba 20 21 22
68
-46
-30
--68
-46
-30
FIG. 3. In vitro translation of total cytoplasmic RNA extracted from PRV-infected cells. Total RNA from mock-infected and PRV-infected cellswasharvested at 16 h postinfection, and equal amounts were translated in a rabbit reticulocyte system asdescribedin Materials and Methods.Aportion of these products wasimmunoprecipitated with polyvalent 282 serum. Both the total translation mixture (left panel) and theimmunoprecipitate (right panel) of each sample were fractionated on a 10% polyacrylamide-SDS gel.[35S]methionine-labeledpolypeptides werevisualized by fluorography. The dot adjacent to bands from the total translation mixture indicates a novel,non-glll-relatedtranslation productdiscussed in the text. Lanes: C, mock-infected control; Be, PRV-Be infected; Ka, PRV-Ka infected; Ba, PRV-Ba infected; 20, PRV20 infected; 21, PRV21 infected; 22, PRV22 infected. Numbers on the sides show molecular mass in kilodaltons.
compared with PRV-Be or PRV-Ka. The defect was
elabo-rated, if not more severe, in the isogenic derivative PRV22
(compare PRV22 with PRV20 or PRV21) and therefore is
tightly
linkedtothePRV-BagIl geneitself.Note that in thisexperiment, fewer PRV-Be virions were recovered after purification. Thiscanbededucedby examining the intensity
of the bandsinFig.4D, the controlprofiles ofthegll protein immunoprecipitated fromthe samesample, or, from Fig. 4E,
thetotalglucosamine-labeled profilesofpurifiedvirus parti-cles.
It hasbeen reportedthat PRV-Ba-encoded gIII does not reactwithasecond
glll-specific
monoclonalantibody, M16(2).This lack of reactivity isclearlydemonstratedinFig. 4B for both parental PRV-Baandthe isogenic strain, PRV22.
Figure 4C shows the reactivity with polyvalent 282 anti-serum. Note that PRV-Ba anditsisogenic derivativePRV22
reacted with thisserum,althoughthe amountofgIIIprotein precipitated wasreduced. Thisreductionisbest seen in the
comparison oftheisogenic strains.
Figure 4D is a control immunoprecipitation of the gIl family of glycoproteins from the same virion samples to
indicate efficiency of infection andsample recovery.
TheexperimentinFig.4Bindicated that thePRV-Ba gIII
protein lacked the M16 epitope. This observation could
indicate that the PRV-Ba glll protein, rather than being poorly expressed, was simply less immunoreactive in gen-eralwithgIII-specific antisera. However,the data inFig.4E
strongly suggested that the gIII protein of PRV-Ba was present inphysically lower amounts in purified virions. In this experiment, purified [3H]glucosamine-labeled virions werefractionated directlyon a10%polyacrylamide-SDS gel
withno immunoprecipitation. Comparethe last three lanes
containing the isogenic strains and note the reduction of
intensityof the92-kDaspecies for PRV22 and PRV-Ba.
We conclude that the apparent reduced expression phe-notype ofgIll seenfor PRV-Ba is tightlylinked to thegIII gene, and in addition, we corroborate the observations of Ben-Porat et al. (2) that the PRV-Ba gIII gene encodes a mutant glycoprotein with altered immunoreactivity to the M16monoclonal antibody.
Immunoprecipitation ofglucosamine-labeled, PRV-specific proteins from infected PK15 cells. In the previous experi-ment, we showed that PRV-Ba glll protein is reduced in virus particles. The next set of experiments examined the infected cells from the sameinfection. The experimentwas doneasdescribedabove. Afterremoval ofthemedium, the infected cells were lysed (see Materials and Methods) and reacted with gIII- orgll-specific antibodies. The immuno-precipitateswerecollected andsubsequently fractionatedon a10%polyacrylamide-SDS gel. The 3H-labeled polypeptides detectedby fluorography aredisplayedinFig.5.
Figure5A shows that allvirusesproduced matureglllin infected cells reactive with Ml monoclonal antibody, al-though PRV-Ba and PRV22 consistently gave lower amounts. The faster-migrating band of 62 kDa, more pre-dominant inPRV-Ba-infected cells(indicatedbyadot),was seenin theother infectionsaswell afterlongerexposures.It was not seen with M16 monoclonal antibody or with 282 polyvalent serum. It could also be precipitated from [35S]cysteine-labeled cell extracts by usingonly Ml mono-clonalantibody(seebelow). ItsrelationshiptogIll,ifany,is
currently notunderstood.
The M16 monoclonal antibody recognizes not only the mature92-kDa form ofgIII, but also the 74-kDaprecursor (18). Although PRV-Be, PRV-Ka, and theirisogenic deriv-ativesgaveindistinguishablepatternsofimmunoprecipitated
glllprotein,PRV-Ba andPRV22hadno detectable
reactiv-itywith thisantibody (shown inFig. SB).
so
so.40
*R.
0on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.140.467.75.310.2]13: Ka F:a:1 :" i
.... Ci a
f.;.1P
Be Ka Baz 20 21 22
--i10
_~~~~~~~~~~~~~- 1.
Wa! .WW.
::....C..
.,Wm
74--92..
.2
-AA. kllis0 _1
t@,, :!
[image:5.612.70.310.76.373.2]9-o
3
:iie-u
FIG. 4. Immunoprecipitation of [3H]glucosamine-labeled,
gIll-specific proteins from purified virions. PK15 cellswereinfected with
PRV-Be, PRV-Ka, PRV-Ba, PRV20, PRV21, or PRV22 in the
presence of[3H]glucosamine asdescribed in Materials and
Meth-ods.Extracellularvirus particleswerepurified by centrifugation and
analyzed with glll- and gll-specific antibodies. (A) Ml monoclonal antibody. (B) M16 monoclonal antibody. (C) 282 polyvalentanti-glll
serum.(D) M3 monoclonalantibody (gll specific, control). (E) Total
profile ofglucosamine-labeled virions priortoimmunoprecipitation. Immune complexes were collected by adsorption to Pansorbin (Staphylococcus aureus) and fractionated by electrophoresison a
10% polyacrylamide-SDS gel. 3H-labeled polypeptides were
de-tected by fluorography. Thevirus strain is indicated above the lanes in panels A and D and is in thesameorder in panels B, C, and E.
Numbers showmolecularmassinkilodaltons.
The polyvalent glll antiserum 282 reacts with both the precursor andthe matureform ofgIII fora variety ofgIll
mutants(6, 18; Ryanetal., inpress). The data given in Fig. 5C demonstrate that all viruses produced precursor and matureforms ofglll reactivewith thissera, although PRV-Ba and PRV22 clearly produced less 74-kDa protein. The criticalcomparisonis of theisogenic strainsinthe lastthree lanes of Fig. 5C. It is clear that PRV22 produced less immunoreactive precursor and mature glycosylated gIII proteinthan eitherPRV20orPRV21.
The control immunoprecipitations of gll glycoprotein (Fig. 5D) were similar, indicating that all infections had progressed essentially to the same state. The 100-kDa
pre-cursor form, not seen in purified virions (Fig. 4D), was
precipitated for all viruses. Wenotethat thegll profile from PRV-Ba, butnotPRV22,was slightly different qualitatively
andquantitatively from all other virusesasobserved
previ-ously (2), suggesting that PRV-Ba has mutations ingll as
well(see Discussion).
Immunoprecipitation of PRV-specific proteins from [35S] cysteine-labeled infected PK15cells. Asimilar setof
steady-FIG. 5. Immunoprecipitation of [3H]glucosamine-labeled,
gIll-specific protein from infected cellextracts.PK15 cellswereinfected withPRV-Be, PRV-Ka, PRV-Ba,PRV20, PRV21, orPRV22 in the
presence of[3H]glucosamine asdescribed in Materials and Meth-ods. The infected cell extracts were reacted with gIII- and
gIl-specific antibodies. (A) Ml monoclonal antibody. (B) M16
mono-clonal antibody. (C) 282 polyvalent anti-gIll serum. (D) M3 monoclonal antibody (gIl-specific control). Immune complexes
werecollectedby adsorptiontoPansorbin(S. aureus) and fraction-ated by electrophoresis on a 10% polyacrylamide-SDS gel.
3H-labeled polypeptides were detected by fluorography. The virus strain is indicated abovepanels A and D and is in thesameorderfor panels B and C. The 92- and 74-kDa forms ofgIll are indicated. Numbers on the right side of panel D show molecular mass in kilodaltons.
state labeling experiments was done with [35S]cysteine rather than [3H]glucosamine as the label. The experiment willdetect all stablegIlI-immunoreactive species, including those that are not glycosylated. Cells were infected with PRV-Be,PRV-Ba, orPRV22, labeled from 6to 16 h postin-fection as described in Materials and Methods, and subse-quentlyfractionated into released virions and infectedcells. Extracts of infected cells or virions were reacted with anti-gIII-specific antibodies, and immunoprecipitates were collected and then fractionated on a 10%
polyacrylamide-SDS gel. The 35S-labeled polypeptides were detected by
fluorography. The results with infected cell extracts are shownin Fig. 6.
Ml monoclonal antibodyimmunoprecipitated the mature 92-kDa form ofgIll from each infected cell extract. How-ever, both PRV-Ba- and PRV22-infected cells clearly con-tained lessimmunoreactiveprotein,consistent withprevious observationsfor[3H]glucosamine-labeled infected cells(Fig. 4). (The sharpband ofapproximately 92 kDapresentin Ml and M16 immunoprecipitates of infected cell extracts is a
coprecipitated, nonglycosylated, non-gIll-related protein [datanotshown].It is notseeniftheextracts aredenatured
priorto immunoprecipitation, aswasdone with 282 serum.
However, asnoted inMaterials andMethods, Ml andM16 monoclonal antibodies do not react with denatured gIll
protein, hence nondenatured extracts must be used in this experiment.)Abandof about 62 kDawas morepredominant
for PRV-Ba, but could be seen in other infections upon
longerexposures.Aspecies ofthesameapparentmolecular
mass was observed previously in [3H]glucosamine-labeled
-- 68
55 iii;; /.% 1. -)
Aik. .0":
...00
.4 40 A-m,
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.323.565.78.272.2]___ .A --M
e Ba £2 Ile Ba 2121 Be Ba 22
94-74-
I.
58-A
2 ..533
92
.74
..58
B 0 30 45 60 90 120 240 480 1N.0 I * ,-
-N
v.
.-..FIG. 6. Immunoprecipitation of[35S]cysteine-labeled PRV gly-coproteins. PK15 cells were infected with PRV in the presence of [35S]cysteine as described in Materials and Methods. Extracts of PRV-Be-, PRV-Ba-, or PRV22-infected cells were reacted with monoclonal antibodies Ml and M16 or polyvalent 282 serum as indicated. Immune complexes were collected by adsorption to Pansorbin (StaphA) and fractionated by electrophoresis on a 10% polyacrylamide-SDS gel.I'S-labeledpolypeptides weredetected by fluorography. Extracts from infected cells were denatured prior to immunoprecipitation with polyvalent 282 serum as described in Materials and Methods. Numbers show molecular mass in kilodal-tons.
92-7
4-
58-C
0 30 45 60 90 120 240 480i2
*w ^*infected cells (Fig. 4). As discussed, its relationship, if any, togIII is under study.
M16 monoclonal antibody immunoprecipitated only the
92-kDamatureform ofgIIIand the 74-kDa precursorspecies fromPRV-Be-infectedcells. This same monoclonal antibody
did not immunoprecipitate any gIII-specific proteins from
either PRV-Ba- or PRV22-infected cells, consistent with
observations with [3H]glucosamine-labeled infected cells (Fig. 4).
282 serum immunoprecipitated only the 92-kDa mature
form and the 74-kDaprecursorform ofglllfrom
PRV-Be-infected cells. In contrast, a significant fraction of the
282-reactive
"S-labeled
polypeptides in PRV-Ba- orPRV22-infected cells migrated with an apparent molecular mass of 58kDa. Thisspecieswasnotapparentin similarexperiments
with
[3H]glucosamine
labeling (Fig. 4). The normal 92-kDa matureform and the74-kDa precursorspecieswerepresent,but atreduced levels.
The profiles for [35S]cysteine-labeled virions were also
examined (data not shown). The results were essentially identical to those obtained for [3H]glucosamine-labeled
vi-rions (Fig. 4) in that both PRV-Ba and PRV22 virions
contained lesscysteine-labeled gIII than PRV-Bedid. Weconclude that the [3H]glucosamine-and [35S]cysteine-labeled
gIll
profiles from infected cellsareessentiallyiden-ticalwithonemajor exception.PRV-Ba-andPRV22-infected cells containanew,58-kDamajor specieslabeledonlywith
[35S]cysteine andreactive onlywith 282 serum. We suggest that this species is a product of the PRV-Ba glll gene
because it isproducedbyPRV22 and notPRV-Be.
Pulse-chaseanalysis ofgIllprotein. Thelong-term
labeling
experiment presented above defines stable gIII
species
whoseintensityoflabelingis dictated by synthesisrateand turnoverrate. Thefollowing experimentexamined therates ofsynthesis
andstability
of[35S]cysteine-labeled glll
pro-FIG. 7. Pulse-chase analysis of gIII species produced by PRV-Be,PRV-Ba, andPRV22. PK15cellswereinfectedwith10 PFUof theindicated virus percell andincubatedat37°C. The pulse-chase protocol is described in Materials and Methods.At 4 h postinfec-tion, cellswerepulse-labeled with[35S]cysteinefor2min and then incubated in the presence ofexcess nonradioactive cystine and methionine. Atthe times indicated above each lane (in minutes), cells were harvested. glll species wereimmunoprecipitated from infected cell extracts with polyvalent 282 serum (A, B, and C). Immunoprecipitates were resolved on a10%)o polyacrylamide-SDS gelandvisualized byfluorography. (A)PRV-Be; (B) PRV-Ba; (C) PRV22.
teins produced by thevirus strains and determinedwhether anytransient, processedforms of theprotein existedin the
infected cell that might not be detected under steady-state radiolabeling conditions. Ofparticular interest was the 58-kDa gIll species made by PRV-Ba and PRV22 seen in
steady-state [35S]cysteine labeling (Fig. 6), but not in [3H] glucosamine labeling (Fig.4and5).PK15cellswereinfected with PRV-Be, PRV-Ba, and PRV22. A pulse-chase
experi-mentwith [35S]cysteine wasperformedat4 hpostinfection
asdescribed in Materials and Methods.gIII-specificproteins were immunoprecipitated with polyvalent 282 serum. The resultsareshown inFig. 7.
Asnotedpreviously (6, 18; Ryanetal.,inpress),the initial
product ofglll translation observed in a 2-min pulse for PRV-Be was the 74-kDa species known tobe modified by high-mannose, N-linked
glycosylation
characteristic of aon November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.64.285.72.248.2] [image:6.612.359.511.78.437.2]location in the endoplasmic reticulum. Export of PRV-Be gIIIfrom theendoplasmicreticulum tothe Golgiapparatus
wasrapid and efficient; the 92-kDamatureformappearedas earlyas30min after pulse-labeling, and onlyasmall fraction
of the74-kDaprecursor remained aftera1-h chase.
The pulse-chase profile obtained for PRV-Ba or PRV22
was strikingly different. First, the predominant primary
translation product observed after the 2-min pulse was a
58-kDa form. The 74-kDa form was present but barely detectable. The small quantity of 74-kDa protein chased to themature92-kDaspecies with essentially identical kinetics
as PRV-Be-encoded gIII. The 58-kDa species remained
essentially constant during the first 2 h of the chase period but showed significant loss of signal toward the end of the 8-h chase. We were struck by the similarity between the PRV-Baand PRV22 pulse-chase profiles and those found for definedglll signalsequencemutantsof PRV-Be constructed by Ryanetal. (in press). Certain signalsequence mutations partially blockcotranslationalexportof the gIII protein from the cytoplasm to the endoplasmic reticulum. The 58-kDa form is believed to be the authentic, nonglycosylated -pri-marytranslation productasidentified by in vitro translation (6, 15, 17; Ryan etal., in press).
For PRV-Ba and PRV22, the intense labeling of the 58-kDa form compared with other forms of gIII is notewor-thy. We note that the amount of cysteine available to the cellsduring the 2-min pulse was not in excess (addition of more radioactive cysteine resulted in more incorporation [unpublishedobservations]). Therefore, the intensity of label inthe58-kDa speciessuggeststhat itwasbeingsynthesized
atanincreasedrate compared with the 74-kDaglycosylated precursor.
Weconclude from theseexperiments that the gIIIgeneof PRV-Baand PRV22 contains adefect giving risetoanovel
primary product of translation not characteristic of the membrane-boundglycosylated 74-kDaprecursor.Themajor species of gIIIobserved ina2-minpulseat4hpostinfection is a 58-kDaprotein with the sameapparent massas the in
vitro-translatedgeneproduct.This species is translatedatan
increasedrate compared with the normal74-kDaprecursor.
The reduced amount of normal 74-kDa precursor made is converted to mature 92-kDa product at the same rate as
normal gIllandappearswith time in virusparticles (datanot shown). The PRV-Ba 58-kDa aberrant gIII precursor is a
dead-end product and slowly turns over. We cannot com-paredirectly theamountof 58-kDaprecursor seenin
steady-statelabeling (Fig. 6) and the apparent amount seen in the
pulse-chase experiment. In addition, wehave notmeasured the half-life of the aberrant precursor at late times of infection.
DNA sequence analysis of promoter and amino-terminal coding regions of PRV-Ba gIll. The datafrom the pulse-chase experimentswere characteristic of the profiles observed for
certain defined signal sequencemutantsin the PRV-Be gIll gene (6; Ryan et al., in press). We examined the DNA sequenceof thepromoter and 5'region ofthe PRV-Baglll gene to determine whether we could identify a potential
signal sequence mutation. The gIll signal sequence was
localizedtothe amino-terminal 22amino acidsbyEnquistet
al. (6) and Ryanet al. (inpress).The DNA sequenceof the
PRV-Ba gIII promoter region and signal sequence was
determined from -60to +93 (+1 is the A ofthe initiator ATG codon)asdescribed in Materials andMethods (datanot shown). Theupstreamregions ofPRV-Be andPRV-BagIll including the predicted promoter were identical. However, we notedasingle base changeofaTtoaCatnucleotide 41
ofthe
gIll
gene.Thischange
in PRV-Ba convertedaleucinecodon
(CTC)
to aproline
codon(CCC)
in thepredicted
hydrophobic
coreoftheglll signal
sequence. Thiswas theonly base
change
in the signal sequence. We have notsequenced thewhole PRV-Ba
glll
gene,butnotethat there arefurther sequencechanges
in thebodyof the PRV-Baglll
geneoutsideof thesignal
sequenceregion.
Forexample,the nextchange identified inoursequencewas anA-to-Cchange
at nucleotide89ofthe
glll
gene. This wouldchange
codon 30 from anasparagine
codon(GAC)
to aglycine
codon(GCC).
DISCUSSION
PRV-Ba isanattenuated vaccinestrain
harboring
multiple
mutations. Many ofthese
changes
and their effecton PRVvirulence have been documented (3, 5, 7,
8,
10, 11, 13,14,
20,21).Onewell-knownbutpoorly characterized
phenotype
ofthePRV-Bastrain isthe apparentlow-level
expression
ofglycoprotein
gIll
(2).We used the
technique
ofgenereplacement
to compare the PRV-BagIIIgenewithtwonon-vaccine strainalleles in anisogenic
background.
Inthis way, wewere abletostudy
only those mutations
tightly
linkedto thegIII gene. Three PRV-BagIII defectswere definedin this manner. Thefirst,
a lack of
reactivity
with the M16 monoclonalantibody,
is well documented (2). Clearly, there are mutations in thebodyof the PRV-Ba
gIll
geneaffecting
the M16epitope.
We know little about the natureofthesemutations. The seconddefectis anapparenttwo- tothreefold-reduced
efficiency
ofin vitro translation ofPRV-Ba
glll
mRNA. At the moment, it is unclear whether this defect is manifested in vivo or ispeculiar
topurified
RNA translated in vitro. We note that DNA sequenceanalysis
revealed no alterations in the pro-moterregion
and 5'region
oftheglll
mRNA exceptasingle
basechange
at codon 14.The most
striking
defect of PRV-Baglll,
however, was novel precursorsynthesis
andsubsequent
effectsonprotein
localization.
Theprotein
had been shownpreviously
to be present in reduced amounts in virusenvelopes
(2).
Weconfirmed these observations and also demonstrated that there was no apparent defect in
expression
ofgIII-specific
mRNA. Akey
observation was that thepredominant
PRV-BagIll
translationproduct
detectedina2-minlabeling
with[35S]cysteine
was58kDa,
thepredicted
size oftheprimary,
unmodifiedgene
product.
This 58-kDaspecies
wasreason-ably
stable and couldeasily
be detectedaftera9-h,
steady-state
labeling experiment
with[35S]cysteine.
In contrast,the58-kDa
species
was notdetectedduring
a16-h, steady-state
labeling
experiment
with[3H]glucosamine.
Moreover, thepulse-chase analysis
indicated that the 58-kDaspecies
was translated at ahigher
rate than the normal 74-kDa glycosy-lated precursor. The 58-kDagIll
proteinisthereforenongly-cosylated
andsynthesized by
moreefficienttranslationma-chinery.
This phenotype can be best explained by another mutation in the PRV-BagIll
gene, the leucine-to-prolinechange
atposition
14 ofthesignal
sequence. We noted inpulse-chase experiments (Fig.
7)that theprofile forPRV-BagIll
corresponded remarkablywelltothatseenforadefinedsignal
sequence mutantdescribed by Ryanetal. (inpress). This mutation, 12R, altered theleucine atposition 12to anarginine.
Codons12and 14areboth in thehydrophobic
core of theglll signal
sequence. Both mutations wereleaky, in the sense that a fraction of thegIll
protein entered theprotein
export pathway to be processed and matured like normalglll
protein. However,themajority of pulse-labeledon November 10, 2019 by guest
http://jvi.asm.org/
12RorPRV-Ba
gIll
protein remained unglycosylated within the cell, most likely inthe cytoplasm. The small amount of maturegIIIproduced appeared on the cell surface, and some was incorporated into envelopes of egressing virions. There-fore, there is indeed a lower amount of gIll protein in PRV-Ba and PRV22 virus envelopes and cell membranes, but the cause is not reduced gene expression. Rather, because the gIII signal sequence is mutated, the primary gene product is localized improperly, and reduced amounts ofmaturegIll
protein are available for virion assembly. This mutation represents, to our knowledge, the first reported natural signal sequence mutation ina herpesvirus glycopro-tein.It is important to note that the site-directed gIll signal sequence mutation at codon 12 described by Ryan et al. (in press)affects the hydrophobic domain of the signal sequence by introducing a charged amino acid. The change in PRV-Ba
gIll
at codon 14 introduces proline, a neutral amino acid that will alter the predicted alpha-helix structure of this domain, but should have little effect on the hydrophobic content. Work is in progress to determine the role of the hydrophobic region ingIII signal sequence function (J. P. Ryan, M. E. Whealy, A. K. Robbins, and L. W. Enquist, manuscript in preparation).Why would such a specific defect occur during the process of making the attenuated vaccine strain? Is the signal se-quence mutation due to chance occurrence or could there be some rationale for its appearance? Most work must be done tounderstand the role ofgIIIin the life cycle of PRVbefore these questions can be answered. Nevertheless, it is clear that a signal sequence mutation represents one simple way to lower, but not totally deplete, the amount ofgIll protein in virus envelopes or on the surface of infected cells without affecting the mature protein structure. Reducing the amount of intracellulargIll that can enter the protein export pathway clearly leads to production of virions with reduced gIll in their envelopes. With defined signal sequence mutations, we can determine whether simply lowering the amount of ma-turegIII is sufficient for decreased virulence. By correcting the signal sequence mutation in PRV-Ba gIll, it will be possible todetermine whether the additional mutation(s) in the body of the gene affecting antibody recognition plays a role in virus release and virulence.
Schreurs et al. (20) suggested that roles in virus adsorption and release from infected cells constituted separate func-tions of thegIll protein. One line of evidence supportingthis hypothesis was that PRV-Ba, despite its reduced comple-ment of
gIll
in virions, absorbed normally to cells yet was defective in release of virus. The implication was that the PRV-Ba gIII protein was defective in one domain affecting release, while the adsorption domain remained intact. Our finding that the PRV-BagIII gene harbors a signal sequence mutation suggests an alternative testable hypothesis. Per-haps the processes of adsorption and virusreleasedifferwith respect to their concentration dependence on mature gIII. For example, the process of efficient virus absorption to cells could require very little gIll protein per virion, while the process of virus release could be more dependent on gIll concentration or could involve moregIll-dependent steps. A test of this hypothesis would be that reversion of the codon 14 mutation in PRV-Ba gIll (if it restored normal levels of gIII in the virus envelope) would result in a virus with normal kinetics of virus release.Our studies support and extend previous observations indicating that other alterations exist in PRV-Ba. Ingeneral, we found that PRV22 was more defective than the parental
PRV-BaorPRV-Bestrains in theglll phenotypesmeasured.
This suggests that the PRV-Ba glll protein functions well
only in the PRV-Ba background, perhaps indicating that
PRV-Ba has developed compensatory mechanisms to deal with its alteredgIIIprotein.As suggested by Ben-Porat etal.
(2),ourobservationsontheapparentincreasein theamount
of gIl present in PRV-Ba virions but not in PRV22 may reflect a compensatory alteration of the gIl protein to ac-commodate the lower level ofgIIIprotein (and the absence ofgI and gp63).
These observations are circumstantial evidence that PRV
glycoproteins are involved in multiple protein-protein inter-actions. Perhaps some of the alterations made specifically
affecting virulence in PRV-Ba resulted in compensatory
changesinotherproteinstomaintain functionalglycoprotein complexes. The gene replacement technology used in this report and the virulence rescue techniques used by Ben-Porat and co-workers (13) provide tools to test this idea.
ACKNOWLEDGMENTS
We thank the members of the Molecular Genetics group for advice and discussion; in particular, Logan Buckley provided criti-cal advice and encouragement.
LITERATURE CITED
1. Bartha, A. 1961. Experimental reduction of virulence of Au-jeszky's disease virus. Magy. Allatorv. Lapja 16:42-45. 2. Ben-Porat, T., J. M. DeMarchi, B. Lomniczi, and A. S. Kaplan.
1986. Role of glycoproteins of pseudorabies virus in eliciting neutralizing antibodies. Virology 154:325-334.
3. Ben-Porat, T., J.DeMarchi, J. Pendrys, R. A. Veach, and A.S. Kaplan. 1986. Proteins specified by the short unique region of the genome of pseudorabies virus play a role inthe release of virions from certain cells. J. Virol. 57:191-196.
4. Ben-Porat, T., and A. S. Kaplan. 1985. Molecular biology of pseudorabies virus, p. 105-173. In B. Roizman (ed.), The herpesviruses. Plenum Publishing Corp., New York.
5. Berns, A., A.Van Den Ouweland, W. Quint, J. van. Oirschot, and A. Gielkens. 1985. Presence of markers for virulence inthe unique short region or repeat region or both of pseudorabies hybrid viruses. J. Virol. 53:89-93.
6. Enquist, L. W.,C. L. Keeler, Jr., A.K. Robbins, J. P. Ryan, and M. E. Whealy. 1988. An amino-terminal deletion mutation of pseudorabies virus glycoprotein gIll affects protein localization and RNA accumulation. J. Virol. 62:3565-3573.
7. Geilkens, A. L., J. T. van Oirschot, and A. J. M. Berns. 1985. Genome differences among field isolates and vaccine strainsof pseudorabies virus. J. Gen. Virol. 66:69-82.
8. Gustafson, D. P. 1975. Pseudorabies, p. 391-410. In H. D. Dunne and A. D. Leman (ed.), Disease ofswine,4thed. Iowa State University Press, Ames.
9. Hampl, H., T. Ben-Porat, L. Ehrlicher, K. 0. Habermehl, and A. S. Kaplan. 1984. Characterization oftheenvelopeproteinsof pseudorabies virus. J. Virol. 52:583-590.
10. Lomniczi, B., S. Watanabe, T. Ben-Porat, and A. S. Kaplan. 1987. Genome location and identification of functions defective in the Bartha vaccine strainofpseudorabies virus. J.Virol. 61: 796-801.
11. Mettenleiter, T. C., N. Lukacs, andJ.-J. Rziha. 1985. Pseudo-rabies virus avirulent strains fail to express amajor glycopro-tein. J. Virol. 56:307-311.
12. Mettenleiter, T. C., C. Schreurs, F. Zuckermann, and T. Ben-Porat. 1987. Role of pseudorabies virusglycoprotein gI in virus release from infected cells. J. Virol. 61:2764-2769.
13. Mettenleiter, T. C., L. Zsak, A. S.Kaplan, T.Ben-Porat, andB. Lomniczi. 1987. Role of a structuralglycoprotein of pseudora-bies in virus virulence. J. Virol. 61:4030-4032.
14. Petrovskis, E. A., J. G.Timmins, T. M.Gierman,and L. E. Post. 1986. Deletions in vaccine strains of pseudorabies virus and their effect onsynthesisof glycoproteingp63. J. Virol.
on November 10, 2019 by guest
http://jvi.asm.org/
1169.
15. Robbins,A.K., R. J. Watson, M. E.Whealy,W. W.Hays, and L. W. Enquist. 1986. Characterization ofa pseudorabies virus
glycoproteingenewith homologytoherpessimplexvirustype1 andtype2glycoprotein C.J. Virol.58:339-347.
16. Robbins, A.K.,J. H. Weis,L. W. Enquist, andR.J.Watson. 1984. Construction of E. coli expression plasmid libraries: localization ofapseudorabies virus glycoproteingene.J. Mol.
Appl. Genet.2:485-496.
17. Robbins, A.K., M. E.Whealy,R. J. Watson,and L. W. Enquist. 1986. Pseudorabies virusgeneencodingglycoproteingIIIisnot
essential forgrowth in tissue culture. J.Virol.59:635-645. 18. Ryan, J. P., M.E.Whealy,A. K. Robbins, and L. W. Enquist.
1987.Analysis of pseudorabies virus glycoproteingIll
localiza-tion and modification by using novel infectious viral mutants carryinguniqueEcoRI sites. J. Virol. 61:2962-2972.
19. Sanger, F., S. Nicklen, and A. R. Coulson. 1977.DNA
sequenc-ing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 74:5463-5467.
20. Schreurs, C., T.C. Mettenleiter, F. Zuckermann, N. Sugg, and T. Ben-Porat. 1988. Glycoprotein gIII ofpseudorabies virus is multifunctional. J. Virol. 62:2251-2257.
21. vanOirschot, J. T., and A. L. J. Gielkens. 1984. Some
charac-teristics of four attenuatedvaccinevirus strainsandavirulent
strain ofAujeszky'sdisease virus.Vet.Q. 6:225-229. 22. Whealy, M. E., A. K. Robbins, and L. W. Enquist. 1988.
Pseudorabies virus glycoprotein gIII is required for efficient virus growthin tissue culture.J. Virol. 62:2512-2515.


![FIG. 4.tectedods.analyzedPRV-Be,presenceantibody.Immune(StaphylococcusinspecificprofileNumbersserum.10% panels Immunoprecipitation of [3H]glucosamine-labeled,gIll- proteins from purified virions](https://thumb-us.123doks.com/thumbv2/123dok_us/1327456.86670/5.612.70.310.76.373/tectedods-analyzedprv-presenceantibody-staphylococcusinspecificprofilenumbersserum-immunoprecipitation-glucosamine-proteins-purified.webp)

