0022-538X/10/$12.00 doi:10.1128/JVI.02215-09
Copyright © 2010, American Society for Microbiology. All Rights Reserved.
Insertion Mutations in Herpes Simplex Virus 1 Glycoprotein H
Reduce Cell Surface Expression, Slow the Rate of Cell Fusion,
or Abrogate Functions in Cell Fusion and Viral Entry
䌤
Julia O. Jackson, Erick Lin, Patricia G. Spear, and Richard Longnecker*
Department of Microbiology and Immunology, the Feinberg School of Medicine, Northwestern University, Chicago, Illinois 60611
Received 20 October 2009/Accepted 24 November 2009
Of the four required herpes simplex virus (HSV) entry glycoproteins, the precise role of gH-gL in fusion remains the most elusive. The heterodimer gH-gL has been proposed to mediate hemifusion after the inter-action of another required glycoprotein, gD, with a receptor. To identify functional domains of HSV-1 gH, we generated 22 randomized linker-insertion mutants. Analyses of 22 gH mutants revealed that gH is relatively tolerant of insertion mutations, as 15 of 22 mutants permitted normal processing and transport of gH-gL to the cell surface. gH mutants that were not expressed well at the cell surface did not function in fusion or viral entry. The screening of gH mutants for function revealed the following: (i) for wild-type gH and some gH mutants, fusion with nectin-1-expressing target cells occurred more rapidly than with herpesvirus entry mediator (HVEM)-expressing target cells; (ii) some gH mutants reduced the rate of cell fusion without abrogating fusion completely, indicating that gH may play a role in governing the kinetics of fusion and may be responsible for a rate-limiting first stage in HSV-1 fusion; and (iii) only one gH mutant, located within the short cytoplasmic tail, completely abrogated function, indicating that the gH cytoplasmic tail is crucial for cell fusion and viral infectivity.
Herpes simplex virus (HSV), an enveloped neurotropic vi-rus, infects target cells via membrane fusion, a process exe-cuted by viral fusion proteins capable of inserting into target membranes. Unlike many enveloped viruses that induce fusion through the activity of a single viral fusion protein, HSV re-quires four glycoproteins, glycoprotein B (gB), glycoprotein D (gD), glycoprotein H (gH), and glycoprotein L (gL), to execute fusion (6, 40, 42). The focus of this study, gH, is expressed as a heterodimer with gL (gH-gL). HSV gH and gL rely on one another for proper folding, posttranslational processing, and transport to the cell and virion surface (5, 23, 35).
A sequential model of entry is the prevailing working hy-pothesis of HSV entry (1–3, 28, 32, 41). Viral attachment is mediated by the binding of glycoprotein C (gC) or gB to cell surface glycosaminoglycans such as heparan sulfate (38). The subsequent fusion between the virion envelope and host cell membrane is thought to result from a series of concerted events. First, gD binds to one of its host cell receptors. These receptors include herpesvirus entry mediator (HVEM), a member of the tumor necrosis factor (TNF) receptor family; nectin-1 and nectin-2, cell adhesion molecules of the Ig superfamily; and heparan sulfate modified by specific 3-O-sulfotransferases (39).
It was previously proposed that gD binding a receptor in-duces a conformational change that allows for interactions between gD, gB, and/or gH-gL (1, 2, 8, 10, 16, 25, 32). It is thought that while gD functions primarily in receptor binding, gB and gH-gL function as the core fusion machinery of HSV.
Based on its crystal structure, gB has structural features typical of viral fusion proteins in general and is structurally similar to vesicular stomatitis virus (VSV) glycoprotein G, the fusion protein of VSV (22, 34). In addition to its resemblance to other viral fusogens, gB also binds its own receptor, paired immunoglobulin-like receptor (PILRalpha) (36, 37). Impor-tantly, HSV gB does not successfully execute fusion in the absence of gD or gH-gL (41). Compared to the other required HSV entry glycoproteins, relatively little is known about the specific roles of gH-gL during fusion. The structure of gH-gL
is unknown, althoughin silicoanalyses and studies of synthetic
gH peptides suggested that gH also has fusogenic properties (12, 13, 17–20).
gD, a gD receptor, and gH-gL have been shown to be suf-ficient for inducing hemifusion, the mixing of the proximal leaflets of the viral and host cell bilayers (41). Several lines of research suggest that the subsequent step in fusion is an inter-action between gH-gL and gB, with the latter glycoprotein being required for a committed and expanding fusion pore (1–3, 16, 28, 41). However, it is still unclear whether the gB and gH-gL interaction requires that gD first bind a receptor (1, 3), indicating that another viable model of HSV entry may be nonsequential gD-gB-gH-gL complex formation.
Several domains important for fusion within HSV gH have been discerned. The only function associated with the N-ter-minal domain of HSV gH, to date, is gL binding. Residues 377 to 397 within a predicted alpha-helix in the gH ectodomain are required for cell-cell fusion and complementation of a gH-null virus (18). The mutation of a predicted heptad repeat region spanning residues 443 to 471 abrogated cell-cell fusion (17). Insertion mutations within what has been termed the pretrans-membrane region of gH have also been shown to abrogate fusion and viral entry (11). The glycine residue at position 812 within the predicted gH transmembrane domain was shown
* Corresponding author. Mailing address: Department of Microbiology and Immunology, the Feinberg School of Medicine, Northwestern Uni-versity, 303 East Chicago Avenue, Ward 6-231, Chicago, IL 60611. Phone: (312) 503-0467. Fax: (312) 503-1339. E-mail: r-longnecker@northwestern .edu.
䌤Published ahead of print on 9 December 2009.
2038
on November 8, 2019 by guest
http://jvi.asm.org/
previously to be important for fusion (21). Finally, although the deletion of the final six residues of gH (residues 832 to 838), which are within its short cytoplasmic tail, has no effect on fusion, further deletions were shown to decrease polykaryo-cyte formation by a syncytial HSV strain (4, 43).
We used a transposon-based comprehensive random linker-insertion mutagenesis strategy to generate a library of mutants spanning the entire length of HSV-1 gH, an 838-amino-acid type I membrane protein. A panel of 22 insertion mutants was generated, 15 of which were expressed at near-normal levels on the cell surface. Interestingly, some insertions reduced the rate of cell fusion rather than abrogating cell fusion activity alto-gether, suggesting that gH may have a role in governing the kinetics of fusion and may be responsible for a rate-limiting first stage in HSV-1 fusion. Additionally, one insertion muta-tion that completely abrogated cell fusion and viral infectivity is located within the gH cytoplasmic tail, indicating that the short C-terminal tail of gH is critical for cell fusion and entry mediated by HSV-1.
MATERIALS AND METHODS
Cells and viruses.Cell lines used were CHO cells, CHO cells stably expressing human HVEM (29) (CHO-HVEM) or nectin-1 (15) (CHO-nectin-1), Vero cells, and Vero-VgHC4 cells carrying the HSV-1 gH gene, which were used for the propagation and titration of the gH-negative mutant HSV-1 (KOS) virus named KOS gH87 (30). The CHO lines were grown in Ham’s F12 medium supple-mented with 10% fetal bovine serum (FBS). The Vero lines were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS.
Random linker-insertion mutagenesis of HSV-1 gH.The GPS-LS linker scan-ning system (New England Biolabs, Ipswich, MA) was used as recommended by the manufacturer for the insert excised from pPEP100 (33) to generate random linker-insertion mutations of HSV-1 gH. After the religation of the library of inserts into the pCAGGS vector and transformation of competent bacteria, 87 unique gH linker-insertion mutants were isolated. After the removal of the transposon, each mutant plasmid was fully sequenced to verify the position of each insert. Plasmids containing inserts outside of the gH open reading frame (ORF), stop codon insertional mutations, and duplicates of identical inserts were dropped from further study.
CELISA.CHO cells seeded into 96-well plates grown to approximately 80% confluence were transfected with 20 ng of empty vector or a plasmid expressing wild-type (WT) gH or mutant gH, 20 ng each of WT gD and gL, and 40 ng of gB with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) diluted in Opti-MEM (Gibco, Carlsbad, CA). After a 2-h transfection, the cells were washed once with Opti-MEM and then incubated with serum-containing medium for 10 h. Cell-based enzyme-linked immunosorbent assays (CELISAs) were performed, as described elsewhere previously (14), by using anti-gH monoclonal antibody (MAb) 52S or gH-gL monoclonal antibody 53S at a 1:10,000 dilution. For time course experiments, CHO cells seeded into 6-well plates grown to approximately 80% confluence were transfected with 400 ng of empty vector or a plasmid expressing WT gH or mutant gH by using Lipofectamine 2000. After a 4-h transfection, cells were washed once and then incubated with serum-containing medium for the indicated times. In addition, CHO cells were transfected with all of the plasmids used to prepare effector cells for the cell fusion assay described below, and those effector cell populations were seeded for both fusion and CELISAs so that gH expression could be assessed in the same cell populations used for each fusion assay.
Cell fusion assay.For the original cell fusion assay performed (Table 1), CHO cells (effectors) were seeded into 96-well plates 18 h before transfection. Effec-tors were transfected with 30 ng of plasmid expressing gB and 20 ng each of plasmids expressing the T7 RNA polymerase, gD, gL, and either empty vector, WT gH, or mutant gH by using Lipofectamine 2000. In parallel, target cells were seeded into 6-well plates and transfected with 400 ng of plasmid carrying the firefly luciferase gene under the control of the T7 promoter and 1.8g empty vector with Lipofectamine 2000. After 2 h of transfection, cells were washed, and target cells were detached and overlaid onto effectors and coincubated for 10 h before lysis and quantification of luciferase signals as described elsewhere pre-viously (26). For the fusion time course assay, effector and target cells were seeded into 6-well plates 18 h prior to transfection. Effector cells were
trans-fected with 600 ng of plasmid containing gB and 400 ng each of plasmids containing T7 polymerase, gD, gL, and either empty vector, WT gH, or mutant gH. Target cells were transfected with 400 ng of plasmid containing luciferase and 1.8g of empty vector. After 4 h of transfection, cells were washed, de-tached, and overlaid as described elsewhere previously (26). After coincubation for the indicated times, cells were washed and lysed, and luciferase activity was measured as described above.
Viral complementation assay.The viral complementation assay was done as described previously for the complementation of a gD-negative virus by WT gD and mutants (27) except that Vero cells were transfected with 1.0g of plasmid expressing WT or mutant gH and later infected with gH-negative mutant HSV-1 (30). Virus stocks were prepared, and titrations were performed on Vero-VgHC4 cells. A blue plaque assay was performed after 2 days of incubation at 37°C. Cells were fixed (2% paraformaldehyde [PFA] and 0.2% glutaraldehyde in phosphate-buffered saline [PBS]) and then permeabilized (2 mM MgCl2, 0.01% deoxy-cholate, 0.02% NP-40) for 5 min and washed twice. Ferricyanide (FeCN) buffer containing 0.5 mg X-gal (5-bromo-4-chloro-3-indolyl--D-galactopyranoside) per ml was added to cells and incubated about 1 h until blue plaques developed. Plaques were counted, using a dissection microscope.
Western blot analysis.For analysis of selected mutants, CHO cells seeded into 25-cm2
flasks were transfected with 5g each of plasmid expressing WT gL and either empty vector or a plasmid expressing WT gH or a gH mutant and 20l Lipofectamine 2000. After 4 h of transfection, cells were washed and incubated with serum-containing medium for 18 h. Cells were then washed with PBS, detached with versene, and counted. Cells were resuspended in 1 ml of lysis buffer (50 mM Tris [pH 8], 150 mM NaCl, 1% Nonidet P-40) containing protease inhibitor per 10⫻106
cells, mixed with sample buffer for SDS-PAGE, and boiled for 5 min. Samples were separated by electrophoresis on 4 to 20% gels, and immunoblots were performed by using rabbit anti-gH-gL antiserum R137 at a 1:10,000 dilution (31), which was kindly provided by Gary H. Cohen and Roselyn J. Eisenberg. For analysis of complemented virus, the supernatant was centri-fuged at 48,000⫻gfor 1 h at 4°C over a 10% sucrose cushion. The pellet was dissolved in sample buffer and separated by SDS-PAGE on a 4 to 20% gel under reducing conditions. Western blot analysis was performed by using R137 at a 1:10,000 dilution and VP5 MAb (EastCoast Bio) at a 1:5,000 dilution.
RESULTS
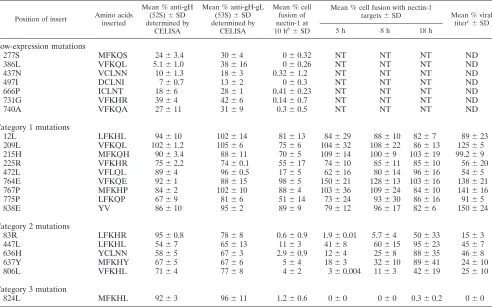
Cell surface expression of gH insertion mutants. The 22 linker-insertion gH mutants are listed in Table 1, along with summary data for each mutant. The number and name of the
amino acid 5⬘of the 5-amino-acid insertion denote the position
of each insertion. To assess expression levels of gH mutants on the cell surface, plasmids encoding each mutant were trans-fected into CHO cells along with plasmids encoding wild-type (WT) HSV-1 gD, gB, and gL. The transfected cells were incu-bated with two monoclonal antibodies against HSV-1 gH for the detection of gH by cell-based enzyme-linked immunosor-bent assay (CELISA). One antibody (52S) recognizes gH re-gardless of the presence of gL, while the other (53S) is a conformational antibody that needs both gH and gL in order to bind. Figure 1A shows the locations of the mutants along a linear representation of gH and its proposed or predicted functional domains and levels of cell surface gH detected for each mutant as a percentage of WT gH expression.
Of the 22 mutants screened, 15 were detected on cell
sur-faces atⱖ60% of WT gH levels. The 277S, 386L, 437N, 497I,
666P, 731G, and 740A mutants were detected at lower levels,
⬍40% of WT gH levels (Fig. 1A). Interestingly, the 386L
mutant showed higher levels of expression detected by anti-gH-gL antibody than detected by the anti-gH antibody, sug-gesting that the 386L insertion site may alter the anti-gH (52S) antibody epitope. For all other mutants, cell surface levels detected by the two antibodies were not significantly different, indicating that complexes with gL were formed even if overall levels of the cell surface complexes were reduced.
on November 8, 2019 by guest
http://jvi.asm.org/
Lysates of cells transfected with selected mutants were also probed with anti-gH-gL polyclonal antibody by Western blot-ting to determine if gH was translated despite low levels of cell surface expression. Figure 1B shows that all mutants with low expression levels at the cell surface were translated, indicating that their insertions disrupted the posttranslational processing required for transport to and/or expression at the cell surface.
Effects of insertions into gH on cell fusion. To assess the function of the gH mutants, we first used a quantitative lucif-erase-based cell-cell fusion assay. All 22 gH mutants were coexpressed with WT HSV-1 gD, gB, and gL in CHO cells (effectors) and mixed with nectin-1-expressing or HVEM-ex-pressing CHO cells (targets). Initially, mutants were screened by the direct transfection of effectors seeded into 96-well plates. This served as a primary screen for the function of mutants and revealed that a number of mutants had cell fusion activity greater than 80% of that of WT gH; some mutants had de-creased fusion activity despite near-normal levels of cell sur-face expression, and all mutants that were expressed on the cell
surface at low levels (⬍40% of WT gH levels) lacked
signifi-cant cell fusion activity (Table 1). These mutants with reduced cell surface expression and no fusion activity were not investi-gated further.
For all other mutants, our secondary screen for function was
the same luciferase-based fusion assay performed over a time course. Effector and target cells were transfected as described above, but transfections were scaled to a 6-well plate. After transfection, effector cells were washed, detached, and seeded into 96-well plates in triplicate. Transfected target cells were overlaid onto effector cells at a 1:1 ratio. Coincubation mix-tures of effectors and targets were harvested at 5, 8, and 18 h after cell mixing, and the luciferase activity was determined as a measure of cell fusion that had occurred up to each time point.
[image:3.585.48.540.81.388.2]The first significant observation with this assay was that cells expressing WT HSV-1 glycoproteins induced fusion with nec-tin-1-expressing target cells at a much higher rate than that observed with HVEM-expressing target cells. Maximal or near-maximal cell fusion was observed with nectin-1-expressing tar-get cells by around 8 h after cell mixing but, with HVEM-expressing target cells, not until some time after 18 h. The decline in luciferase activity at 18 h with nectin-1-expressing targets may be due to the decay of luciferase mRNA or protein after cell fusion had gone to completion. It is not clear whether this receptor-dependent difference in rates relates to the na-ture of each receptor or to the effective quantity of each re-ceptor expressed on the cell surface of the target cells that we used. However, in a direct comparison of nectin-1 and HVEM,
TABLE 1. Linker-insertion gH mutantsa
Position of insert Amino acids inserted
Mean % anti-gH (52S)⫾SD determined by
CELISA
Mean % anti-gH-gL (53S)⫾SD determined by
CELISA
Mean % cell fusion of nectin-1 at 10 hb⫾SD
Mean % cell fusion with nectin-1
targets⫾SD Mean % viral titerc⫾SD 5 h 8 h 18 h
Low-expression mutations
277S MFKQS 24⫾3.4 30⫾4 0⫾0.32 NT NT NT ND
386L VFKQL 5.1⫾1.0 38⫾16 0⫾0.26 NT NT NT ND
437N VCLNN 10⫾1.3 18⫾3 0.32⫾1.2 NT NT NT ND
497I DCLNI 7⫾0.7 13⫾2 0⫾0.3 NT NT NT ND
666P ICLNT 18⫾6 28⫾1 0.41⫾0.23 NT NT NT ND
731G VFKHR 39⫾4 42⫾6 0.14⫾0.7 NT NT NT ND
740A VFKQA 27⫾11 31⫾9 0.3⫾0.5 NT NT NT ND
Category 1 mutations
12L LFKHL 94⫾10 102⫾14 81⫾13 84⫾29 88⫾10 82⫾7 89⫾23
209L VFKQL 102⫾1.2 105⫾6 75⫾6 104⫾32 108⫾22 86⫾13 125⫾5
215H MFKQH 90⫾3.4 88⫾11 70⫾5 109⫾14 100⫾9 103⫾19 99.2⫾9
225R VFKHR 75⫾2.2 74⫾0.1 55⫾17 74⫾10 85⫾11 85⫾10 56⫾20
472L VFLQL 89⫾4 96⫾0.5 17⫾5 62⫾16 80⫾14 96⫾16 54⫾5
764E VFKQE 92⫾1 88⫾15 98⫾5 150⫾21 128⫾13 103⫾16 138⫾21
767P MFKHP 84⫾2 102⫾10 88⫾4 103⫾36 109⫾24 84⫾10 141⫾16
775P LFKQP 67⫾9 81⫾6 51⫾14 73⫾24 93⫾30 86⫾16 91⫾5
838E YV 86⫾10 95⫾2 89⫾9 79⫾12 96⫾17 82⫾6 150⫾24
Category 2 mutations
83R LFKHR 95⫾0.8 78⫾8 0.6⫾0.9 1.9⫾0.01 5.7⫾4 50⫾33 15⫾3
447L LFKHL 54⫾7 65⫾13 11⫾3 41⫾8 60⫾15 95⫾23 45⫾7
636H YCLNN 58⫾5 67⫾3 2.9⫾0.9 12⫾4 25⫾8 88⫾35 46⫾8
637Y MFKHY 67⫾5 67⫾6 5⫾4 18⫾3 32⫾10 89⫾41 24⫾10
806L VFKHL 71⫾4 77⫾8 4⫾2 3⫾0.004 11⫾3 42⫾19 25⫾10
Category 3 mutation
824L MFKHL 92⫾3 96⫾11 1.2⫾0.6 0⫾0 0⫾0 0.3⫾0.2 0⫾0
a
All percentages shown are a percentage of the WT. b
Refers to the original fusion experiments with nectin-1-expressing target cells. Target and effector cells were transfected for 2 h, overlaid, and coincubated for 10 h before fusion was assessed by measuring luciferase activity. gH mutants that that were not expressed at levels that were⬎40% of WT levels by anti-gH CELISA and did not show any fusion activity in these experiments were not tested (NT) in the fusion time course.
c
Cell-associated viral titers are reported. See Fig. 4A for extracellular viral titers. Viral titers equal to or less than background are reported as being not detected (ND).
on November 8, 2019 by guest
http://jvi.asm.org/
nectin-1 was previously shown to be more efficient at promot-ing entry than HVEM (24).
gH mutants consistently clustered into three categories based on their fusion activity (Fig. 2A and B). Category 1 mutants
were defined as having⬎50% of the cell fusion activity of WT
gH at the 5-h time point with either receptor. These mutants were nearly indistinguishable from WT gH with regard to the kinetics of cell fusion with either receptor (Fig. 2A). Also, the levels of cell fusion activity at each time point were similar although not identical to those of WT gH (either higher or lower but not lower than 70%). Category 2 mutants were
defined as having⬍50% of the cell fusion activity of WT gH at
the 5-h time point. The kinetics of cell fusion for category 2 mutants were distinct from those of WT gH (Fig. 2A and B). These mutants achieved the highest level of fusion with cells expressing either receptor at the 18-h time point (with the exception of the 447L mutant for nectin-1) and the highest level of cell fusion, as a percentage of WT gH cell fusion, at the 18-h time point (Fig. 2B). The sole category 3 mutant (824L)
was incapable of supporting cell fusion with either receptor at any of the time points.
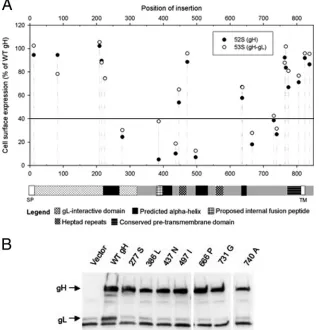
In order to ensure that the decreased rate of fusion for category 2 mutants was not due to a lower rate of cell surface expression, we performed an expression time course. CHO cells were transfected as they were for cell fusion, washed, and allowed to incubate for 5, 8, and 18 h before expression levels were read by CELISA. As shown in Fig. 3, only one gH mutant, the 775P mutant, which belonged to category 1 and resembled WT gH in fusion kinetics, exhibited a moderate stepwise in-crease in expression levels relative to those of WT gH over time. For all category 2 mutants, reduced fusion rates were not due to reduced rates of expression at the cell surface. Similarly, the expression levels of the 824L fusion-defective mutant were similar to those of WT gH at all time points, indicating that the abrogation of functional activity caused by the 824L insertion is not due to decreased expression levels.
Viral complementation correlates with fusion function. In order to explore the effect of the insertions on gH function in
FIG. 1. (A) Effects of insertional mutations of HSV-1 gH on cell surface expression. CHO cells were transfected with plasmids expressing WT gL, gD, and gB and either empty vector (pCAGGS), WT gH, or mutant gH. The cell surface expression of gH or gH mutants was quantified by CELISA using anti-gH (52S) and anti-gH-gL (53S) monoclonal antibodies. The values presented for cell surface expression are means for two experiments expressed as a percentage of WT gH values. A linear representation of gH is shown below the graph. Signal peptide (SP) and transmembrane (TM) regions are indicated. Predicted structural domains and proposed functional domains are also noted. (B) Analysis of lysates of transfected cells by Western blotting for the presence of gH mutants poorly expressed by CELISA. CHO cells were harvested 18 h posttransfection with plasmids encoding WT gL and either empty vector (pCAGGS), WT gH, or mutant gH. After separation by SDS-PAGE and transfer onto polyvinylidene difluoride (PVDF) blots, lysates were probed with rabbit anti-gH-gL antiserum R137. Proteins of interest are indicated with arrows.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:4.585.134.453.72.402.2]FIG. 2. (A) Cell fusion activities of the gH mutants. CHO cells were transfected with HSV-1 WT gB, gD, and gL; either WT or mutant gH; and T7 polymerase for 4 h. CHO-nectin-1 and CHO-HVEM cells were transfected with T7 luciferase for 4 h. Cells were washed and detached, and effector cells were reseeded with target cells in 96-well plates. At the indicated times after replating, fusion activity was assessed by the
on November 8, 2019 by guest
http://jvi.asm.org/
[image:5.585.44.541.73.693.2]viral entry and plaque formation, we used a viral complemen-tation assay in which cells were transfected with WT or mutant gH expression constructs and subsequently infected with gH-null HSV-1. Complemented virus was harvested from the cell lysate (cell-associated virus) and supernatant (extracellular vi-rus). Complemented viral titers were determined by plaque assay, and the level of gH incorporation into virions was de-termined by Western blotting.
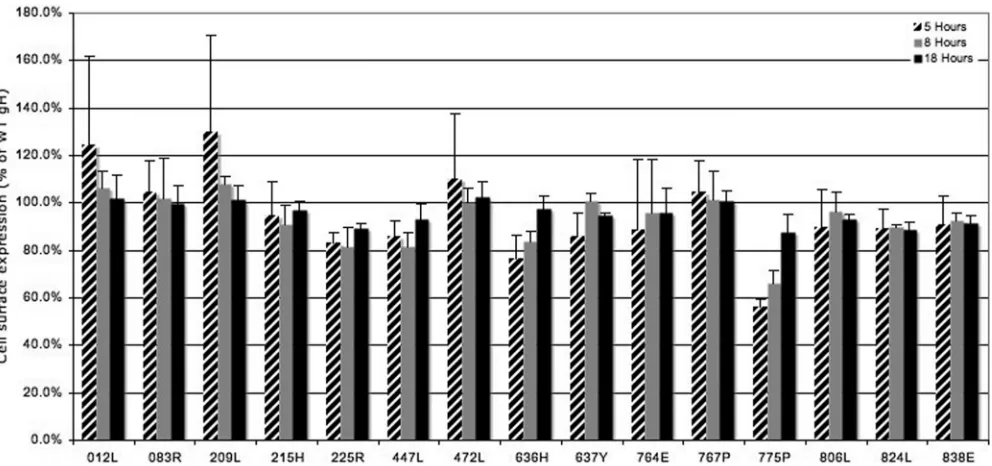
Of particular interest were the mutants that were defective in fusion or that exhibited a reduced rate of cell fusion. As shown in Fig. 4A for the category 2 mutants, a decreased rate of fusion correlated with decreased, but not absent, viral in-fectivity. Furthermore, the reduced viral infectivity does not seem to be due to a decreased incorporation of gH into the virions (Fig. 4B). The nonfunctional 824L mutant was as de-fective for viral infectivity as the gH mutants that were not properly processed (Table 1) despite normal levels of gH in-corporation into virions. Collectively, these data suggest that the regions of gH identified as described above to be important for rates of cell-cell fusion, or for detectable cell fusion activity, are also important for viral infectivity.
DISCUSSION
We generated a panel of HSV-1 gH insertion mutants in order to further identify gH functional domains. Fifteen gH
mutants were expressed at the cell surface at levels greater than or equal to 60% of WT gH levels. We screened for the function of our gH mutants with a luciferase-based cell-cell fusion assay standard to the field but modified the assay by monitoring the extent of cell fusion as a function of time after mixing the effector and target cells. The first interesting finding revealed by our time course was that fusion with nectin-1-expressing target cells occurred more rapidly than that with HVEM-expressing target cells. Nectin-1-triggered fusion is so rapid that the standard effector and target cell coincubation time of 18 h or more is likely too long to accurately reflect the function of mutations with intermediate phenotypes. It is not clear whether the receptor-dependent difference in the rate of fusion that we observed relates to the nature of each receptor or to the effective quantity of each receptor expressed on our target cells.
Our study also revealed that a number of insertion muta-tions lowered the rate of cell-cell fusion without abrogating fusion altogether. Importantly, the decreased rate of cell-cell fusion was not due to a decreased rate of cell surface expres-sion. The decreased rate of fusion observed for these mutants suggests that gH may have a role in governing the kinetics of fusion and may be responsible for a rate-limiting first stage in HSV-1 fusion. However, we acknowledge that our experiments measure fusion events within a population of cells and not the rate of individual fusion events. If we embrace the current
[image:6.585.46.542.68.301.2]quantification of luciferase activity. A representative experiment is shown. Each plate contained WT and empty vector controls to which mutants were always compared. Left panels show the category 1 mutants tested with each receptor. Right panels show the category 2 and 3 mutants also tested with each receptor. (B) Cell fusion activity expressed as a function of time and as a percentage of WT gH activity at each time point for selected mutants. The category 1 012L mutant is shown with all six category 2 and 3 mutants. The results shown are the means and standard deviations for three independent experiments.
FIG. 3. Cell surface expression of gH as a function of time. CHO cells were transfected as described in the legend of Fig. 2 for 4 h, washed, and incubated for the indicated times before expression levels were read by CELISA. For each time point, expression is compared to that of WT gH. Results shown are the means and standard deviations for three independent experiments.
on November 8, 2019 by guest
http://jvi.asm.org/
working model of HSV fusion in which gH is responsible for hemifusion, our data suggest that the hemifusion process may be more easily slowed than shut down altogether. It is also possible that the mutations that affect rate of fusion, all of which are located in the gH ectodomain, interrupt but do not totally abolish the gH-gD and/or gH-gB interactions necessary for fusion.
It is interesting that of the quartet of required entry glyco-proteins, gD and gH are more tolerant of insertion mutations than the presumed fusogen gB. In the linker-insertion mu-tagenesis analysis reported here, 68% of mutants were expressed at the cell surface. A previous study also reported a panel of gH insertional mutants in which between 57 and 76% of mu-tants were expressed normally, depending on the antibody used to detect expression (11). In similar studies done with gD (7) and gB (26), cell surface expression was achieved for 86% and 33% of mutants, respectively. This difference between the panels of gH and gB insertion mutants generated in an iden-tical fashion may indicate that gH, unlike gB, has a more flexible conformation and performs functions that are not so
dependent on intrachain interfaces or dramatic conformational changes.
One of our insertion mutations, 824L, was incapable of per-forming cell fusion or complementing a gH-null virus despite normal cell surface expression and viral incorporation. The 824L gH mutant clearly indicates a crucial role for the short cytoplasmic tail of gH in both cell fusion and viral entry. Two previous studies demonstrated the importance of 831V, also located within the gH cytoplasmic tail, downstream of our 824L insertion mutant, to the fusion activity of a syncytial strain of HSV-1 (4, 43). An alanine substitution at 831V de-creased the viral entry efficiency of a syncytial strain of com-plemented virus but did not abrogate viral fusion altogether, as our 824L mutant did. It is possible that our 824L insertion mutant and the previously identified 831A mutant are disrupt-ing the same required functional domain of gH, although they may also be distinct functional domains. Regardless, the pre-cise role of the short cytoplasmic tail of gH in fusion and viral entry remains to be defined. Since soluble gD is sufficient for triggering fusion, we can conclude that interactions between
FIG. 4. (A) Complementation of a gH-null virus with selected gH mutants. Vero cells were transfected with the gH mutants indicated and infected with a gH-null HSV-1 recombinant virus that expresses beta-galactosidase from the gH locus. Complemented virus was harvested from the cell lysate and supernatant. Viral titers were determined by plating the samples on complementing Vero cells. For gH-null virus complemented with WT gH, cell lysate viral titers (2.0⫻107PFU/ml) were 320 times greater than extracellular viral titers (7.0⫻104PFU/ml) (average of three
experiments). (B) Incorporation of mutant gH into virions. Virions were isolated from the supernatants of cells that were transfected and infected for the complementation assay. Virion proteins were solubilized for SDS-PAGE, transferred onto PVDF blots, and probed with an anti-gH antiserum and anti-VP5 MAb, as a loading control. The positions of gH and VP5 are marked on the blot. Densitometries were calculated, and ratios of gH to VP5 normalized to WT gH were as follows: 83R, 1.5; 447L, 2.5; 636H, 1.6; 637Y, 1.1; 806L, 1.5; 824L, 1.7.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:7.585.111.474.77.407.2]gH and gD via their cytoplasmic domains are not required for fusion (8). Similarly, it seems unlikely that the cytoplasmic domain of gH is interacting with gB, although perhaps an inter-action between gH and gB via their cytoplasmic tails is required for fusion. It seems more likely that the gH cytoplasmic tail is mediating the multimerization of gH or the gH association with soluble proteins or is required for gH conformation and/or stability. Based on the normal cell surface expression and viral incorporation of the 824L mutant, it seems unlikely that the functional defect of 824L is due entirely to decreased protein stability.
It may be significant that the 824L mutation abrogated membrane fusion entirely, whereas the 831A mutation did not. Perhaps the most membrane-proximal region of the cyto-plasmic tail, which is disrupted in the 824L mutant but not in the 831A mutant, is interacting with viral envelope or plasma membrane lipids and plays a direct role in the perturbation of the inner leaflet of the bilayer that is required for fusion to proceed. As others noted previously (43), gH proteins of the alphaherpesviruses HSV-1 and -2, equine herpesviruses 1 and 4, bovine rhinotracheitis virus type 1, varicella-zoster virus, and pseudorabies virus all contain two neutral, nonaromatic amino acids (ML or VL) in the most-membrane-proximal region of the cytoplasmic tail. Based on sequence prediction, the 824L mutation may extend the transmembrane of gH by two amino acids (9), thus perhaps pushing the conserved membrane-prox-imal VL amino acids downstream. Further studies are needed to define the functional regions of the gH cytoplasmic tail and their specific functions in membrane fusion.
ACKNOWLEDGMENTS
We thank Gary H. Cohen and Roselyn J. Eisenberg for the generous gift of the R137 antibody and Nanette Susmarski for providing cells and her cell line expertise. We thank the members of the Longnecker, Spear, and Jardetzky laboratories for help and support.
This research was supported by grants CA117794 (R.L.) and CA021776 (P.G.S. and R.L.) from the National Cancer Institute.
REFERENCES
1.Atanasiu, D., J. C. Whitbeck, T. M. Cairns, B. Reilly, G. H. Cohen, and R. J. Eisenberg.2007. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc. Natl. Acad. Sci. U. S. A.104:18718–18723.
2.Avitabile, E., C. Forghieri, and G. Campadelli-Fiume.2007. Complexes between herpes simplex virus glycoproteins gD, gB, and gH detected in cells by complementation of split enhanced green fluorescent protein. J. Virol.
81:11532–11537.
3.Avitabile, E., C. Forghieri, and G. Campadelli-Fiume.2009. Cross talking among the glycoproteins involved in herpes simplex virus entry and fusion: the interaction between gB and gH/gL does not necessarily require gD. J. Virol.83:10752–10764.
4.Browne, H. M., B. C. Bruun, and A. C. Minson.1996. Characterization of herpes simplex virus type 1 recombinants with mutations in the cytoplasmic tail of glycoprotein H. J. Gen. Virol.77(Pt. 10):2569–2573.
5.Cairns, T. M., L. S. Friedman, H. Lou, J. C. Whitbeck, M. S. Shaner, G. H. Cohen, and R. J. Eisenberg.2007. N-terminal mutants of herpes simplex virus type 2 gH are transported without gL but require gL for function. J. Virol.81:5102–5111.
6.Campadelli-Fiume, G., M. Amasio, E. Avitabile, A. Cerretani, C. Forghieri, T. Gianni, and L. Menotti.2007. The multipartite system that mediates entry of herpes simplex virus into the cell. Rev. Med. Virol.17:313–326. 7.Chiang, H. Y., G. H. Cohen, and R. J. Eisenberg.1994. Identification of
functional regions of herpes simplex virus glycoprotein gD by using linker-insertion mutagenesis. J. Virol.68:2529–2543.
8.Cocchi, F., D. Fusco, L. Menotti, T. Gianni, R. J. Eisenberg, G. H. Cohen, and G. Campadelli-Fiume.2004. The soluble ectodomain of herpes simplex virus gD contains a membrane-proximal pro-fusion domain and suffices to mediate virus entry. Proc. Natl. Acad. Sci. U. S. A.101:7445–7450. 9.Emanuelsson, O., S. Brunak, G. von Heijne, and H. Nielsen.2007. Locating
proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc.
2:953–971.
10.Fusco, D., C. Forghieri, and G. Campadelli-Fiume.2005. The pro-fusion domain of herpes simplex virus glycoprotein D (gD) interacts with the gD N terminus and is displaced by soluble forms of viral receptors. Proc. Natl. Acad. Sci. U. S. A.102:9323–9328.
11.Galdiero, M., A. Whiteley, B. Bruun, S. Bell, T. Minson, and H. Browne.
1997. Site-directed and linker insertion mutagenesis of herpes simplex virus type 1 glycoprotein H. J. Virol.71:2163–2170.
12.Galdiero, S., A. Falanga, M. Vitiello, M. D’Isanto, C. Collins, V. Orrei, H. Browne, C. Pedone, and M. Galdiero. 2007. Evidence for a role of the membrane-proximal region of herpes simplex virus type 1 glycoprotein H in membrane fusion and virus inhibition. Chembiochem8:885–895. 13.Galdiero, S., A. Falanga, M. Vitiello, L. Raiola, R. Fattorusso, H. Browne, C.
Pedone, C. Isernia, and M. Galdiero.2008. Analysis of a membrane inter-acting region of herpes simplex virus type 1 glycoprotein H. J. Biol. Chem.
283:29993–30009.
14.Geraghty, R. J., C. R. Jogger, and P. G. Spear.2000. Cellular expression of alphaherpesvirus gD interferes with entry of homologous and heterologous alphaherpesviruses by blocking access to a shared gD receptor. Virology
268:147–158.
15.Geraghty, R. J., C. Krummenacher, G. H. Cohen, R. J. Eisenberg, and P. G. Spear.1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science280:1618–1620. 16.Gianni, T., M. Amasio, and G. Campadelli-Fiume.2009. Herpes simplex
virus gD forms distinct complexes with fusion executors gB and gH/gL in part through the C-terminal profusion domain. J. Biol. Chem.284:17370–17382. 17.Gianni, T., R. Fato, C. Bergamini, G. Lenaz, and G. Campadelli-Fiume.
2006. Hydrophobic alpha-helices 1 and 2 of herpes simplex virus gH interact with lipids, and their mimetic peptides enhance virus infection and fusion. J. Virol.80:8190–8198.
18.Gianni, T., P. L. Martelli, R. Casadio, and G. Campadelli-Fiume.2005. The ectodomain of herpes simplex virus glycoprotein H contains a membrane alpha-helix with attributes of an internal fusion peptide, positionally con-served in the Herpesviridae family. J. Virol.79:2931–2940.
19.Gianni, T., L. Menotti, and G. Campadelli-Fiume.2005. A heptad repeat in herpes simplex virus 1 gH, located downstream of the alpha-helix with attributes of a fusion peptide, is critical for virus entry and fusion. J. Virol.
79:7042–7049.
20.Gianni, T., A. Piccoli, C. Bertucci, and G. Campadelli-Fiume.2006. Heptad repeat 2 in herpes simplex virus 1 gH interacts with heptad repeat 1 and is critical for virus entry and fusion. J. Virol.80:2216–2224.
21.Harman, A., H. Browne, and T. Minson.2002. The transmembrane domain and cytoplasmic tail of herpes simplex virus type 1 glycoprotein H play a role in membrane fusion. J. Virol.76:10708–10716.
22.Heldwein, E. E., H. Lou, F. C. Bender, G. H. Cohen, R. J. Eisenberg, and S. C. Harrison.2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science313:217–220.
23.Hutchinson, L., H. Browne, V. Wargent, N. Davis-Poynter, S. Primorac, K. Goldsmith, A. C. Minson, and D. C. Johnson.1992. A novel herpes simplex virus glycoprotein, gL, forms a complex with glycoprotein H (gH) and affects normal folding and surface expression of gH. J. Virol.66:2240–2250. 24.Krummenacher, C., F. Baribaud, M. Ponce de Leon, I. Baribaud, J. C.
Whitbeck, R. Xu, G. H. Cohen, and R. J. Eisenberg.2004. Comparative usage of herpesvirus entry mediator A and nectin-1 by laboratory strains and clinical isolates of herpes simplex virus. Virology322:286–299.
25.Krummenacher, C., V. M. Supekar, J. C. Whitbeck, E. Lazear, S. A. Con-nolly, R. J. Eisenberg, G. H. Cohen, D. C. Wiley, and A. Carfi.2005. Struc-ture of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J.24:4144–4153.
26.Lin, E., and P. G. Spear.2007. Random linker-insertion mutagenesis to identify functional domains of herpes simplex virus type 1 glycoprotein B. Proc. Natl. Acad. Sci. U. S. A.104:13140–13145.
27.Manoj, S., C. R. Jogger, D. Myscofski, M. Yoon, and P. G. Spear.2004. Mutations in herpes simplex virus glycoprotein D that prevent cell entry via nectins and alter cell tropism. Proc. Natl. Acad. Sci. U. S. A.101:12414– 12421.
28.Maurer, U. E., B. Sodeik, and K. Grunewald.2008. Native 3D intermediates of membrane fusion in herpes simplex virus 1 entry. Proc. Natl. Acad. Sci. U. S. A.105:10559–10564.
29.Montgomery, R. I., M. S. Warner, B. J. Lum, and P. G. Spear.1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/ NGF receptor family. Cell87:427–436.
30.Novotny, M. J.1996. A structural and functional analysis of herpes simplex virus type 1 glycoprotein L, an essential component of the viral fusogenic complex. Ph.D. thesis. Northwestern University, Chicago, IL.
31.Peng, T., M. Ponce-de-Leon, H. Jiang, G. Dubin, J. M. Lubinski, R. J. Eisenberg, and G. H. Cohen.1998. The gH-gL complex of herpes simplex virus (HSV) stimulates neutralizing antibody and protects mice against HSV type 1 challenge. J. Virol.72:65–72.
32.Perez-Romero, P., A. Perez, A. Capul, R. Montgomery, and A. O. Fuller.
on November 8, 2019 by guest
http://jvi.asm.org/
2005. Herpes simplex virus entry mediator associates in infected cells in a complex with viral proteins gD and at least gH. J. Virol.79:4540–4544. 33.Pertel, P. E., A. Fridberg, M. L. Parish, and P. G. Spear.2001. Cell fusion
induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology279:313–324. 34.Roche, S., S. Bressanelli, F. A. Rey, and Y. Gaudin.2006. Crystal structure
of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science
313:187–191.
35.Roop, C., L. Hutchinson, and D. C. Johnson.1993. A mutant herpes simplex virus type 1 unable to express glycoprotein L cannot enter cells, and its particles lack glycoprotein H. J. Virol.67:2285–2297.
36.Satoh, T., and H. Arase.2008. HSV-1 infection through inhibitory receptor, PILRalpha. Uirusu58:27–36. (In Japanese.)
37.Satoh, T., J. Arii, T. Suenaga, J. Wang, A. Kogure, J. Uehori, N. Arase, I. Shiratori, S. Tanaka, Y. Kawaguchi, P. G. Spear, L. L. Lanier, and H. Arase.
2008. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell132:935–944.
38.Shukla, D., and P. G. Spear.2001. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J. Clin. Invest.108:503–510. 39.Spear, P. G., R. J. Eisenberg, and G. H. Cohen.2000. Three classes of cell
surface receptors for alphaherpesvirus entry. Virology275:1–8.
40.Spear, P. G., and R. Longnecker.2003. Herpesvirus entry: an update. J. Vi-rol.77:10179–10185.
41.Subramanian, R. P., and R. J. Geraghty.2007. Herpes simplex virus type 1 mediates fusion through a hemifusion intermediate by sequential activity of glycoproteins D, H, L, and B. Proc. Natl. Acad. Sci. U. S. A.104:2903–2908. 42.Turner, A., B. Bruun, T. Minson, and H. Browne.1998. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J. Virol.72:873– 875.
43.Wilson, D. W., N. Davis-Poynter, and A. C. Minson.1994. Mutations in the cytoplasmic tail of herpes simplex virus glycoprotein H suppress cell fusion by a syncytial strain. J. Virol.68:6985–6993.