0022-538X/96/$04.0010
Copyrightq1996, American Society for Microbiology
Mutational Analysis of ICP0R, a Transrepressor Protein
Created by Alternative Splicing of the ICP0 Gene
of Herpes Simplex Virus Type 1
STEPHEN J. SPATZ, ERIC C. NORDBY,ANDPETER C. WEBER*
Infectious Diseases Section, Parke-Davis Pharmaceutical Research, Division of Warner-Lambert Company, Ann Arbor, Michigan 48105
Received 10 May 1996/Accepted 26 July 1996
The immediate-early protein ICP0 (infected-cell polypeptide 0) of herpes simplex virus type 1 (HSV-1) is a promiscuous transactivator of both viral and nonviral promoters in transient expression assays. Failure to splice the second of two introns in the ICP0 gene results in the utilization of an alternate stop codon that generates a truncated form of ICP0 called ICP0R. This protein exists in low levels in HSV-1-infected cells and functions as a dominant negative repressor of ICP0-mediated transactivation in transient expression assays. To conduct a detailed structure-function analysis of ICP0R, a series of insertion and deletion mutants of this protein were generated and analyzed in transfection assays. These studies indicated that segments of ICP0R that were rich in acidic amino acid residues (amino acids 9 to 76 and 233 to 241) or glycine residues (amino acids 242 to 262) were dispensable for the dominant negative phenotype. In contrast, the RING finger domain (amino acids 116 to 156) and surprisingly the sequences carboxy terminal to it (amino acids 157 to 232) were absolutely essential for transdominant repression. Consistent with these findings, the amino acid sequences of these two regions were conserved among other alphaherpesvirus ICP0 homologs. A construct containing only amino acids 76 to 232 inhibited ICP0-mediated transactivation almost as efficiently as wild-type ICP0R and represented the minimal sequences necessary for the dominant negative phenotype. These results demon-strated that the critical functional domain shared by both ICP0R and ICP0 is much more complex than a simple RING finger motif. Western blot (immunoblot) analyses of transfected cell lysates revealed that nearly all of the mutant constructs directed the expression of stable ICP0R proteins of the predicted molecular weight. However, there was a striking inverse correlation between the ability of a mutant construct to mediate transrepression and the amount of protein that it synthesized, indicating that dominant negative inhibition is achieved through the action of very little ICP0R protein.
ICP0 (infected-cell polypeptide 0) of is one of five immedi-ate-early polypeptides encoded by herpes simplex virus type 1 (HSV-1). Mutant viruses containing deletions of the gene en-coding ICP0 are capable of replication on noncomplementing cell lines, indicating that the function provided by this polypep-tide is not absolutely essential for infection in cell culture. However, these null mutants do exhibit an impaired ability to replicate during low-multiplicity infections, resulting in reduc-tions in both plaque size and virus yield and the inefficient expression of all classes of viral genes (44, 50). These defects are augmented in infections from which the virion transactiva-tor protein VP16 is absent, such as transfection of naked viral DNA into cells or replication of HSV-1 in vivo (3, 4, 8, 27). In the latter situation, ICP0 null mutants display a marked reduc-tion in neurovirulence in mice and reactivate inefficiently from latent infections in trigeminal ganglion explant cocultivation experiments (4, 8, 27). This potential role of ICP0 in HSV-1 latent infections is further underscored in experiments using in vitro latency models; in such studies, ICP0 null mutants were shown to be completely defective for reactivation, and super-infection with a recombinant ICP0-expressing adenovirus was capable of reactivating latent HSV-1 (23, 56).
The behavior of ICP0 null mutant viruses indicates that this
protein plays an important role in upregulating viral genes, particularly during reactivation from latency. Consistent with this interpretation is the demonstration that ICP0 behaves as a potent transactivator in transfection assays: it can activate a wide variety of promoters in a manner that is independent of specific promoter elements (11, 22, 36, 40, 41, 43, 46, 47). Many of these transient expression studies also demonstrated that ICP0 can transactivate in synergy with the HSV-1 immediate-early protein ICP4. Direct interaction between these two trans-activators in vitro, as well as colocalization of the two proteins in discrete sites of the nuclei of infected and transfected cells, has been reported (37, 38, 55, 57). The mechanism by which ICP0 mediates transactivation is not known but is likely to involve interactions with one or more host cell proteins. Re-cently, ICP0 has been shown to specifically interact with a 135-kDa cellular protein (33, 34). Moreover, during infection it associates with and eventually disrupts the ND10 domain, a complex nuclear structure that contains a large number of cellular proteins including the acute promyelocytic leukemia oncoprotein PML (17, 30, 31). Although the nature of these interactions is unclear, they are likely to be critical for ICP0 function, as mutants which fail to either bind the 135-kDa protein or associate with and disperse ND10 are defective for transactivation and virus replication (17, 30, 33).
The gene encoding ICP0 is unusual in that it is one of the few HSV-1 genes that expresses a spliced transcript; its pro-tein-coding sequences are contained in three exons (42). Effi-cient splicing from the primary mRNA typically occurs during productive infection, so that translation of the processed
tran-* Corresponding author. Mailing address: Parke-Davis Pharmaceu-tical Research, Division of Warner-Lambert Company, Infectious Diseases Section, 2800 Plymouth Rd., Ann Arbor, MI 48105. Phone: (313) 998-3295. Fax: (313) 998-3318. Electronic mail address: [email protected].
7360
on November 9, 2019 by guest
http://jvi.asm.org/
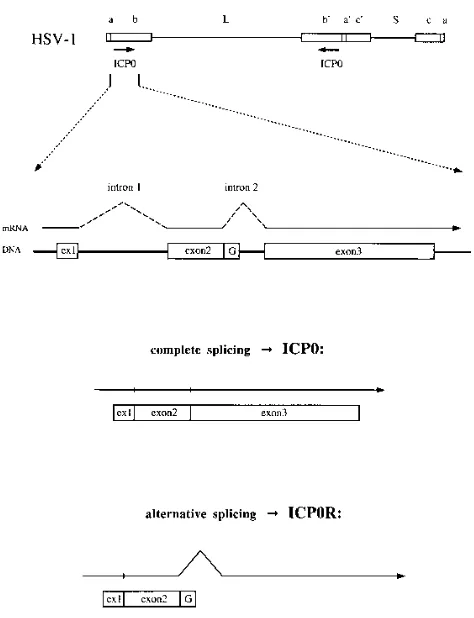
script yields a full-length 110-kDa polypeptide containing 775 amino acids (Fig. 1). However, retention of intron 2 by alter-native splicing of the primary mRNA results in the appearance of a novel truncated form of ICP0 that has been designated ICP0R (53). This 41-kDa polypeptide contains all 241 amino acids encoded by exons 1 and 2 of the ICP0 gene plus an additional 21 amino acids resulting from readthrough into the second intron (Fig. 1). ICP0R was first characterized in tran-sient expression assays, in which it was found to behave as a potent dominant negative repressor of ICP0 transactivation (53). It was subsequently detected in HSV-1-infected cells at levels that were significantly lower than those of the full-length ICP0 protein (16). The functional role of ICP0R in the HSV-1 life cycle is unclear, as a mutant virus which failed to express ICP0R as a result of a deletion of both ICP0 introns exhibited both normal replication in cell culture and normal establish-ment and reactivation from latency in mouse trigeminal gan-glion explant cocultivation experiments (14, 39). However, this virus has not yet been examined in more relevant animal model systems which assess the capability for in vivo reactivation from latent infections.
The mechanism by which ICP0R exerts dominant negative inhibition over ICP0 in transient expression assays is unknown, but several obvious possibilities can be ruled out. It is unlikely
that ICP0R interacts directly with ICP0 to form nonfunctional ICP0-ICP0R heterodimers, since promiscuous repression has also been observed between dominant negative mutants of ICP0 and other viral transactivator proteins (54). Moreover, exon 3-encoded sequences which have been shown to be nec-essary for multimerization in ICP0 (7, 33, 55) are absent in ICP0R. A more likely possibility is that ICP0 and ICP0R pos-sess a common functional domain which interacts with a host cell factor and that the domain present in ICP0R is capable of titrating or sequestering this factor from ICP0. The identity of this putative cellular protein is unknown, but it is presumably distinct from the 135-kDa protein and the ND10 nuclear do-main described above, since ICP0R lacks the exon 3-encoded sequences required for interactions with these proteins (17, 33).
Several interesting features are present in the amino acid sequences common to ICP0 and ICP0R. These include two regions rich in acidic amino acid residues (amino acids 1 to 115 and 233 to 241), two consensus casein kinase II phosphoryla-tion sites, and a cysteine-rich RING finger motif (amino acids 116 to 156). The RING finger domain is of particular interest because it represents the only region of the protein that is conserved between the ICP0 homologs of the alphaherpesvirus family and because it has been found on an increasing number of cellular proteins of diverse function (21, 28). For this reason, it is perhaps not surprising that the RING finger domain has been identified as the region of the ICP0 protein most sensitive to amino acid changes in every mutational analysis carried out to date (3, 5, 12, 13).
To better understand how the ICP0R protein is able to mediate dominant negative repression of transactivation by ICP0, an extensive mutational study of the ICP0R gene was undertaken in this work. The effects of disrupting specific do-mains of ICP0R on both repression of ICP0-mediated gene activation and stable ICP0R protein expression were analyzed in transient expression assays. The results obtained indicated that much of the ICP0R protein is dispensable for both of these activities, but that the RING finger domain and unex-pectedly a stretch of amino acids carboxy terminal to the RING finger were both essential for normal ICP0R function. One transrepression-competent mutant contained both of the latter regions in a polypeptide that was only 157 amino acids in length and therefore represented the minimal domain required for the dominant negative phenotype.
MATERIALS AND METHODS
Cell culture, transfection procedures, and CAT assays.Vero (African green monkey kidney) cells were used in all transfection experiments and were prop-agated in Dulbecco’s minimum essential medium supplemented with 10% fetal bovine serum. Cells were transfected in triplicate with equimolar amounts of plasmid constructs by using Lipofectin reagent (GIBCO-BRL Life Technologies, Gaithersburg, Md.). Briefly, 5mg of total plasmid DNA was resuspended in 2.5 ml of serum-free medium and mixed with 2.5 ml of serum-free medium contain-ing 75ml of Lipofectin. After incubation at 258C for 15 min, the liposome-DNA complex was added to overnight cultures of cells (106in 60-mm-diameter dishes)
that had been prewashed with serum-free medium. At 24 h posttransfection, the medium was replaced with fresh medium containing serum, and the cells were allowed to grow for another 24 h. The cells were then lysed, and extracts were prepared by using a detergent extraction procedure (54). The extracts were analyzed in chloramphenicol acetyltransferase (CAT) assays, in which then -butyrylated chloramphenicol products were extracted with mixed xylenes and quantified by liquid scintillation (54).
Western blot (immunoblot) analysis of mutant ICP0R polypeptides. Over-night cultures of Vero cells (106in 60-mm-diameter dishes) were transfected with
[image:2.612.60.297.65.374.2]5mg of mutant ICP0R-expressing plasmid DNA by using Lipofectin as described above. At 48 h posttransfection, the medium was removed and the cells were lysed directly in sodium dodecyl sulfate (SDS)-polyacrylamide gel loading buffer (125 mM Tris-HCl, 20% [vol/vol] glycerol, 2% [wt/vol] SDS, 25mg of bromo-phenol blue per ml, 5% [vol/vol]b-mercaptoethanol) to eliminate degradation of FIG. 1. Generation of the ICP0R protein through alternative splicing of the
primary mRNA transcript of the HSV-1 ICP0 gene. The map of the HSV-1 genome shown at the top identifies the long (L) and short (S) components; the inverted repeat sequencesa,b, andc; and the locations of the two copies of the ICP0 gene in thebsequences. The intron-exon structure of the ICP0 gene is shown below, as are the two potential splicing products of the primary mRNA transcript of the ICP0 gene. Complete splicing of the mRNA yields the full-length protein ICP0, which is encoded by all three exons of the gene. A failure to splice out intron 2 yields the truncated protein ICP0R, which is encoded by the first two exons of the gene plus a short stretch of unspliced intron 2 sequence that is rich in glycine codons (G). ex1, exon 1.
on November 9, 2019 by guest
http://jvi.asm.org/
ICP0R proteins by endogenous protease activities present in Vero cells. An amount of extract corresponding to 104
cells was loaded onto each lane of Tris-glycine-containing 8 to 16% polyacrylamide gels. Fractionated polypeptides were then electroblotted onto polyvinylidene difluoride membranes for use in Western blot analysis. Mouse monoclonal antibody 11060 (16), specific for exon 2 sequences of both ICP0 and ICP0R, was a generous gift from R. Everett (MRC Virology Unit, Glasgow, United Kingdom) and was used as the primary antibody (1:5,000 dilution). A goat anti-mouse immunoglobulin G alkaline phosphatase-conjugated antibody (Promega, Madison, Wis.) was used as the secondary anti-body (1:5,000 dilution) with a 5-bromo-4-chloro-3-indolylphosphate toluidinium-–nitroblue tetrazolium substrate (Kirkegaard & Perry Laboratories).
Construction of plasmids.All transfections were performed with an equimolar mixture of three plasmids: pTAAT-CAT, pKST or one of its mutant derivatives, and p111. pTAAT-CAT contains the promoter of the HSV-1 immediate-early gene ICP4 fused to the CAT gene and represented the reporter plasmid; its construction was described in an earlier work (54). pKST expressed the wild-type ICP0R protein of HSV-1; it contained an ICP0 gene which was truncated at the
Sau3AI site of intron 2 (53). To facilitate the insertion of mutated ICP0R coding sequences into pKST, theEcoNI site in the vector portion of this plasmid was destroyed by fill-in with the Klenow fragment of DNA polymerase I. This version of pKST was used as the parental plasmid for all ICP0R insertion and deletion mutants. p111 expressed the full-length ICP0 protein (12). As outlined below, a panel of p111 mutant plasmids constructed in previous studies on the ICP0 polypeptide (12, 13) served as the source of many of the linker insertion and deletion mutations that were incorporated into pKST.
The linker insertion mutants R1I, E25I, E3I, and pKST-E13I were created by replacing the 2.2-kbNotI-KpnI fragment of pKST with the 2.2-kbNotI-KpnI fragments of p111-R1, p111-E25, p111-E3, and p111-E13 (12, 13), respectively. Similarly, pKST-F1I, pKST-R2I, pKST-E8I, pKST-R3I, and pKST-E32I were created by replacing the 0.5-kbXhoI-EcoNI fragment of pKST with the 0.5-kbXhoI-EcoNI fragments of p111-F1, p111-R2, p111-E8, p111-R3, and p111-E32 (12, 13), respectively. pKST-A2I was created by replacing the 0.9-kbBamHI-EcoNI fragment of pKST with the 0.9-kbBamHI-EcoNI fragment of p111-A2 (12, 13).
The internal deletion mutants pKST-FXE and pKST-D21 were created by replacing the 2.2-kbNotI-KpnI fragment of pKST with the 2.1-kbNotI-KpnI fragments of p111-FXE and p111-D21 (12, 13), respectively. pKST-R2/E8, pKST-E8/E13, pKST-E13/R3, and pKST-R3/E32 were created by cloning the 0.6- to 0.8-kbBamHI-EcoRI fragments of p111-R2, p111-E8, p111-E13, and p111-R3 (12, 13), respectively, into theBamHI andEcoRI sites of pKST-E8I, pKST-E13I, pKST-R3I, and pKST-E32I, respectively. This resulted in the gen-eration of four new in-frame deletions, since theEcoRI linkers at the R2, E8, E13, R3, and E32 sites are all in frame with each other (12, 13).
The construction of the carboxy-terminal truncation mutations pHKT and pHXT has been described previously (54). pD2T was created by cloning the 0.8-kbBamHI-EcoRI fragment of p111-D2 (12, 13) into theBamHI andEcoRI sites of pHKT. pENT was created by removing the 2.5-kbHindIII-EcoNI frag-ment from pKST, converting itsEcoNI tail to a blunt end by fill-in with the Klenow fragment of DNA polymerase I, and inserting it into theHindIII and
SmaI sites of pSG424 (45). The presence of translation termination codons adjacent to theSmaI site of this vector resulted in the introduction of a trunca-tion mutatrunca-tion at theEcoNI site of the ICP0R gene. pnewT was constructed from a 12-bpEcoRI linker insertion mutation (59-CCCGAATTCGGG-9) that was engineered into the codon for amino acid 233 of ICP0R by using PCR. One of the PCR primers that was used mapped upstream of theKpnI site of the ICP0R gene (59-ATCCCGATCGTGAACGACC-39), while the other introduced a 12-bpEcoRI linker into the ICP0R sequence at the desired codon (59-CCCGA ATTCGGGCCGTGGTGGGCTCCGGGTGG-39). The resulting 183-bp ampli-fied product was digested withKpnI andEcoRI to yield a 75-bp fragment that was inserted into theKpnI andEcoRI sites of pHKT to create pnewT. The presence of translation termination codons adjacent to theEcoRI site of this plasmid resulted in the introduction of a truncation mutation at theEcoRI linker of the ICP0R gene.
The amino-terminal truncation mutants pKST-D1, pD19T-D1, and pKST-D1/ newT were created by replacing the 1.9-kbNotI-XhoI fragments of pKST, pD19T (54), and pnewT, respectively, with the 1.1-kbNotI-XhoI fragment of p111-D1 (12, 13). pKST-R1/X was created in two steps. First, the 0.5-kb XhoI-XbaI fragment of pKST was cloned into theSalI andXbaI sites of pSG424. The presence of anEcoRI site in the vector sequences immediately adjacent to the ligatedXhoI-SalI site enabled this to be cloned back out as a 0.5-kbEcoRI-XbaI fragment and inserted into theEcoRI andXbaI sites of pKST-R1I. This intro-duced an in-frame deletion between the R1 linker in exon 1 and theXhoI site in exon 2 of the ICP0R gene. pKST-R1/M was created by cloning the 0.3-kb
MunI-SstI fragment of pKST into theEcoRI andSstI sites in pKST-R1I, thereby introducing an in-frame deletion between the R1 linker in exon 1 and theMunI site in exon 2 of the ICP0R gene. The specific amino acid residues of the ICP0R protein that were altered or deleted by each of the 26 mutations introduced into the pKST plasmid are summarized in Table 1.
RESULTS
Analysis of linker insertion and internal deletion mutants of
ICP0R in a transient transrepression assay. To examine the
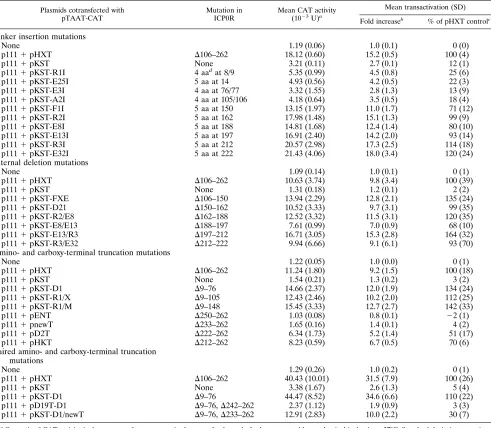
ability of ICP0R derivatives carrying defined mutations to me-diate dominant negative inhibition of transactivation by ICP0, a transient transrepression assay developed in an earlier work (54) was used. This involved cotransfection of three plasmids into Vero cells: the reporter plasmid pTAAT-CAT, which con-tains the promoter of the HSV-1 immediate-early gene ICP4 fused to the CAT gene; the ICP0R-expressing plasmid pKST; and the ICP0-expressing plasmid p111. As a control transfec-tion to determine the level of transactivatransfec-tion by ICP0 that occurs in the absence of a functional ICP0R protein, the ICP0 null mutant-encoding plasmid pHXT was substituted for pKST. The concentrations of plasmid and Lipofectin transfec-tion reagent that were used in the assay were varied until optimization of both transactivation by ICP0 and transrepres-sion by ICP0R were achieved. The final protocol routinely yielded a 10- to 30-fold induction of the ICP4 promoter by cotransfected pHXT and p111 plasmids; this induction was typically reduced by greater than 95% in transfections using cotransfected plasmids pKST and p111 (Table 1).
To use this transrepression assay in a structure-function analysis of the ICP0R protein, a series of mutant derivatives of pKST were substituted for wild-type pKST in the transfection mixture to ascertain the effect of specific mutations on the dominant negative phenotype. The initial panel of mutants studied represented a series of 10 linker insertion mutations which created small four- or five-amino-acid insertions at spe-cific sites in the ICP0R polypeptide (Fig. 2 and Table 1); these were originally created in a mutational analysis of the ICP0 protein (12, 13) and were incorporated into the ICP0R-coding sequences by simply swapping homologous restriction frag-ments. In the analysis of these mutants and of the ones de-scribed later, the results are presented both as the fold increase in CAT activity over that of a control transfection lacking the ICP0 transactivator and as a normalized percentage of the optimal CAT activity obtained in a control transfection con-taining pHXT instead of pKST (Table 1).
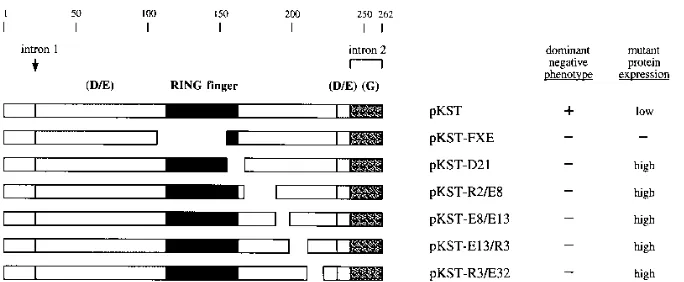
Four of the linker insertion mutations had little effect on the transrepression phenotype of ICP0R, in that polypeptides bearing these alterations still promoted a reduction in ICP0-mediated transactivation of greater than 75% (Table 1). These mutations, in plasmids pKST-R1I, pKST-E25I, pKST-E3I, and pKST-A2I, all mapped to the amino-terminal region of the protein between amino acids 9 and 105 (Fig. 2). In contrast, the six remaining linker insertion mutations greatly impaired trans-repression by ICP0R, in that polypeptides bearing these alter-ations reduced ICP0-mediated transactivation by less than 30% and in some cases not at all (Table 1). One of these mutations, in plasmid pKST-F1I, mapped at amino acid 150 within the RING finger domain of ICP0R; the remaining mu-tations, in plasmids R2I, E8I, E13I, pKST-R3I, and pKST-E32I, mapped carboxy terminal to the RING finger domain between amino acids 162 and 222 (Fig. 2).
The observation that the RING finger domain mutation in pKST-F1I crippled transrepression in ICP0R was not surpris-ing, given the importance of this region to transactivation by ICP0 (3, 5, 12, 13) and its conservation among other alphaher-pesvirus ICP0 homologs (6, 9, 20, 32, 42, 51). However, the finding that an extended stretch of sequences carboxy terminal to the RING finger domain was exquisitely sensitive to muta-tions was unexpected. Therefore, the contribution of this re-gion of ICP0R to transrepression was examined further by using a panel of in-frame deletions (Fig. 3 and Table 1); these
on November 9, 2019 by guest
http://jvi.asm.org/
removed short sections of the ICP0R polypeptide 10 to 45 amino acids in length which mapped within the RING finger domain and the region carboxy terminal to it.
Consistent with the results obtained with the pKST-F1I linker insertion mutation, those constructs which possessed deletions that removed portions of the RING finger domain (pKST-FXE and pKST-D21 [Fig. 3]) were completely defec-tive for transrepression (Table 1). More importantly, all of the constructs containing deletions that mapped downstream of the RING finger domain between amino acids 162 and 222 (pKST-R2/E8, pKST-E8/E13, pKST-E13/R3, and pKST-R3/ E32 [Fig. 3]) were either critically impaired or totally defective for repression of ICP0-mediated transactivation (Table 1). These results were in complete agreement with those obtained in the analysis of the linker insertion mutants spanning the
same region (Table 1) and demonstrated that aside from the obvious requirement for the RING finger domain, an adjacent stretch of 66 amino acids was also essential for ICP0R func-tion.
Definition of the minimal domain of ICP0R necessary for
transrepression. The results of the mutational analysis
de-scribed above had indicated that a segment of the ICP0R polypeptide containing the RING finger domain and se-quences downstream to amino acid 222 were sensitive to even minor changes in protein structure, resulting in the loss of transrepression capability. It was of interest to ascertain what portion of the sequences flanking this essential region could be deleted without affecting ICP0R function, in order to deter-mine whether the same domain that was necessary for transre-pression would also be sufficient for it. Therefore, pKST
de-TABLE 1. Dominant negative inhibition of ICP0 transactivation by mutant ICP0R derivatives
Plasmids cotransfected with pTAAT-CAT
Mutation in ICP0R
Mean CAT activity (1023U)a
Mean transactivation (SD)
Fold increaseb
% of pHXT controlc
Linker insertion mutations
None 1.19 (0.06) 1.0 (0.1) 0 (0)
p1111pHXT D106–262 18.12 (0.60) 15.2 (0.5) 100 (4)
p1111pKST None 3.21 (0.11) 2.7 (0.1) 12 (1)
p1111pKST-R1I 4 aadat 8/9 5.35 (0.99) 4.5 (0.8) 25 (6)
p1111pKST-E25I 5 aa at 14 4.93 (0.56) 4.2 (0.5) 22 (3)
p1111pKST-E3I 4 aa at 76/77 3.32 (1.55) 2.8 (1.3) 13 (9)
p1111pKST-A2I 4 aa at 105/106 4.18 (0.64) 3.5 (0.5) 18 (4)
p1111pKST-F1I 5 aa at 150 13.15 (1.97) 11.0 (1.7) 71 (12)
p1111pKST-R2I 5 aa at 162 17.98 (1.48) 15.1 (1.3) 99 (9)
p1111pKST-E8I 5 aa at 188 14.81 (1.68) 12.4 (1.4) 80 (10)
p1111pKST-E13I 5 aa at 197 16.91 (2.40) 14.2 (2.0) 93 (14)
p1111pKST-R3I 5 aa at 212 20.57 (2.98) 17.3 (2.5) 114 (18)
p1111pKST-E32I 5 aa at 222 21.43 (4.06) 18.0 (3.4) 120 (24)
Internal deletion mutations
None 1.09 (0.14) 1.0 (0.1) 0 (1)
p1111pHXT D106–262 10.63 (3.74) 9.8 (3.4) 100 (39)
p1111pKST None 1.31 (0.18) 1.2 (0.1) 2 (2)
p1111pKST-FXE D106–150 13.94 (2.29) 12.8 (2.1) 135 (24)
p1111pKST-D21 D150–162 10.52 (3.33) 9.7 (3.1) 99 (35)
p1111pKST-R2/E8 D162–188 12.52 (3.32) 11.5 (3.1) 120 (35)
p1111pKST-E8/E13 D188–197 7.61 (0.99) 7.0 (0.9) 68 (10)
p1111pKST-E13/R3 D197–212 16.71 (3.05) 15.3 (2.8) 164 (32)
p1111pKST-R3/E32 D212–222 9.94 (6.66) 9.1 (6.1) 93 (70)
Amino- and carboxy-terminal truncation mutations
None 1.22 (0.05) 1.0 (0.0) 0 (1)
p1111pHXT D106–262 11.24 (1.80) 9.2 (1.5) 100 (18)
p1111pKST None 1.54 (0.21) 1.3 (0.2) 3 (2)
p1111pKST-D1 D9–76 14.66 (2.37) 12.0 (1.9) 134 (24)
p1111pKST-R1/X D9–105 12.43 (2.46) 10.2 (2.0) 112 (25)
p1111pKST-R1/M D9–148 15.45 (3.33) 12.7 (2.7) 142 (33)
p1111pENT D250–262 1.03 (0.08) 0.8 (0.1) 22 (1)
p1111pnewT D233–262 1.65 (0.16) 1.4 (0.1) 4 (2)
p1111pD2T D222–262 6.34 (1.73) 5.2 (1.4) 51 (17)
p1111pHKT D212–262 8.23 (0.59) 6.7 (0.5) 70 (6)
Paired amino- and carboxy-terminal truncation mutations
None 1.29 (0.26) 1.0 (0.2) 0 (1)
p1111pHXT D106–262 40.43 (10.01) 31.5 (7.9) 100 (26)
p1111pKST None 3.38 (1.67) 2.6 (1.3) 5 (4)
p1111pKST-D1 D9–76 44.47 (8.52) 34.6 (6.6) 110 (22)
p1111pD19T-D1 D9–76,D242–262 2.37 (1.12) 1.9 (0.9) 3 (3)
p1111pKST-D1/newT D9–76,D233–262 12.91 (2.83) 10.0 (2.2) 30 (7)
a
One unit of CAT activity is the amount of enzyme required to transfer 1 nmol ofn-butyrate to chloramphenicol in 1 min at 378C. Standard deviations are given in parentheses.
b
Transactivation of pTAAT-CAT by the ICP0 gene in p111 as measured by the fold increase in CAT activity over that of pTAAT-CAT-transfected alone.
c
Transactivation of pTAAT-CAT by the ICP0 gene in p111 interpolated from a scale whereby the control transfections of pTAAT-CAT plus p111 and pHXT and of pTAAT-CAT alone represent 100 and 0% transactivation, respectively.
d
aa, amino acids.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.65.556.82.510.2]rivatives containing amino-terminal and carboxy-terminal truncations in the ICP0R coding sequences were constructed and assayed for the ability to inhibit ICP0-induced transacti-vation (Fig. 4 and Table 1).
Unlike the four linker insertion mutations mapping between amino acids 9 and 105 that had little effect on transrepression (Table 1), the three deletions within the amino-terminal end of the ICP0R protein that were analyzed (pKST-D1, pKST-R1/X, and pKST-R1/M [Fig. 4]) completely abolished the dominant negative phenotype (Table 1). However, as discussed below, the ability of the deletion in pKST-D1 to affect protein func-tion varied significantly with the type of carboxy-terminal se-quences present in a given ICP0R derivative. In contrast, two of the four carboxy-terminal truncation mutants tested (pENT and pnewT [Fig. 4]) exhibited wild-type levels of transrepres-sion (Table 1), while the remaining two (pD2T and pHKT) possessed residual levels of transrepression (only a 30 to 50% reduction in ICP0-induced transactivation [Table 1]). These results indicated that removal of the carboxy-terminal 30
amino acids of ICP0R had no effect on protein function but that deletion of an additional 12 to 22 amino acids had a significant effect on the dominant negative phenotype of these polypeptides. In fact, the reduction in transrepression activity in the latter two mutants was consistent with the results ob-tained earlier with pKST-R3I, pKST-E32I, and pKST-R3/E32 (Table 1), since all of these plasmids possessed mutations within a short stretch of essential sequences from amino acids 212 to 222.
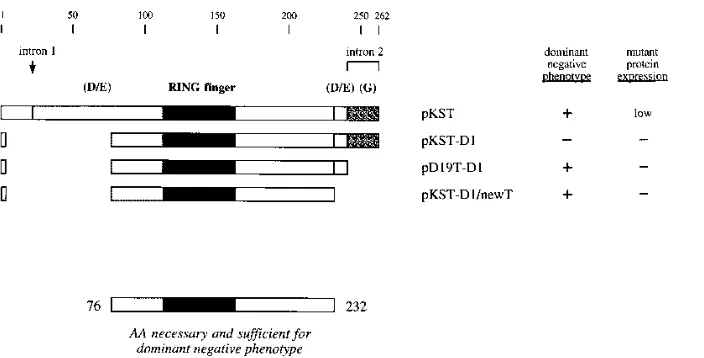
[image:5.612.141.477.67.267.2]The finding that the D1 deletion encompassing amino acids 9 to 76 destroyed the ability of pKST to mediate transrepres-sion (pKST-D1 [Table 1]) was somewhat unexpected, since all three linker insertion mutations mapping within this same re-gion had little effect on the dominant negative phenotype (pKST-R1I, pKST-E25I, and pKST-E3I [Table 1]). However, the same D1 deletion had little effect on transrepression after it was incorporated into pD19T and pnewT, two pKST deriv-atives that already contained truncations of the carboxy-termi-nal 21 and 30 amino acids of ICP0R, respectively (pD19T-D1
FIG. 2. Structures of linker insertion mutants of the ICP0R-expressing plasmid pKST. Features of the wild-type ICP0R polypeptide encoded by pKST that are identified at the top include amino acid numbers, the site of intron 1 splicing, the amino acids encoded by unspliced intron 2 sequences, the RING finger motif (dark box), two regions rich in acidic amino acid residues [light boxes labeled (D/E)], and the region rich in glycine residues [stippled box labeled (G)]. The locations of linker insertion mutations that introduce four- or five-amino-acid insertions into the ICP0R polypeptide sequence are denoted by small black boxes. The presence or absence of a dominant negative phenotype was based on the data in Table 1 and is summarized at the right;1and2indicate that the mutant promoted a reduction in ICP0-mediated transactivation of greater than 75% and less than 50%, respectively. The ability of each plasmid to promote high- or low-level expression of ICP0R is based on the Western blot results in Fig. 6 and is listed at the far right.
FIG. 3. Structures of internal deletion mutants of the ICP0R-expressing plasmid pKST. The indicated features of the ICP0R protein and the summaries of the phenotypic properties of its mutants are explained in the legend to Fig. 2. Deleted amino acid residues are denoted by gaps in each polypeptide map.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.139.481.566.707.2]and pKST-D1/newT [Fig. 5 and Table 1]). These results indi-cated that the negative effect of the D1 deletion on a given pKST derivative could be effectively nullified by second-site mutations present in the carboxy-terminal end of the protein. More importantly, the finding that the pKST-D1/newT mutant retained nearly all of the transrepression activity of wild-type pKST plasmid (Table 1) served to delineate the minimal do-main required for ICP0R function. Together with the results of the mutational analysis described above, the behavior of the pKST-D1/newT mutant demonstrated that the amino acids from 76 to 232 represented the minimal sequence of ICP0R that was both necessary and sufficient for transrepression (Fig. 5).
Analysis of ICP0R polypeptides synthesized in cells
trans-fected with mutant derivatives of pKST. The mutations in
pKST that eliminated the ability of this plasmid to mediate transrepression could do so through two obvious mechanisms: they may disrupt a domain of ICP0R that is essential for function, or they may create an unstable ICP0R polypeptide that is quickly degraded. To distinguish between these two possibilities, extracts from Vero cells transfected with each of
[image:6.612.142.484.69.238.2]the pKST mutant plasmids were examined for the levels of ICP0R protein detectable by Western blot analysis (Fig. 6). The blotted lysates were probed with the ICP0 exon 2-specific monoclonal antibody 11060, which had been previously shown to recognize the ICP0R protein in HSV-1-infected cells (16). Analysis of extracts from cells that had been transfected with wild type pKST revealed specific recognition of a 41 kDa protein by antibody 11060 (Fig. 6); the molecular weight of this species was identical to that reported previously for ICP0R in HSV-1-infected cells (16). Nearly all of the pKST mutants synthesized a protein that was detectable by antibody 11060 (Fig. 6). In mutants containing linker insertions or small inter-nal deletions, the molecular weight of ICP0R was not appre-ciably altered; however, in mutants containing larger carboxy-terminal truncations, the proteins detected exhibited increases in mobility that were consistent with the reductions in molec-ular weight predicted for the mutants. Only six of the pKST mutants failed to express a polypeptide that was detectable by antibody 11060. Five of these, D1, R1/X, pKST-R1/M, pD19T-D1, and pKST-D1/newT, contained deletions that partially or completely spanned amino acids 20 to 105 of
FIG. 4. Structures of amino-terminal and carboxy-terminal truncation mutants of the ICP0R-expressing plasmid pKST. The indicated features of the ICP0R protein and the summaries of the phenotypic properties of its mutants are explained in the legend to Fig. 2. Deleted amino acid residues are denoted by gaps in each polypeptide map.
FIG. 5. Structures of paired amino-terminal and carboxy-terminal truncation mutants of the ICP0R-expressing plasmid pKST. The indicated features of the ICP0R protein and the summaries of the phenotypic properties of its mutants are explained in the legend to Fig. 2. Deleted amino acid (AA) residues are denoted by gaps in each polypeptide map. The sequences present in the minimal core domain of ICP0R are designated at the bottom.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.141.493.518.697.2]ICP0R. Since this region of the protein was previously shown to be essential for recognition by antibody 11060 (16), it is likely that mutant polypeptides were synthesized by these five plasmids but remained undetected because of the specificity of the antibody used. This conclusion is supported by the obser-vation that two of these mutants, pD19T-D1 and pKST-D1/ newT, exhibited strong transrepression in transfection assays (Fig. 5 and Table 1), which indicated that they had indeed produced a functional ICP0R derivative. The sixth pKST mu-tant which failed to express a polypeptide that was detectable by antibody 11060 was pKST-FXE; since the deletion in this mutant mapped outside the ICP0R sequences necessary for antibody recognition, it likely represented the only construct that synthesized an unstable polypeptide.
One unexpected observation in these experiments was the dramatic differences in the levels of expression between differ-ent pKST mutants; the range of this variance was estimated by densitometric analysis to be greater than 15-fold (49). Inspec-tion of the sequences involved in this phenomenon indicated a striking correlation between the ability of a given mutation to abrogate transrepression by pKST and its ability to permit high-level expression of ICP0R (summarized in Fig. 2 to 5). Since these experiments had used transfection of pKST mutant constructs in the absence of other plasmids, it was of interest to determine whether this apparent relationship between func-tional activity and reduced protein synthesis was also manifest under those conditions in which the transient transrepression assays had originally been carried out. Western blot analyses were therefore carried out on lysates of cells transfected with equimolar mixtures of pTAAT-CAT, p111, and various pKST mutants; representative results for two of these mutants,
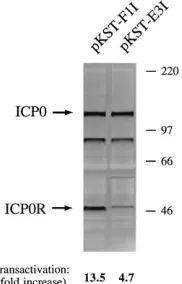
pKST-E3I and pKST-F1I, are shown in Fig. 7. The relative levels of expression of pKST mutants in the transrepression assay were comparable to those observed in the Western blot analysis (Fig. 6), since the transrepression-competent mutant pKST-E3I directed the synthesis of considerably lower amounts of protein than the transrepression-defective mutant pKST-F1I (Fig. 7). In contrast, there was no significant differ-ence in the levels of ICP0 protein that were produced by the cotransfected plasmid p111 in these experiments. The poten-tial implications of these observations are discussed below.
Thus, nearly all of the pKST mutants produced stable pro-teins whose existence could be directly confirmed by Western blot analysis or inferred from transient transrepression assay results. Those mutants which were defective for transrepres-sion synthesized considerably larger amounts of protein than those which retained this capability. This result demonstrated that mutations in ICP0R which abolished transrepression did so by disrupting a domain of the polypeptide that was essential for function, rather than creating unstable or poorly expressed versions of an otherwise active protein.
DISCUSSION
[image:7.612.392.483.71.213.2]Several years ago, mutational studies of the ICP0 gene of HSV-1 demonstrated that removal of coding sequences of the third exon resulted in mutants which possessed a dominant negative phenotype in transient expression assays (54). Since alternative splicing of the second intron of the ICP0 mRNA during HSV-1 infection was predicted to create a protein that would be structurally similar to these mutants (Fig. 1), a plas-mid expressing this hypothetical polypeptide was characterized in transient expression assays. Like the engineered dominant negative mutants, this protein was found to act as a potent repressor of ICP0-induced transactivation and was therefore named ICP0R (53). It was subsequently detected in HSV-1-infected cells, but at much lower levels than the full-length ICP0 protein (16). Since it acts as an antagonist of the ICP0 protein, which plays a critical role in the activation of HSV-1 genes during lytic replication and reactivation from latency (4,
[image:7.612.59.296.73.264.2]FIG. 6. Western blot analysis of ICP0R polypeptides expressed in transfected Vero cells. Extracts from Vero cells transfected with each of the pKST mutant plasmids were prepared, blotted, and probed with the ICP0 exon 2-specific monoclonal antibody 11060 (16). The mutant plasmid used in each transfection is indicated above each lane; plasmids bearing linker insertion or internal dele-tion mutadele-tions are included in the upper panel, while plasmids containing amino-or carboxy-terminal truncation mutations are presented in the lower panel. The molecular mass of ICP0R and most of the mutant polypeptides is 41 kDa, which is indicated by an arrow on the molecular mass scale on the right. The proteins expressed by those plasmids bearing carboxy-terminal truncation mutations have a markedly reduced molecular mass and are indicated in the lower panel by black dots within the lanes. Two additional mutants not shown on these blots, pD19T-D1 and pKST-D1/newT, failed to yield detectable protein (49). Like pKST-D1, pKST-R1/X, and pKST-R1/M, they lacked the coding sequences for amino acids 9 to 76, which are required for detection by antibody 11060 (16).
FIG. 7. Western blot analysis of ICP0 and ICP0R polypeptides expressed during transient transrepression assays. Extracts from Vero cells transfected with equimolar amounts of pTAAT-CAT, p111, and the pKST mutant plasmids indicated above the lanes were prepared, blotted, and probed with the ICP0 exon 2-specific monoclonal antibody 11060 (16). The ICP0 and ICP0R proteins are indicated by arrows on the left; a molecular mass scale (in kilodaltons) is in-cluded on the right. The corresponding levels of transactivation that were ob-served in the various experiments are indicated below the lanes. These values were determined by quantifying CAT activity in lysates prepared from additional sets of cells that had been transfected at the same time as those used in the Western blot analyses and were calculated as described in Table 1, footnotesa
andb.
on November 9, 2019 by guest
http://jvi.asm.org/
8, 27), it is tempting to speculate that ICP0R acts to down-regulate viral gene expression. However, the true contribution of this protein to the infectious cycle of HSV-1 remains un-known.
In an attempt to identify domains of the ICP0R protein that are critical for its transrepression phenotype, a series of linker insertion, internal deletion, and amino- and carboxy-terminal truncation mutations were introduced into the ICP0R-express-ing plasmid pKST (Fig. 2 to 5). The ability of these mutants to repress transactivation by the wild-type ICP0 protein was then examined in transient expression assays using cotransfection of pKST, the ICP0-expressing plasmid p111, and a CAT reporter construct (Table 1). This analysis revealed that the ICP0R could be clearly delineated into (i) a central core domain (amino acids 76 to 232) that was both necessary and sufficient for transrepression and (ii) flanking amino-terminal and car-boxy-terminal domains that were not required for activity.
Sequences of ICP0R that were dispensable for
transrepres-sion.One of the two nonessential domains of ICP0R mapped
to the first 76 amino acids of the protein, as determined by the finding that mutants containing linker insertions at amino acids 8 (pKST-R1), 14 (pKST-E25), and 76 (pKST-E3), or a deletion of the entire region (pD19T-D1 and pKST-D1/newT), exhib-ited normal transrepression (Table 1). Interestingly, this do-main was found to exert an anomalous effect on ICP0R func-tion when analyzed in combinafunc-tion with different carboxy-terminal truncations: in pKST-D1, which contained the normal carboxy-terminal sequences of ICP0R, it was essential for trans-repression, while in the carboxy-terminal truncation mutants pD19T-D1 and pKST-D1/newT, it was dispensable (Table 1). The simplest interpretation of these observations is that dele-tion of the first 76 amino acids of ICP0R resulted in a delete-rious effect on function, but that the introduction of short deletions at the opposite end of the protein relieved this inhi-bition. The carboxy-terminal truncations therefore acted as second-site suppressor mutations that fortuitously revealed the dispensability of the amino-terminal segment of ICP0R for transrepression. Alternatively, the first 76 amino acids of ICP0R could still constitute a functional domain which re-quired the presence of the carboxy-terminal sequences of the protein in order to mediate transrepression. This possibility is considered to be somewhat less attractive, since these amino-terminal sequences were shown to be dispensable for both transactivation in ICP0 (12, 13, 49) and transrepression in the pKST-D1/newT mutant of ICP0R (Table 1). As discussed be-low, the latter mutant already defines the minimal functional domain of ICP0R, making it highly improbable that its deleted amino-terminal sequences represented a redundant second functional domain. Moreover, the majority of the ICP0R mu-tants which failed to mediate transrepression in this study did so despite the presence of this putative domain (Table 1).
These results indicated that several interesting features in the amino acid sequence of the amino-terminal region of ICP0R, including two consensus casein kinase II phosphoryla-tion sites and a segment containing an overrepresentaphosphoryla-tion of charged amino acid residues, are apparently not required for transrepression. The latter is part of a larger region from amino acids 1 to 114 that contains 9 basic and 30 acidic resi-dues, which is reminiscent of the acidic activation domains found in a large number of eukaryotic transcription factors (25, 29, 52). However, deletion of amino acids 9 to 76 in mutants pD19T-D1 and pKST-D1/newT, which removes all but 3 basic and 10 acidic residues from this region, has little effect on transrepression (Table 1). These results indicated that this portion of the ICP0R polypeptide is unlikely to function as an acidic activation domain, a conclusion which is supported by
the inability of these same sequences to activate transcription in mammalian cells when fused to the DNA binding domain of the yeast protein GAL4 (49).
The other nonessential domain mapped to the last 30 amino acids of ICP0R, as judged from the finding that mutants con-taining a deletion of this region (pnewT and pKST-D1/newT) exhibited normal transrepression (Table 1). Like its amino-terminal counterpart, the amino acid sequence of this dispens-able region also contains several interesting features that are apparently not required for transrepression, including short stretches that are rich in either acidic residues (amino acids 233 to 241) or glycine residues (amino acids 242 to 262). The latter domain was of particular interest, as it contained a strik-ing overrepresentation of glycine (13 residues out of a total of 21), and it represented the only portion of ICP0R that was unique from ICP0 since it was derived exclusively from the coding sequences of intron 2. However, the finding that these regions were dispensable for transrepression was in fact con-sistent with results from two previous studies. In the first, deletion of the short acidic region at amino acids 233 to 241 from ICP0 was found to have no effect on transactivation or stimulation of virus replication by this protein (14). In the second, substitution of the glycine-rich region at amino acids 242 to 262 of ICP0R with various lengths of unrelated se-quences derived from exon 3 of the ICP0 gene did not alter the transrepression competence of these derivatives (54).
Sequences in ICP0R that comprised the core functional
domain.Analysis of the pKST-D1/newT mutant revealed that
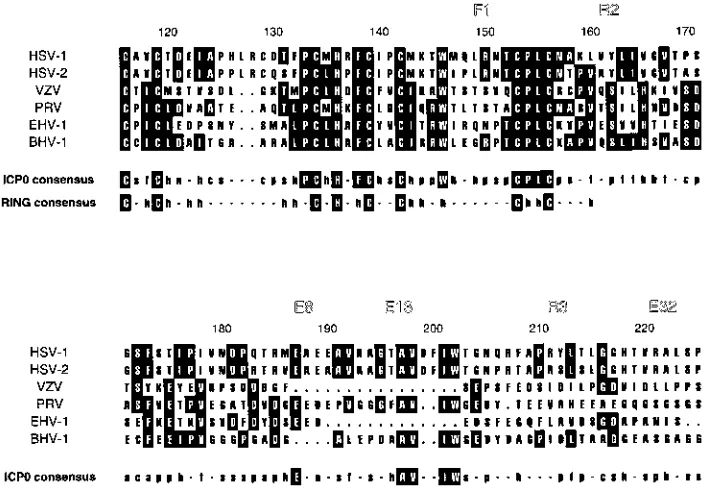
both the amino- and carboxy-terminal dispensable domains could be removed from ICP0R with minimal loss of function (Table 1). Moreover, this mutant demonstrated that ICP0R could be reduced to a core domain of amino acids 76 to 232 that was both necessary and sufficient for transrepression. The most prominent feature of this minimal functional domain was the RING finger homology at amino acids 116 to 156. Since this sequence motif is highly conserved in all known ICP0 homologs of alphaherpesviruses (Fig. 8) and represents the most mutation-sensitive region of the ICP0 protein (3, 5, 12, 13), its inclusion within the core domain of ICP0R was not unexpected. Nevertheless, the minimal functional domain also contained an extended stretch of amino acids (157 to 232) that mapped carboxy terminal to the RING finger; the importance of this region to transrepression was further underscored by its extreme sensitivity to every linker insertion, internal deletion, and carboxy-terminal truncation mutation that was tested (Ta-ble 1). The essential nature of this portion of ICP0R was somewhat surprising, since previous homology searches failed to detect any obvious relatedness between alphaherpesvirus ICP0 homologs within this region (28). However, if homology analyses are broadened to include identification of matches of amino acid residues with similar functional groups, a consid-erable amount of sequence similarity is uncovered in the re-gion downstream of the RING finger domain of alphaherpes-virus ICP0 homologs (Fig. 8).
Thus, the core domain of ICP0R possesses two predominant features: a 41-amino-acid RING finger motif that is highly conserved within not only the ICP0 gene family but also a variety of other cellular proteins (21, 28) and a 76-amino-acid region which exhibits a more modest level of sequence conser-vation that falls exclusively within the ICP0 gene family. Clearly, much more is known about the former component, to the extent that three-dimensional structures of two different RING finger domains have even been elucidated (1, 2, 15). However, the results of this study suggest that this structure, at least in the case of ICP0 and ICP0R, is actually just a small portion of a much larger functional domain. Interestingly,
on November 9, 2019 by guest
http://jvi.asm.org/
since the original identification of dominant negative mutants of ICP0 (54), similar mutants have been found in other RING
finger proteins, including the oncoprotein PML and the
Dro-sophila apoptosis inhibitor proteins DIAP1 and DIAP2 (24, 26). The common feature of all of these mutants is that they have lost one or more functional domains but retained their RING finger motifs, suggesting that transdominance may be an intrinsic feature of RING fingers when isolated from their parental proteins.
The finding that sequences which flank the RING finger were also essential for function may help to explain two puz-zling observations from recent experiments involving this struc-tural entity. First, attempts to swap RING finger domains between different proteins and still retain function have met with little success (10, 18). However, exchanging amino acids 1 to 245 of ICP0 with the corresponding sequences of the ICP0 homolog of varicella-zoster virus (VZV) did result in func-tional chimeric proteins (35). The results of this study suggest that the latter swap was possible because the sequences that were interchanged included not only the RING finger domain but also the entire core domains of ICP0 and ICP0R. Second, studies with the ICP0 homolog in VZV have revealed that like HSV-1 ICP0, many carboxy-terminal truncation mutations will impart a dominant negative phenotype on this protein (35). However, these truncations could extend up to amino acid 79 of the VZV ICP0 homolog, a residue which corresponds to amino acid 177 of ICP0R (Fig. 8), without abrogating transre-pression (35). The shortest carboxy-terminal truncation mu-tant in ICP0R which retained transrepression mapped to only amino acid 232 (pnewT [Table 1]), which implied that the
minimal functional domain of the ICP0 homolog in VZV was 56 amino acids shorter than that of ICP0R. It is likely that the significant amino acid sequence heterology between these two proteins in both the RING finger domain and its flanking sequences (Fig. 8) influences the overall tertiary structure and therefore the minimal length of the core domain. Such differ-ences have been shown to have a profound effect on the three-dimensional structure of just the RING finger in the absence of these additional sequences (1, 2, 15), which again underscores the potential contribution of amino acid residues outside this region to the overall integrity of the core domain.
Implications for the mechanism of transrepression by
ICP0R. It is of interest that all of the linker insertion and
internal deletion mutations that mapped within the minimal functional domain invariably abolished not only transrepres-sion in ICP0R (Table 1) but also transactivation when analyzed in the context of the coding sequence of ICP0 (12, 13, 49). Thus, the 157 amino acids which comprise the core domain of ICP0R are also essential for function in ICP0, suggesting that a similar if not identical domain exists within the ICP0 protein. It has been previously postulated that ICP0R exerts its domi-nant negative effect over ICP0 by titrating an essential cellular factor (53, 54). If this interpretation is correct, then the core domain common to ICP0 and ICP0R is likely to act as the interface through which the binding of this factor occurs. This interaction would be nonproductive in the case of ICP0R but would result in transactivation in the case of ICP0, presumably as a result of the presence of an additional exon 3-encoded domain.
[image:9.612.130.483.71.315.2]There are additional mechanisms which could explain the
FIG. 8. Multiple alignment of RING finger motifs and adjacent downstream sequences in alphaherpesvirus ICP0 homologs. The sequences of amino acids 116 to 225 of HSV-1 ICP0 (42), amino acids 126 to 235 of HSV-2 ICP0 (32), amino acids 19 to 110 of VZV gene 61 (9), amino acids 46 to 150 of pseudorabies virus (PRV) EP0 (6), amino acids 8 to 100 of equine herpesvirus 1 (EHV-1) gene 63 (51), and amino acids 13 to 114 of bovine herpesvirus 1 (BHV-1) BICP0 (20) were aligned by using the PILEUP program from The University of Wisconsin’s sequence analysis package GCG. The amino acid numbers above the alignment correspond to those of the HSV-1 ICP0 sequence, and the locations of six linker insertion mutations analyzed in this study are also indicated above the alignment for reference purposes. Amino acid residues which represent perfect matches in at least three of the six alphaherpesvirus ICP0 homologs are identified by black boxes. The consensus sequence derived from a comparison of these homologs includes invariant residues present in all six sequences (uppercase letters in black boxes), as well as residues with similar functional groups in five of six, five of five, or four of four sequences (lower case unboxed letters, where h5hydrophobic, f5aliphatic, a5aromatic, p5polar, n5 negatively charged, s5small side chain, and c5single carbon functional groups). The consensus RING finger motif (28) is included below the ICP0 consensus sequence for comparison.
on November 9, 2019 by guest
http://jvi.asm.org/
phenomenon of transrepression by ICP0R, some of which are not mutually exclusive from the one proposed above. One intriguing possibility is that ICP0 and ICP0R compete for the same cellular target, but that ICP0R is able to sequester this factor away from ICP0 at a different site in the cell. Evidence for this possibility is provided by the observation that the two proteins appear to have very distinct sites of localization in the cell: ICP0 exists primarily in punctate nuclear granules, whereas ICP0R maintains a diffuse distribution in the cyto-plasm (17). Another possibility, that both proteins compete for binding sites on the viral DNA, seems less likely. Although it has been widely assumed in the literature that RING finger proteins associate with DNA by virtue of their sequence sim-ilarities to classical zinc finger DNA binding domains, such an activity has yet to be demonstrated for even a single member of
this large family. In fact,1H nuclear magnetic resonance
spec-troscopy studies of the RING finger itself have revealed fea-tures that are structurally distinct from those of the DNA binding domains of other zinc finger proteins (1, 15). More-over, highly purified preparations of ICP0 have been shown to be deficient in DNA binding activity (19), and examples of RING finger proteins that possess exclusively cytoplasmic functions have recently been identified (48).
Western blot analyses of lysates from cells transfected with plasmids expressing ICP0R mutants revealed a dramatic vari-ability in their levels of polypeptide expression (Fig. 6). Inter-estingly, there was a nearly perfect correlation between the ability of a given mutation to abrogate transrepression and its ability to permit high-level expression of ICP0R in a plasmid transfection assay (Fig. 2 to 5). Two obvious mechanisms could account for the decreased levels of protein synthesized by transrepression-competent mutants of pKST. First, ICP0R and its functional derivatives might simply be toxic to the trans-fected cell, resulting in a general reduction in the levels of protein that would be detectable by Western blot analyses; alternatively, the expression of these mutants could be tightly controlled through a negative autoregulatory loop. However, neither of these possibilities was consistent with the observa-tion that normal levels of ICP0 protein were expressed from a p111 plasmid cotransfected into the same cells (Fig. 7), partic-ularly since this construct contains the same promoter-regula-tory sequences as pKST and was transfected at the same molar concentrations. Thus, perhaps the simplest explanation for the remarkable differences in the level of proteins expressed by the panel of pKST mutants is that those polypeptides which were expressed by the transrepression-competent mutants are less stable than those of the transrepression-defective mutants; this possibility is now being explored.
The differences in the relative levels of the ICP0 and ICP0R proteins in transrepression assays are also significant from a mechanistic standpoint. For example, they indicate that ICP0R-mediated transrepression can occur in the presence of abundant quantities of the ICP0 protein. Although this sug-gests that ICP0R could abrogate transactivation through a direct physical interaction with ICP0, the previous demonstra-tion that dominant negative mutants of ICP0 were also able to inhibit transactivation by other unrelated regulatory proteins (54) argues against this possibility. It is more likely that the expression of ICP0R within a transfected cell creates an envi-ronment that is no longer conducive to transactivation, even in the presence of high concentrations of ICP0. The route by which such a transformation would occur is unknown but could involve any of the mechanisms described above. Moreover, these results demonstrate that ICP0R-mediated transrepres-sion is effected through a protein that is expressed only at very low levels in the cell and which may even be inherently
unsta-ble. This finding agrees with previous studies which showed that levels of a ICP0 dominant negative mutant plasmid as low as 1/20 of the concentration of a cotransfected wild-type ICP0 plasmid were still capable of efficient inhibition (53). The mo-lecular mechanism by which ICP0R mediates such potent tran-srepression is currently under further investigation.
ACKNOWLEDGMENTS
We are indebted to R. Everett for his gift of antiserum 11060 and his panel of mutant ICP0 plasmids, and we also thank M. Ptashne for providing vector pSG424.
REFERENCES
1.Barlow, P. N., B. Luisi, A. Milner, M. Elliott, and R. Everett.1994. Structure of the C3HC4 domain by H-1-nuclear magnetic resonance spectroscopy—a new structural class of zinc-finger. J. Mol. Biol.237:201–211.
2.Borden, K. L. B., M. N. Boddy, J. Lally, N. J. Oreilly, S. Martin, K. Howe, E. Solomon, and P. S. Freemont.1995. The solution structure of the RING finger domain from the acute promyelocytic leukaemia proto-oncoprotein PML. EMBO J.14:1532–1541.
3. Cai, W., and P. A. Schaffer.1989. Herpes simplex virus type 1 ICP0 plays a critical role in the de novo synthesis of infectious virus following transfection of viral DNA. J. Virol.63:4579–4589.
4. Cai, W. H., T. L. Astor, L. M. Liptak, C. Cho, D. M. Coen, and P. A. Schaffer.
1993. The herpes simplex virus type 1 regulatory protein ICP0 enhances virus replication during acute infection and reactivation from latency. J. Virol.
67:7501–7512.
5. Chen, J., X. Zhu, and S. Silverstein.1991. Mutational analysis of the se-quence encoding ICP0 from herpes simplex virus type 1. Virology180:207– 220.
6. Cheung, A. K.1991. Cloning of the latency gene and early protein 0 gene of pseudorabies virus. J. Virol.65:5260–5271.
7. Ciufo, D. M., M. A. Mullen, and G. S. Hayward.1994. Identification of a dimerization domain in the C-terminal segment of the IE110 transactivator protein from herpes simplex virus. J. Virol.68:3267–3282.
8. Clements, G. B., and N. D. Stow.1989. A herpes simplex virus type 1 mutant containing a deletion within immediate early gene 1 is latency-competent in mice. J. Gen. Virol.70:2501–2506.
9. Davison, A. J., and J. E. Scott.1986. The complete DNA sequence of varicella-zoster virus. J. Gen. Virol.67:1759–1816.
10. Everett, R., A. Orr, and M. Elliott.1995. The equine herpesvirus 1 gene 63 RING finger protein partially complements Vmw110, its herpes simplex virus type 1 counterpart. J. Gen. Virol.76:2369–2374.
11. Everett, R. D.1984. Transactivation of transcription by herpes virus prod-ucts: requirement for two HSV-1 immediate early polypeptides for maxi-mum activity. EMBO J.3:3135–3141.
12. Everett, R. D.1987. A detailed mutational analysis of Vmw110, a trans-acting transcriptional activator encoded by herpes simplex virus type 1. EMBO J.
6:2069–2076.
13. Everett, R. D.1988. Analysis of functional domains of herpes simplex virus type 1 immediate early polypeptide Vmw110. J. Mol. Biol.202:87–96. 14. Everett, R. D.1991. Construction and characterization of herpes simplex
virus type 1 viruses without introns in immediate early gene 1. J. Gen. Virol.
72:651–659.
15. Everett, R. D., P. Barlow, A. Milner, B. Luisi, A. Orr, G. Hope, and D. Lyon.
1993. A novel arrangement of zinc-binding residues and secondary structure in the C3HC4 motif on an alpha herpes virus protein family. J. Mol. Biol.
234:1038–1047.
16. Everett, R. D., A. Cross, and A. Orr.1993. A truncated form of herpes simplex virus type-1 immediate-early protein Vmw110 is expressed in a cell type dependent manner. Virology197:751–756.
17. Everett, R. D., and G. G. Maul.1994. HSV-1 IE protein Vmw110 causes redistribution of PML. EMBO J.13:5062–5069.
18. Everett, R. D., G. G. Maul, A. Orr, and M. Elliott.1995. The cellular RING finger protein PML is not a functional counterpart of the herpes simplex virus type 1 RING finger protein Vmw110. J. Gen. Virol.76:791–798. 19. Everett, R. D., A. Orr, and M. Elliott.1991. High level expression and
purification of herpes simplex virus type 1 immediate early polypeptide Vmw110. Nucleic Acids Res.19:6155–6161.
20. Fraefel, C., J. Zeng, Y. Choffat, M. Engels, M. Schwyzer, and M. Ackermann.
1994. Identification and zinc dependence of the bovine herpesvirus 1 trans-activator protein Bicpo. J. Virol.68:3154–3162.
21. Freemont, P. S., I. M. Hanson, and J. Trowsdale.1991. A novel cysteine-rich sequence motif. Cell64:483–484.
22. Gelman, I. H., and S. Silverstein.1985. Identification of immediate early genes from herpes simplex virus that transactivate the virus thymidine kinase gene. Proc. Natl. Acad. Sci. USA82:5265–5269.
23. Harris, R. A., R. D. Everett, X. Zhu, S. Silverstein, and C. M. Preston.1989. Herpes simplex virus type 1 immediate-early protein Vmw110 reactivates
on November 9, 2019 by guest
http://jvi.asm.org/
latent herpes simplex virus type 2 in an in vitro latency system. J. Virol.
63:3513–3515.
24. Hay, B. A., D. A. Wassarman, and G. M. Rubin.1995. Drosophila homologs of baculovirus inhibitor of apoptosis proteins function to block cell death. Cell83:1253–1262.
25. Hope, I. A., and K. Struhl.1986. Functional dissection of a eukaryotic transcriptional activator protein, GCN4 of yeast. Cell46:885–894. 26. Le, X. F., P. Yang, and K. S. Chang.1996. Analysis of the growth and
transformation suppressor domains of promyelocytic leukemia gene, PML. J. Biol. Chem.271:130–135.
27. Leib, D. A., D. M. Coen, C. L. Bogard, K. A. Hicks, D. R. Yager, D. M. Knipe, K. L. Tyler, and P. A. Schaffer.1989. Immediate-early regulatory gene mutants define different stages in the establishment and reactivation of herpes simplex virus latency. J. Virol.63:759–768.
28. Lovering, R., I. M. Hanson, K. L. B. Borden, S. Martin, N. J. Oreilly, G. I. Evan, D. Rahman, D. J. C. Pappin, J. Trowsdale, and P. S. Freemont.1993. Identification and preliminary characterization of a protein motif related to the zinc finger. Proc. Natl. Acad. Sci. USA90:2112–2116.
29. Ma, J., and M. Ptashne. 1987. Deletion analysis of GAL4 defines two transcriptional activating segments. Cell48:847–853.
30. Maul, G. G., and R. D. Everett.1994. The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J. Gen. Virol.75:1223–1233.
31. Maul, G. G., H. H. Guldner, and J. G. Spivack.1993. Modification of discrete nuclear domains induced by herpes simplex virus type-1 immediate early gene-1 product (ICP0). J. Gen. Virol.74:2679–2690.
32. McGeoch, D. J., C. Cunningham, G. McIntyre, and A. Dolan.1991. Com-parative sequence analysis of the long repeat regions and adjoining parts of the long unique regions in the genomes of herpes simplex viruses types 1 and 2. J. Gen. Virol.72:3057–3075.
33. Meredith, M., A. Orr, M. Elliott, and R. Everett.1995. Separation of se-quence requirements for HSV-1 Vmw110 multimerisation and interaction with a 135-kDa cellular protein. Virology209:174–187.
34. Meredith, M., A. Orr, and R. Everett.1994. Herpes simplex virus type 1 immediate-early protein Vmw110 binds strongly and specifically to a 135-kDa cellular protein. Virology200:457–469.
35. Moriuchi, H., M. Moriuchi, and J. I. Cohen.1994. The RING finger domain of the varicella-zoster virus open reading frame 61 protein is required for its transregulatory functions. Virology205:238–246.
36. Mosca, J. D., D. P. Bednarik, N. B. K. Raj, C. A. Rosen, J. G. Sodroski, W. A. Haseltine, G. S. Hayward, and P. M. Pitha.1987. Activation of human immunodeficiency virus by herpesvirus infection: identification of a region with in the long terminal repeat that responds to a trans-acting factor en-coded by herpes simplex virus 1. Proc. Natl. Acad. Sci. USA84:7408–7412. 37. Mullen, M. A., D. M. Ciufo, and G. S. Hayward.1994. Mapping of intracel-lular localization domains and evidence for colocalization interactions be-tween the IE110 and IE175 nuclear transactivator proteins of herpes simplex virus. J. Virol.68:3250–3266.
38. Mullen, M. A., S. Gerstberger, D. M. Ciufo, J. D. Mosca, and G. S. Hayward.
1995. Evaluation of colocalization interactions between the IE110, IE175, and IE63 transactivator proteins of herpes simplex virus within subcellular punctate structures. J. Virol.69:476–491.
39. Natarajan, R., S. Deshmane, T. Valyi-Nagy, R. Everett, and N. W. Fraser.
1991. A herpes simplex virus type 1 mutant lacking the ICP0 introns
reac-tivates with normal efficiency. J. Virol.65:5569–5573.
40. O’Hare, P., and G. S. Hayward.1985. Evidence for a direct role for both the 175,000- and 110,000-molecular-weight immediate-early proteins of herpes simplex virus in the transactivation of delayed early promoters. J. Virol.
53:751–760.
41. Ostrove, J. M., J. Leonard, K. E. Weck, A. B. Rabson, and H. E. Gendelman.
1987. Activation of the human immunodeficiency virus by herpes simplex virus type 1. J. Virol.61:3726–3732.
42. Perry, L. J., F. J. Rixon, R. D. Everett, M. C. Frame, and D. J. McGeoch.
1986. Characterization of the IE110 gene of herpes simplex virus type 1. J. Gen. Virol.67:2365–2380.
43. Quinlan, M. P., and D. M. Knipe.1985. Stimulation of expression of a herpes simplex virus DNA-binding protein by two viral functions. Mol. Cell. Biol.
5:957–963.
44. Sacks, W. R., and P. A. Schaffer.1987. Deletion mutants in the gene encod-ing the herpes simplex virus type 1 immediate-early protein ICP0 exhibit impaired growth in cell culture. J. Virol.61:829–839.
45. Sadowski, I., and M. Ptashne.1989. A vector for expressing GAL4(1-147) fusions in mammalian cells. Nucleic Acids Res.17:7539.
46. Sekulovich, R. E., K. Leary, and R. M. Sandri-Goldin.1988. The herpes simplex virus type 1aprotein ICP27 can act as atrans-repressor ortrans -activator in combination with ICP4 and ICP0. J. Virol.62:4510–4522. 47. Shapira, M., F. L. Homa, J. C. Glorioso, and M. Levine.1987. Regulation of
the herpes simplex virus type 1 late (gamma2) glycoprotein C gene: se-quences between base pairs234 to129 control transient expression and responsiveness to transactivation by the products of the immediate early (alpha) 4 and 0 genes. Nucleic Acids Res.15:3097–3111.
48. Song, H. Y., and D. B. Donner.1995. Association of a RING finger protein with the cytoplasmic domain of the human type-2 tumour necrosis factor receptor. Biochem. J.309:825–829.
49. Spatz, S. J., E. C. Nordby, and P. C. Weber.Unpublished data.
50. Stow, N. D., and E. C. Stow.1986. Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate early polypeptide Vmw 110. J. Gen. Virol.67:2571–2585. 51. Telford, E. A., M. S. Watson, K. McBride, and A. J. Davison.1992. The DNA
sequence of equine herpesvirus-1. Virology189:304–316.
52. Triezenberg, S. J., R. C. Kingsbury, and S. L. McKnight.1988. Functional dissection of VP16, the transactivator of herpes simplex virus immediate early gene expression. Genes Dev.2:718–729.
53. Weber, P. C., J. J. Kenny, and B. Wigdahl.1992. Antiviral properties of a dominant negative mutant of the herpes simplex virus type-1 regulatory protein-ICP0. J. Gen. Virol.73:2955–2961.
54. Weber, P. C., and B. Wigdahl.1992. Identification of dominant-negative mutants of the herpes simplex virus type 1 immediate-early protein ICP0. J. Virol.66:2261–2267.
55. Yao, F., and P. A. Schaffer.1994. Physical interaction between the herpes simplex virus type 1 immediate-early regulatory proteins ICP0 and ICP4. J. Virol.68:8158–8168.
56. Zhu, X., J. Chen, C. S. H. Young, and S. Silverstein.1990. Reactivation of latent herpes simplex virus by adenovirus recombinants encoding mutant IE-0 gene products. J. Virol.64:4489–4498.
57. Zhu, Z. M., W. Z. Cai, and P. A. Schaffer.1994. Cooperativity among herpes simplex virus type 1 immediate-early regulatory proteins ICP4 and ICP27 affect the intracellular localization of ICP0. J. Virol.68:3027–3040.