Common determinants in DNA melting and helicase-catalysed DNA unwinding by papillomavirus replication protein E1
Full text
Figure
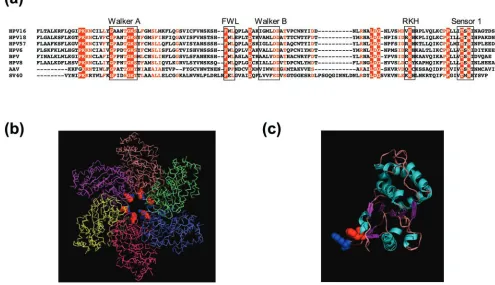



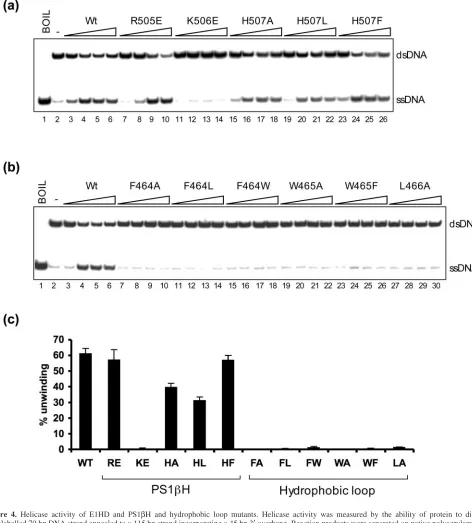



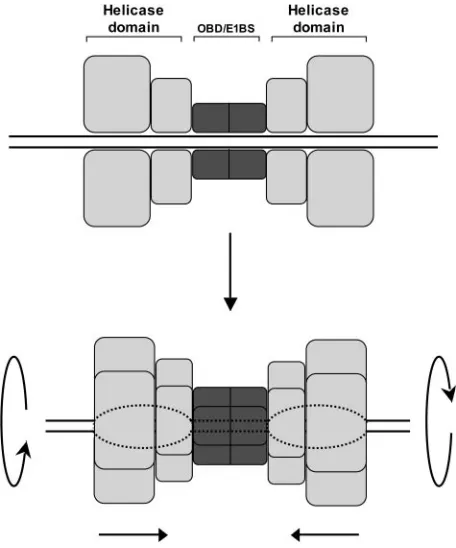
Related documents
The free surface profiles of internal water are similar in the two studied cases, suggesting that the flooding effect on the free surface motion inside the
These hernias may occur after large surgeries such as intestinal or vascular (large arteries, and veins) surgery, or after smaller surgeries such as an appendectomy or an
While a high number of rAAV sequences were detected during the first 5 days following the injection, only a few percent of these sequences was retained in the animals analyzed after
Supershift assays failed to identify any GATA-related factor binding to the 5 9 -end site (data not shown). 1), were shown upon EMSA analysis to bind not only the core binding
Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension; Final results of the Systolic Hypertension in the Elderly
To accurately estimate the potencies of these inhibitors, we carried out titration experiments of enzymatic activity as a function of inhibitor concentration by using the same assay
Thus, to address the possible role of inflammatory cytokines in vertical HIV transmission from women to their newborns, we measured the expression of IL-1 b , IL-6, and TNF- a , at
Viral RNA was synthesized in vitro at the permissive temperature (30 8 C [S 30]) or the restrictive temperature (37 8 C [S37]) after reconstitution of polymerase in vivo at
