Copyright © 2000, American Society for Microbiology. All Rights Reserved.
Conditional Site-Specific Integration into Human Chromosome 19 by
Using a Ligand-Dependent Chimeric Adeno-Associated
Virus/Rep Protein
DANIELA RINAUDO, STEFANIA LAMARTINA, GIUSEPPE ROSCILLI, GENNARO CILIBERTO,
ANDCARLO TONIATTI*
Department of Genetics, Istituto di Ricerche di Biologia Molecolare, I.R.B.M.—Piero Angeletti,
00040 Pomezia (Rome), Italy
Received 7 May 1999/Accepted 21 September 1999
It is of great interest for gene therapy to develop vectors that drive the insertion of a therapeutic gene into
a chosen specific site on the cellular genome. Adeno-associated virus (AAV) is unique among mammalian
viruses in that it integrates into a distinct region of human chromosome 19 (integration site AAVS1). The
inverted terminal repeats (ITRs) flanking the AAV genome and the AAV-encoded nonstructural proteins Rep78
and/or Rep68 are the only viral elements necessary and sufficient for site-specific integration. However, it is
also known that unrestrained Rep activity may cause nonspecific genomic rearrangements at AAVS1 and/or
have detrimental effects on cell physiology. In this paper we describe the generation of a ligand-dependent form
of Rep, obtained by fusing a C-terminally deleted Rep68 with a truncated form of the hormone binding domain
of the human progesterone receptor, which does not bind progesterone but binds only its synthetic antagonist
RU486. The activity of this chimeric protein, named Rep1-491/P, is highly dependent on RU486 in various
assays: in particular, it triggers site-specific integration at AAVS1 of an ITR-flanked cassette in a
ligand-dependent manner, as efficiently as wild-type Rep68 but without generating unwanted genomic rearrangement
at AAVS1.
One of the major goals of gene therapy is to develop safe
and reliable systems for the prolonged expression of a
thera-peutic gene (9). This result can be achieved by promoting
integration of the desired DNA sequence into a predetermined
site on the host genome (14). Therefore, considerable interest
has been raised by the observation that the adeno-associated
virus (AAV) integration machinery can be used for directing
the integration of a transgene into a specific site on the human
genome (31, 44).
AAV is a human defective parvovirus whose single-stranded
genome, 4.7 kb long, is flanked by two inverted terminal
re-peats (ITRs) and comprises two open reading frames (ORFs)
called
rep
and
cap
, which code for nonstructural and structural
proteins, respectively (4). AAV replicates only in cells
coin-fected by a helper virus, such as adenovirus (Ad) or herpes
simplex virus, or undergoing genotoxic stress, such as UV
treatment or X-ray irradiation (4). Under conditions which are
not permissive for replication, AAV establishes a latent
infec-tion in which the viral genome integrates stably and efficiently
into a defined region, AAVS1, of human chromosome 19
(q13.3-qter) (4, 26, 27, 44, 46).
The precise molecular mechanisms underlying AAV
inte-gration have not yet been fully elucidated, but it has been
clearly established that two viral elements are required: the
145-bp ITRs and the Rep78 and/or Rep68 protein encoded by
the
rep
ORF (14, 24, 31, 65). Rep78 and Rep68, which are
respectively 623 and 536 amino acids (aa) long, are expressed
from alternatively spliced transcripts initiated at the same
pro-moter (the p5 propro-moter) and therefore differ only at their
carboxy termini (4). The two proteins, which are essential not
only for integration but also for AAV replication, have several
biochemical activities in common: they both interact with a
specific DNA sequence (Rep binding site) and they both have
a strand- and site-specific endonuclease activity and an
ATP-dependent DNA-DNA and DNA-RNA helicase activity (22,
23). In addition, they are able to modulate the activity of
endogenous as well as heterologous promoters (21, 29, 39). In
vitro and ex vivo experiments suggest that the earliest step in
the integration process is when Rep78 and/or Rep68 tethers
the two Rep binding sites present in the AAV ITR and in
AAVS1 (8, 9, 15, 60). Following the formation of this
Rep-mediated complex between AAV DNA and its target site in
chromosome 19, Rep78 and/or Rep68 is postulated to
specif-ically nick DNA within the ITR and AAVS1; subsequently,
integration is likely to occur via a nonhomologous
recombina-tion process mediated by replicarecombina-tion of the integrating DNA
with the active participation of host factors responsible for
DNA synthesis (32, 33, 56, 65).
The limited length of the DNA sequence which can be
packaged into AAV particles, coupled with the need to
main-tain the
rep
ORF, precludes the use of AAV itself as a delivery
vector for promoting site-specific integration of a transgene
(14). However, recent results indicate that the AAV
integra-tion machinery works quite efficiently also when incorporated
into a variety of alternative nonviral and viral delivery systems
(31). It has in fact been demonstrated that Rep78 and/or
Rep68, when delivered to cells either as an expression plasmid
or as a recombinant protein, promotes the site-specific
inte-gration of an AAV ITR-flanked cassette contained in the same
or in a separate plasmid (2, 30, 47, 52, 55). Furthermore, it was
recently shown that a baculovirus/AAV hybrid vector, which
carries both an AAV ITR-flanked transgene and a Rep
ex-pression cassette, was capable of driving integration of the
ITR-flanked transgene at AAVS1 (38).
* Corresponding author. Mailing address: Istituto di Ricerche di
Biologia Molecolare, I.R.B.M.—P. Angeletti, Via Pontina Km 30.600,
00040 Pomezia (Rome), Italy. Phone: 91093668. Fax:
39-06-91093654. E-mail: [email protected].
281
on November 9, 2019 by guest
http://jvi.asm.org/
In considering the Rep78/68-based integration system as a
new approach to gene therapy, it would be highly desirable to
restrict Rep78/68 activity in target cells only to the time
re-quired for site-specific integration to occur, in order to
mini-mize additional and possible detrimental effects. In fact, it has
been shown that Rep proteins down-regulate the expression of
human genes such as c-H-
ras
, c-
fos
, c-
myc
, and c-
sis
(18, 19, 62)
and can inhibit the proliferation of some cell lines (4, 68). A
further fact for consideration is that Rep-mediated integration
at AAVS1 can lead to nonspecific genomic rearrangements at
the same locus, and the frequency and severity of these might
be attenuated or suppressed by setting a time limit to the
activity of Rep proteins in target cells (2, 52).
With this in mind, we decided to generate an inducible form
of Rep78/68 whose activity could be controlled by an externally
added small-molecule ligand. In this paper we describe the
construction of a ligand-dependent Rep chimeric protein,
made up of a C-terminal Rep68 deletion mutant fused with a
truncated form of the hormone binding domain (HBD) of the
human progesterone receptor (PR), known to interact with the
synthetic steroid RU486 but not with endogenous
progester-one (3, 5, 57). This Rep/HBD fusion protein displays strictly
RU486-dependent activity in a wide array of functional assays
and promotes site-specific integration at AAVS1 without
ma-jor nonspecific genomic rearrangements.
MATERIALS AND METHODS
Plasmid construction and site-directed mutagenesis.Expression vectors for Rep78 and Rep68 (plasmids pCMV/Rep78 and pCMV/Rep68) were obtained by cloning the coding regions for Rep78 and Rep68 under the control of the cytomegalovirus (CMV) enhancer-promoter element contained in plasmid pcD-NAIII (30). To obtain the cDNAs coding only for Rep78 or Rep68, therepORF, spanning from nucleotides 321 to 2252 of the AAV-2 genome (51), was subjected to PCR-based mutagenesis with the AAV-2 genome contained in plasmid pTAV2 (17) as a substrate. To generate the cDNA coding for Rep78, the internal start methionine for the small Rep proteins (Rep52 and Rep40) was mutated to glycine (nucleotides 993 to 995, ATG changed to GGA) and the splice donor site required for the expression of the spliced version Rep68 was eliminated by introducing a G-to-A transversion at nucleotide 1907 (30). The cDNA for Rep68 was obtained by first mutating the internal translational AUG as described above and then deleting the entire intron (positions 1907 to 2227) (30). C-terminal Rep68 deletion mutants (Rep1–484, Rep1–491, Rep1–502, and Rep1–520) were obtained by inserting stop codons at appropriate positions in the context of pCMV/Rep68 by PCR-mediated site-specific mutagenesis (1). Expression plas-mid pCMV/Rep contains the wholerepORF, with no mutations, cloned down-stream of the CMV enhancer/promoter in plasmid pcDNAIII and codes for all four species of Rep. The cDNAs coding for the C-terminal deletion mutants were then cloned into pcDNAIII, thus creating expression plasmids pCMV/ Rep1–484, pCMV/Rep1–491, pCMV/Rep1–502 and pCMV/Rep1–520. All Rep/PR fusions (Rep78/PR, Rep78int/PR, PR/Rep78, Rep68/PR, Rep68int/PR, PR/Rep68, Rep1–491/P, and Rep1–484/Pn) were generated by a PCR-based mutagenesis strategy: their corresponding cDNAs were cloned into pcDNAIII downstream of the CMV enhancer/promoter element, thus generating the ex-pression vectors pCMV/Rep78/PR, pCMV/Rep78int/PR, pCMV/PR/Rep78, pCMV/Rep68/PR, pCMV/Rep68int/PR, pCMV/PR/Rep68, pCMV/Rep1– 491/P, and pCMV/Rep1–484/Pn. The sequences of all mutants and fusions were verified by the dideoxy-sequencing method (1). The sequences of all oligonucle-otides used for PCRs are available on request. Plasmid p5/LUC was constructed as follows: the p5 promoter region (nucleotides 1 to 319 of the AAV-2 sequence) (51) was PCR amplified from psub201 and then cloned as anEcoRV-HindIII fragment upstream of thelucgene contained in plasmid pGL2-Basic (Promega). Plasmid ITR/Hook-Neo, containing the expression cassette for the neomycin resistance gene (neo) and for the Hook gene, has been described previously (30). Plasmid pT7bhPRB-891, containing the C-terminal deletion of the HBD of the human progesterone receptor, was a generous gift of B. O’Malley and S. Tsai.
Cell culture and transfections.293, HeLa, and Hep3B cells were propagated in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum plus glutamine and antibiotics at 37°C in 5% CO2. All transfections were
per-formed by the calcium phosphate procedure (1).
Immunoblot analysis of transiently transfected cells and of stable transfor-mants.For analysis of Rep expression, 3⫻105cells (Hela, 293, or Hep3B) were
transfected with 10g of the various expression plasmids. At 48 h after trans-fection, the cells were washed with phosphate-buffered saline (PBS) and total cellular extracts were prepared as described previously with minor modifications (53). Briefly, the cells were lysed in 10 mM Tris-HCl (pH 8.0)–5 mM EDTA–1%
sodium dodecyl sulfate (SDS) by passage through a 1-ml syringe. The lysate was precipitated with 10% trichloroacetic acid at room temperature for 15 min and then left in ice for additional 10 min. After centrifugation at 12,000⫻gfor 15 min, pellets were washed in ice-cold acetone, resuspended in 60l of sample buffer, and incubated at 65°C for 15 min and then at 100°C for 3 min. Equivalent amounts of proteins were fractionated on an 8% polyacrylamide–SDS gel, trans-ferred to nitrocellulose filters, and detected by sequential incubation with a rabbit polyclonal antibody directed against AAV Rep proteins (dilution, 1:1,000) and then with an alkaline phosphatase-conjugated rat polyclonal anti-rabbit immunoglobulin G (IgG) antiserum (Promega no. S3731; dilution, 1:4,000). The polyclonal antiserum against Rep proteins was obtained by immunizing rabbits with purified recombinant Rep68 produced inEscherichia coli(30) and recog-nizes all four species of Rep.
To check the expression of the Hook gene product (sFv/PDGFR fusion pro-tein) (6) in the stable transfectants, total cellular extracts were prepared as described above, fractionated on an SDS–12% polyacrylamide gel, and trans-ferred to nitrocellulose membranes. To detect the sFv/PDGFR fusion protein, the membranes were incubated first with monoclonal antibody 9E10.2 (dilution, 1:500), which recognizes the Myc.1 epitope tag (13) present as a tandem repeat near the transmembrane domain of sFv/PDGFR (6), and then with an alkaline phosphatase-conjugated goat polyclonal anti-mouse IgG antiserum (Sigma no. A7434; dilution, 1:2,000). In all immunoblotting experiments, 5% nonfat dry milk in TBST (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.05% Tween 20) was used as a blocking agent and for diluting the various antibodies.
Immunofluorescence experiments. For immunofluorescence assays, cells (Hep3B, 293, or HeLa) were grown on glass coverslips and transfected with 10
g of the expression vectors for the various Rep derivatives. At 36 h posttrans-fection, the cells were fixed with 3% formaldehyde in PBS at room temperature for 20 min, washed with PBS, and incubated in 0.1 M glycine in PBS at room temperature for 10 min. Subsequently, the cells were permeabilized by incuba-tion at room temperature for 5 min with 0.1% Triton X-100 in PBS, and after a washing step, they were incubated for 20 min with the rabbit polyclonal antibody directed against AAV Rep proteins (diluted 1:200 in 10% goat serum in PBS). They were then washed with PBS and incubated for an additional 20 min with a fluorescein-conjugated goat anti-rabbit IgG (Pierce No. 31572; diluted 1:100 in 10% goat serum in PBS). After sequential washing steps in PBS and distilled water, the coverslips were mounted in Moviol containing 1 mg of para -phe-nylenediamine per ml and photographed by epifluorescence on a Leica Diaplan photomicroscope with fluorescein filters and a 63x planar objective.
p5 promoter repression assay.For analysis of Rep repressing activity, 3⫻105
293 or HeLa cells were seeded in a 60-mm-diameter dish; 20 h later, they were cotransfected with 50 ng of the various expression plasmids and 5g of p5/LUC. At 15 h later, the cells were washed, and 100 nM RU486 was added to some of the cells. After 36 h, the cells were collected in 40 mM Tris–1 mM EDTA–150 mM NaCl (pH 7.5), centrifuged, resuspended in 250 mM Tris (pH 8.0), and lysed by three freeze-thaw cycles. Cell debris was pelleted by centrifugation, and the supernatant was used for quantitation of luciferase activity as described previ-ously (1) by using a Lumat luminometer (Berthold). Luciferase activity in cell lysates was normalized to the corresponding protein concentration.
Rescue-replication assay.A total of 1.2⫻106HeLa, 293, or Hep3B cells were
seeded in a 10-mm-diameter dish and 18 h later were infected with Ad2 at a multiplicity of infection of 10. After 2 h of incubation, the medium was changed and the cells were transfected with 10g of plasmid ITR/Hook-Neo with or without 10g of the expression vectors for wild-type wt Rep78, Rep68, or their various derivatives. At 15 h after transfection, the cells were washed and incu-bated with fresh medium containing or not containing 100 nM RU486. At 48 h later, low-MrDNA samples were isolated from cells by the procedure described
by Hirt (20), digested extensively withDpnI (62), and analyzed by Southern blotting with a32P-labeled DNA probe specific forneosequence (1, 30).
In vitro translations.Rep68, Rep1–484, Rep1–491, Rep1–502, and Rep1–520 were translated in vitro from plasmids pCMV/Rep68, pCMV/Rep1–484, pcDNA/Rep1–491, pcDNA/Rep1–502, and pcDNA/Rep1–520, respectively, with the TnT-T7 coupled reticulocyte lysate system (Promega) as recommended by the manufacturer. The quantity of proteins used for in vitro experiments was normalized by densitometric analysis (with the GS-700 imaging densitometer with Molecular Analyst software [Bio-Rad]) of SDS-polyacrylamide gels loaded with increasing quantities of the various in vitro translation products.
Electrophoretic mobility shift assays.Electrophoretic mobility shift assays were performed with 20,000 cpm of32P-end-labeled AAV ITR. Reaction
mix-tures (10l) contained 10 mM HEPES-NaOH (pH 7.9), 8 mM MgCl2, 40 mM
KCl, 0.2 mM dithiothreitol, 1g of poly(dI-dC), and different amounts of the in vitro-translated proteins. Following a 20-min incubation at room temperature, 2
l of 20% Ficoll was added, samples were loaded on 4% polyacrylamide gels (acrylamide/bisacrylamide ratio, 29:1; 0.5⫻Tris-borate-EDTA [TBE]) and elec-trophoresed at room temperature at 10 V/cm. The gels were dried and subjected to autoradiography for 6 h at⫺80°C.
trsendonuclease assay.Endonuclease assays were performed essentially as previously described, using substrates with a single-stranded terminal resolution site (30, 48). Briefly, the double-strandedXbaI-PvuII fragments from plasmid psub201 (45) containing the AAV ITRs, were dephosphorylated with calf intes-tinal alkaline phosphatase, purified from agarose gels, 5⬘-end labelled with polynucleotide kinase, and loaded on 6% polyacrylamide sequencing gels. The
282
RINAUDO ET AL.
J. V
IROL.
on November 9, 2019 by guest
http://jvi.asm.org/
plus (trs⫹) strand was eluted from the gels in 0.5 M ammonium acetate (pH
8.0)–1 mM EDTA and annealed. For the endonuclease assay, the reaction mixture (20l) contained 25 mM HEPES-KOH (pH 7.5), 5 mM MgCl2, 0.2 mM
EGTA, 1 mM dithiothreitol, 0.4 mM ATP, 0.2g of bovine serum albumin, 1g of poly(dI-dC), 20,000 cpm of32P-end-labeled substrate, and different amounts
of the in vitro-translated proteins. The reaction mixtures were incubated for 1 h at 37°C, the reactions were stopped with proteinase K for 30 min at 65°C, and the products were subjected to phenol-chloroform extraction, ethanol precipitated, and analyzed on an 8% sequencing gel.
PCR assay for site-specific integration.For each assay, 1.2⫻106HeLa cells
were seeded in a 10-mm-diameter dish and 20 h later were cotransfected with 10
g of the ITR/Hook-Neo plasmid alone or with 10g of one of the Rep or Rep/PR fusion expression plasmids. Transfected cells were serially passaged for 14 days. Total genomic DNA was then extracted (1), and 500 ng was used as a template for two consecutive rounds of nested PCR amplifications performed with two matched couples of ITR-specific and AAVS1-specific primers as de-scribed previously (30). The PCR products were resolved on a 1.5% agarose gel, blotted onto a nylon membrane, and hybridized with an AAVS1-specific probe spanning nucleotides 210 to 1207 of the published AAVS1 sequence (27). For molecular cloning of the amplified ITR/AAVS1 junctions, the products of the second round of amplification which were detectable on ethidium bromide-stained agarose gels were purified, filled with Klenow enzyme, cloned into plas-mid pBluescript II SK(⫹) (Stratagene), and sequenced by the dideoxy sequenc-ing method (1).
Isolation and Southern blot analysis of neomycin-resistant clones.A total of 106HeLa cells were seeded in a 10-mm-diameter dish and 20 h later were
cotransfected with 10g of ITR/Hook-Neo plasmid and 10g of plasmid pCMV/Rep68 or pCMV/Rep1–491/P. At 15 h later, after a washing step, the cells cotransfected with pCMV/Rep1–491/P were treated for 12 h with 100 nM RU486 or left untreated. Subsequently, the medium was changed again and the cells incubated in normal medium for an additional 36 h. Selection was then carried out by growing cells in the presence of 700g of G418 per ml (70.6% active; effective concentration, 494.2g/ml). After 14 to 18 days of growth in selective medium, single-cell neomycin-resistant clones were isolated and ex-panded. For Southern blot analysis, genomic DNA was extracted and purified by standard procedures (1), digested with the restriction enzymeBamHI, and blot-ted onto a nylon membrane, which was sequentially hybridized with AAVS1- and neo-specific probes by published methods (1). The AAVS1-specific probe was a DNA fragment (derived from plasmid pRVK [K. Berns, Cornell Medical School, Ithaca, N.Y.]) spanning nucleotides 1 to 3525 of AAVS1, which was labelled with
32P by the random-priming reaction. A DNA fragment of 630 bp was used as a
template in the random-priming reaction for generating theneo-specific probe.
RESULTS
RU486-dependent nuclear localization of full-length Rep78
and Rep68 fused to the HBD of the progesterone receptor.
To
obtain ligand-dependent Rep78 and/or Rep68 proteins, we
decided to generate fusions with a 42-aa C-terminal deletion of
the human PR891-HBD (57). PR891-HBD (aa 642 to 891 of
the human PR) was chosen because it binds synthetic
proges-terone antagonists, such as RU486, but not the progesproges-terone
or other natural steroids (5, 57); therefore, the activity of
heterologous proteins fused to PR891-HBD cannot be affected
by natural steroids (25, 50).
Since it is not possible to exactly anticipate on a rational
basis the position in which the HBD must be fused with the
heterologous moiety to obtain a stable, properly folded and
ligand-dependent chimera, we constructed several different
fu-sions. In four of them, the HBD was cloned either at the N
terminus (PR/Rep78 and PR/Rep68) or at the C terminus
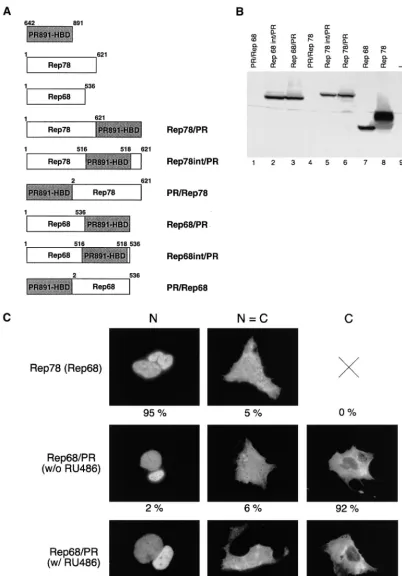
(Rep78/PR and Rep68/PR) of both Rep78 and Rep68 (Fig.
1A). We also constructed two additional chimeras in which the
HBD was introduced at the level of the splicing site
(Rep78int/PR and Rep68int/PR [Fig. 1A]). In fact, evidence
reported in the literature suggests that the splicing site in the
rep
ORF delimits a distinct C-terminal domain of the protein
(12, 21); we therefore reasoned that insertion at this point
should not dramatically affect protein folding and, with respect
to C-terminal fusions, should bring the HBD in closer contact
with the regions of Rep78/68 known to be important for DNA
binding and nicking (11, 34, 37, 55, 61, 67).
Expression vectors for the six fusions were transfected into
human adenocarcinoma-derived HeLa cells, and their
expres-sion levels were assessed by Western blotting experiments. As
shown in Fig. 1B, the N-terminal fusions were undetectable in
transfected cells (Fig. 1B, lanes 1 and 4) while the expression
levels of the other four fusions were comparable to that of wt
Rep68 (Fig. 1B, lanes 2, 3, 5, and 6). Similar results were
obtained in Ad-transformed human embryonic kidney 293 cells
and human hepatoma Hep3B cells (not shown). Notably,
N-terminal fusions were produced by in vitro translation as
effi-ciently as the wt Rep proteins were (data not shown),
suggest-ing that their low expression in cells is due to intracellular
instability. Further analysis was therefore restricted only to the
four chimeric proteins expressed in vivo, namely, Rep78/PR,
Rep78int/PR, Rep68/PR, and Rep68int/PR.
Rep78 and Rep68 are intranuclear proteins (21, 66);
there-fore, before testing the functional activity of our four chimeric
proteins, we first analyzed their intracellular localization.
Ex-pression vectors for Rep78/PR, Rep78int/PR, Rep68/PR, and
Rep68int/PR were thus transfected into Hep3B cells treated or
not treated with 100 nM RU486, and the subcellular
distribu-tion of the fusion proteins was monitored by
immunofluores-cence analysis (see Materials and Methods). Control
experi-ments were performed with Rep78 and Rep68. For each type
of protein, at least 1,000 stained cells were analyzed and
clas-sified into three categories: N, containing cells showing
pre-dominantly nuclear staining; C, containing cells with
predom-inantly cytoplasmic staining; and N
⫽
C, containing cells in
which cytoplasm and nucleus are equally stained. The results
were expressed as the percentages of stained cells in each
category; they are summarized in Table 1, and examples of the
immunocytochemical presentation of cells scored in the three
different categories are shown in Fig. 1C. As expected, Rep78
and Rep68 showed a clear nuclear localization (N
⫽
95%
[Table 1 and Fig. 1C]). In contrast, Rep78/PR, Rep78int/PR,
Rep68/PR, and Rep68int/PR were confined predominantly to
the cytoplasm in the absence of RU486 (C
ⱖ
90% and N
ⱕ
2%
[Table 1]) but migrated into the nuclei following hormone
treatment (N
ⱖ
90% [Table 1]): representative results
ob-tained with the Rep68/PR fusion are shown in Fig. 1C. These
results were also obtained with HeLa and 293 cells (not shown)
and demonstrated that within the sensitivity limits of
immuno-fluorescence, nuclear translocation of the fusion proteins was
under quite stringent hormonal control.
The activity of full-length Rep78 and Rep68 fused to PR
HBD is not hormone dependent.
To determine whether the
activity of the four chimeric constructs was hormone
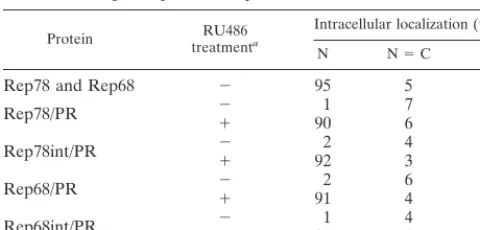
depen-dent, we tested them in a transcription repression assay. Rep78
and Rep68 inhibit transcription starting from the AAV p5
promoter (21, 29, 39). Expression vectors for the Rep/PR
fu-sions were thus cotransfected with a plasmid containing the
luciferase gene under the control of the p5 promoter (plasmid
p5/LUC), in the presence or absence of 100 nM RU486. 293
cells were selected as recipient cells, because the p5 promoter
is known to be highly active in this cell line (29). As expected,
cotransfection of p5/LUC with expression vectors for Rep78
and Rep68 (pCMV/Rep78 and pCMV/Rep68, respectively)
led to the complete inhibition of luciferase activity (98%
re-pression [Fig. 2A]). The four chimeric proteins Rep78/PR,
Rep78int/PR, Rep68/PR, and Rep68int/PR also acted as
strong repressors in the absence of hormone treatment (Fig.
2A). Similar results were observed in HeLa cells, in which the
p5 promoter had a lower but still detectable activity (reference
39 and data not shown).
The four chimeric proteins were further tested in a
rescue-replication assay commonly used to monitor the ability of
Rep78 and Rep68 to promote, in Ad-infected cells, the
exci-sion and replication of an ITR-flanked cassette contained in a
on November 9, 2019 by guest
http://jvi.asm.org/
FIG. 1. Structure, expression, and intracellular distribution of fusion constructs derived from wt Rep78 and Rep68. (A) Diagram of the different chimeras made up of full-length Rep78 or Rep68 fused with the PR891-HBD. For all constructs, the HBD was the same (residues 642 to 891 of the human progesterone receptor). For PR891-HBD, Rep78, and Rep68, the numbers above the diagrams refer to the amino acid positions. For the fusions, the numbers do not refer to the amino acid position in the context of the fusion protein but instead indicate the amino acid positions of the corresponding parental Rep protein. (B) Expression levels of the various fusions in transiently transfected HeLa cells. Whole-cell extracts were prepared from HeLa cells transfected with the expression vectors for wt Rep78, wt Rep68, and the six different chimeras. wt Rep78, wt Rep68, and their derivatives were detected by immunoblotting with a rabbit polyclonal serum against Rep proteins. Lane 9 contains untransfected cells. (C) Representative micrographs of the staining patterns observed in Hep3B cells transfected with Rep78, Rep68, and the various Rep/PR fusions. Hep3B cells were transfected with 10g of the expression vectors pCMV/Rep78, pCMV/Rep68, and pCMV/Rep68/PR. In this last case, cells were treated (w/ RU486) or not (w/o RU486) with 100 nM RU486. The cells were stained with a rabbit polyclonal antibody directed against the Rep moiety (see also Materials and Methods). The staining was classified into three categories: N, predominantly nuclear fluorescence; C, predominantly cytoplasmic staining; N⫽C, equal cytoplasmic and nuclear staining. At least 1,000 stained cells, obtained from a minimum of three experiments, were scored for each protein. The numbers below the micrographs represent the percentage of cells falling into each category.
284
on November 9, 2019 by guest
http://jvi.asm.org/
cotransfected plasmid (45). The plasmid ITR/Hook-Neo (30),
containing the expression cassette for the membrane-bound
single-chain antibody (Hook) and for the neomycin resistance
genes (
neo
) cloned between the AAV ITRs, was cotransfected
with expression vectors for the four Rep/PR fusions in
Ad-2-infected HeLa cells, treated or not treated with RU486.
Low-molecular-weight DNA was isolated 63 h posttransfection (20),
digested with
Dpn
I to degrade input plasmid DNA (62), and
fractionated by electrophoresis on an agarose gel. Monomeric
and dimeric forms of the rescued and replicated ITR-flanked
Hook-
neo
cassette were detected on Southern blots by using a
32
P-labelled
neo
probe: the intensity of the signal was
consid-ered to be a measurement of the activity of the various Rep/PR
fusions in the assay. The autoradiogram of one such
experi-ment is presented in Fig. 2B: Fig. 2C shows the corresponding
agarose gel, which was blotted onto a nylon membrane. No
sig-nal was detected in untransfected cells or in cells transfected
only with ITR/Hook-
neo
(Fig. 2B, lanes 1 and 2). In cells
trans-fected with expression vectors for the Rep78/PR, Rep78int/
PR, Rep68/PR, and Rep68int/PR chimeras, bands
correspond-ing to rescued monomers and dimers were clearly detected in
the absence of RU486 treatment (lanes 3, 5, 7, and 9); their
intensities were similar to those monitored in cells transfected
with wt Rep78 and Rep68 (lanes 11 and 12) and was not
in-creased following RU486 treatment (lanes 4, 6, 8, and 10).
Identical results were obtained with 293 and Hep3B cells (not
shown). In conclusion, we found that the four chimeric Rep/PR
proteins displayed a constitutive rather than
hormone-induc-ible activity in both the p5 promoter repression and
rescue-replication assays. This suggested that the relatively small
amount of protein present in the nuclei of untreated cells
(Table 1) was not only constitutively active but also
suffi-cient to give a full response in our experimental settings (see
also Discussion).
It is known that a constitutively nuclear fusion protein can be
rendered hormone responsive by placing the HBD in close
contact with the active domains of the heterologous protein
(40, 41). We thus decided to generate a new set of fusions in
which the distance of the HBD from the potentially regulatable
activities of Rep was reduced. To do this, a few Rep68 deletion
mutants were constructed to identify the minimal region of
Rep retaining wild-type activity and then fuse it with
PR891-HBD.
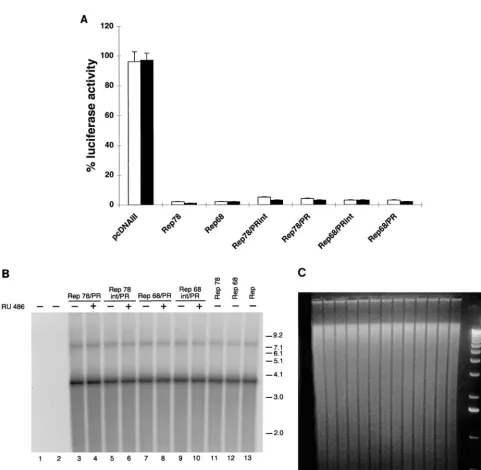
Identification of the minimal region of Rep68 retaining full
activity in vitro and in vivo.
Since the N terminus of Rep68 is
required for DNA binding, deletions were generated starting
from the carboxy terminus of the protein (37). Four
C-termi-nally truncated Rep68 derivatives were thus constructed,
name-ly, Rep1–520, Rep1–502, Rep1–491, and Rep1–484, which lack
the last 17, 35, 46, and 53 C-terminal amino acids, respectively.
Their schematic structure is shown in Fig. 3A. No further
deletions were constructed, because they have been previously
reported to strongly impair DNA binding and endonuclease
activity (34, 61, 67).
The four Rep68 derivatives were first analyzed in vitro.
Rep1–520, Rep1–502, Rep1–491, and Rep1–484, as well as
Rep68, were produced by in vitro translation and then assayed
for their DNA binding and endonuclease activity. As shown in
Fig. 3B and C, Rep1–520, Rep1–502, and Rep1–491 bound
(Fig. 3B) and cleaved (Fig. 3C) a 5
⬘
-end-labelled hairpinned
AAV ITR as efficiently as Rep68 did, while Rep1–484
dis-played a weaker activity in both assays (Fig. 3B and C).
Immunoblotting analysis demonstrated that all four mutants
were expressed at levels comparable to that of Rep68 in HeLa,
Hep3B, and 293 cells (results not shown). The major Rep
nu-clear localization signal (NLS) spans a region between amino
acids 485 to 510 which is highly enriched in positively charged
residues (Lys and Arg) (21, 51, 66): since three of four deletion
mutants lacked at least part of this sequence, we first checked
their intracellular distribution. As shown in Table 2,
progres-sive deletions from the C terminus of Rep68 gradually reduced
the capacity of the mutants to localize into the nucleus. Rep1–
520 behaved like Rep68 (N
⫽
95% [Table 2]), and Rep1–484,
which lacked the entire NLS, was found predominantly in the
cytoplasm of all scored cells (C
⫽
100% [Table 2]). Rep1–502
and Rep1–491 did not display a clear preferential subcellular
compartimentalization but were still capable of localizing into
the nuclei (Table 2).
The in vivo activity of the four mutants was then tested in
both the p5 promoter-repression and the rescue-replication
assays performed with various cell lines. As summarized in
Table 3, Rep1–520, Rep1–502, and Rep1–491 displayed a
wt-like activity in both tests. In contrast, Rep1–484 acted as a poor
repressor and was totally inactive in the rescue-replication
assay, in agreement with its reduced activity in vitro (Fig. 3B
and C) and its mainly cytoplasmic distribution (Table 2).
Fi-nally, we checked the integration competence of the various
Rep68 derivatives by using a recently developed PCR-based
integration assay (30). HeLa cells were cotransfected with ITR/
Hook-Neo and the various mutants and were then serially
passaged for 14 days in the absence of selection. ITR/AAVS1
junctions were then selectively amplified by PCR and revealed
by Southern blotting as described in the footnotes to Table 3
(30, 52, 55). As previously reported, under these experimental
conditions no signal is detected in cells transfected only with
the ITR/Hook-Neo cassette (reference 30 and data not shown). A
positive signal, indicative of site-specific integration events at
the AAVS1 site, was instead detected in cells in which plasmid
ITR/Hook-Neo was cotransfected with the expression vectors
for Rep78 or Rep68 and its derivatives Rep1–520, Rep1–502,
and Rep1–491 (Table 3). Conversely, no site-specific
integra-tion was observed when Rep1–484 was used (Table 3).
[image:5.612.54.294.92.207.2]In summary, the experiments performed with Rep68
C-ter-minally deleted mutants demonstrated that Rep1–491 is the
shortest variant which retains the capacity to localize into the
nuclei, although it does so less efficiently than wt protein, while
still displaying full in vitro and in vivo activity, including
site-specific integration. However, Rep1–484 binds and nicks DNA
with reduced efficiency in vitro and is poorly active in vivo, also
because it lacks a functional NLS.
TABLE 1. Effects of RU486 on the intracellular distribution of
full-length Rep78 and Rep68 fused with PR891-HBD
Protein treatmentRU486a
Intracellular localization (%)b
N N⫽C C
Rep78 and Rep68
⫺
95
5
0
Rep78/PR
⫺
⫹
90
1
7
6
92
4
Rep78int/PR
⫺
⫹
92
2
4
3
94
5
Rep68/PR
⫺
⫹
91
2
6
4
92
5
Rep68int/PR
⫺
⫹
91
1
4
4
95
5
a⫺, transfected cells were untreated;⫹, 100 nM RU486 was added to the
culture medium.
bStained Hep3B cells were classified as described in the legend to Fig. 1C. At
least 1,000 stained cells, obtained in three or more experiments, were analyzed for each protein, and the results are expressed as the percentage of cells included in each category.
on November 9, 2019 by guest
http://jvi.asm.org/
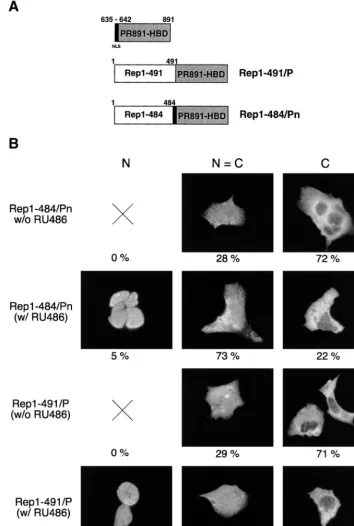
Construction of Rep1–484/Pn and Rep1–491/P.
In an
at-tempt to generate a hormone-dependent Rep protein, we
con-structed two new Rep/PR hybrid proteins (Fig. 4A). In the
first, Rep1–491/P, the PR891-HBD was fused to the C
[image:6.612.62.544.62.532.2]termi-nus of Rep1–491. The second chimera, named Rep1–484/Pn,
was generated by C-terminally fusing Rep1–484 to a slightly
larger segment of the human PR (aa 635 to 891), which
in-cludes the major NLS (aa 638 to 642) of the human PR (16,
FIG. 2. Activity of chimeras derived from wt Rep78 and Rep68. (A) Rep/PR fusions constitutively inhibit p5 promoter activity. 293 cells were transfected with 5
g of plasmid p5/LUC and 50 ng of the expression plasmids pCMV/Rep78, pCMV/Rep68, pCMV/Rep78/PR, pCMV/Rep78int/PR, pCMV/Rep68/PR, and pCMV/ Rep68int/PR (see Materials and Methods). In control experiments, the empty expression vector pcDNAIII was cotransfected with p5/LUC. At 15 h posttransfection, the cells were treated for 36 h or not treated with RU486. The luciferase activity observed in the presence of the different Rep and Rep derivative expression vectors was calculated as the percentage of that (arbitrarily assumed to be 100%) measured in cells transfected with p5/LUC and pcDNAIII. White columns show activity in the absence of RU486 treatment; black columns show activity in the presence of 100 nM RU486. Each column represents the mean and standard deviation for at least three different experiments, performed in duplicate with different plasmid preparations. (B) Constitutive activity of Rep/PR fusions in a rescue-replication assay. Ad-2-infected HeLa cells were cotransfected with 10g of the ITR/Hook-Neo plasmid and 10g of the expression plasmids pCMV/Rep78 (lane 11), pCMV/Rep68 (lane 12), pCMV/Rep78/PR (lanes 3 and 4), pCMV/Rep78int/PR (lanes 5 and 6), pCMV/Rep68/PR (lanes 7 and 8), or pCMV/Rep68int/PR (lanes 9 and 10). After 15 h, the cells were washed and incubated either with normal medium (⫺) or with medium containing 100 nM RU486 (⫹). After 48 h, low-molecular-weight DNA samples were isolated (20), digested withDpnI (62), and analyzed on Southern blots with a32P-labelledneo-derived probe. The two bands corresponding to rescued
monomeric (about 3.7-kb) and dimeric (about 7.5-kb) ITR-flanked cassette are visible. Higher-order multimeric forms were evident after longer exposures (data not shown). In control experiments, cells were transfected only with the ITR/Hook-Neo plasmid (lane 2). Untransfected cells are shown in lane 1. Lane 13 shows results obtained in cells cotransfected with 10g of plasmid ITR/Hook-Neo and 10g of plasmid pCMV/Rep, which express all four species of Rep (see also Materials and Methods). Molecular sizes are shown in kilobases. The autoradiogram shown is representative of five different experiments which all gave similar results. (C) Ethidium bromide staining of the agarose gel which was blotted onto a nylon membrane. Numbering below the lanes is the same as in B.
286
RINAUDO ET AL.
J. V
IROL.
on November 9, 2019 by guest
http://jvi.asm.org/
69); this motif was expected to facilitate the intranuclear
lo-calization of the fusion.
Rep1–484/Pn and Rep1–491/P were cloned into eukaryotic
expression vectors; their expression levels in transfected HeLa,
293, and Hep3B cells were comparable to that of wt Rep68
(results not shown). Immunofluorescence experiments
re-vealed that in untreated cells, both fusions were on average
predominantly cytoplasmic or evenly distributed between the
[image:7.612.122.476.70.548.2]nucleus and cytoplasm (Fig. 4B). RU486 modestly increased
the intranuclear accumulation of Rep1–484/Pn, whereas a
more pronounced effect was observed for Rep1–491 (Fig. 4B).
In this last case, upon hormone treatment there was a
substan-tial increase in the number of cells stained exclusively in the
nuclei (N
⫽
20% [Fig. 4B]) and a drastic reduction in the
number of cells in which Rep1–491/P was localized only in the
cytoplasm (C
⫽
71 and 10% in the absence and presence of
FIG. 3. Structure and in vitro activity of Rep C-terminal deletion mutants. (A) Schematic representation of the Rep deletion mutants. Numbers to the right refer to amino acid positions. (B) Electrophoretic mobility shift assay with wt and mutant Rep proteins. Rep68 and the four deletion mutants were translated in vitro, and equivalent amounts of the various proteins (normalized as described in Materials and Methods) were used in dose-dependent DNA binding assays. Reaction mixtures contained 20,000 cpm of32P-5⬘-end-labeled AAV ITR and either no protein (lane 1) or increasing concentrations of the various proteins indicated above lanes 2 to
21. (C) Nicking activity of wild-type and mutant Rep proteins. A 20,000-cpm sample of32P-5⬘-end-labeled AAV ITR containing a single-stranded trs (trs⫹[48]) was
incubated with increasing concentrations, normalized as in panel B, of in vitro-translated Rep68, Rep1–484, Rep1–491, Rep1–502, and Rep1–520. A standard endonuclease reaction was carried out (30, 48), and the reaction products were resolved on an 8% polyacrylamide sequencing gel. The positions of the substrate (trs
⫹) and of the released 73-bp fragment are indicated. The two labelled fragments shorter than 73 bp are the result of aberrant nicking sometimes observed when single-strandedtrs⫹substrates are used in Rep endonuclease assays (30, 48).
on November 9, 2019 by guest
http://jvi.asm.org/
RU486, respectively [Fig. 4B]). Similar results were obtained
with Hep3B (Fig. 4B), HeLa, and 293 (results not shown) cells.
Ligand-dependent activity of Rep1–484/Pn and Rep1–491/P.
The two fusions were first tested for their capability to
down-regulate the p5 promoter activity in 293 cells. As shown in Fig.
5A, both fusions displayed low repressing activity in the
ab-sence of the hormone (about 20% repression [Fig. 5A]):
how-ever, following RU486 treatment, Rep1–491/P strongly
inhib-ited p5/LUC activity (94% repression [Fig. 5A]). The
repressing activity of Rep1–484/Pn was also stimulated by the
steroid but to a more limited extent (45% repression [Fig.
5A]). Similar results were obtained with HeLa cells (not
shown).
Rep1–484/Pn also displayed hormone dependence in the
rescue-replication assay: in fact, rescue-replication of the
ITR-flanked Hook-
neo
cassette was seen in Ad2-infected HeLa
cells only upon RU486 treatment (Fig. 5B, compare lanes 2
and 3). However, the maximal activity was lower than that of wt
Rep68 (compare lanes 3 and 6). Also, Rep1–491/P was at least
partially hormone dependent in this assay: in fact, basal activity
was detectable in the absence of RU486 (lane 4), but the
activity was strongly enhanced by steroid treatment (compare
lanes 4 and lane 5). The same results were observed with 293
and Hep3B cells (not shown).
The capability of Rep1–484/Pn and Rep1–491/P to mediate
site-specific integration was then examined by using the
PCR-based assay described above. As shown in Fig. 5C, no
site-specific integration could be monitored in cells transfected
with Rep1–484/Pn (Fig. 5C, lanes 6 and 7). In contrast, Rep1–
491/P clearly triggered integration at AAVS1 in the presence
of RU486 (lanes 4 and 5). It is worth noting that in this assay,
the full-length Rep proteins fused with PR891-HBD also
dis-played a constitutive activity (data not shown). The
heteroge-neous size of the amplified AAVS1-positive bands is in
agree-ment with the fact that Rep-mediated integration occurs in a
region spanning more than 500 bp of human chromosome 19
(30, 44, 46, 65). To further verify that positive signals are
indicative of true integration events, we cloned two junctions
amplified from hormone-treated cells transfected with Rep1–
491/P. Their sequences, shown in Fig. 5D, demonstrate that
integration has occurred at nucleotides 842 and at 950 of
AAVS1. In both cases, a complete ITR was not found, in line
with all the ITR/AAVS1 junctions analyzed so far (30, 38, 42,
43, 46, 65).
Rep1–491/P mediates site-specific integration in the absence
of chromosomal rearrangements.
To better characterize the
integration efficacy of Rep1–491/P, we performed Southern
blot analysis of individual clones derived from HeLa cells
co-transfected with the expression vector for Rep1–491/P and
plasmid ITR/Hook-Neo. At 15 h posttransfection, the cells
were incubated for 24 h with 100 nM RU486 and then grown
in the absence of steroid treatment under selection with 700
g
of G418 per ml for 3 weeks. Genomic DNA was then extracted
from individual clones and digested with the restriction
en-zyme
Bam
HI, which has no recognition site in the ITR-flanked
Hook-
neo
cassette and in the region of AAVS1 in which the
great majority of integration events take place (27, 30, 46, 65).
Digested DNA was then subjected to Southern blot analysis
with AAVS1-derived and
neo
-derived probes. Site-specific
in-tegration was assigned to clones displaying an AAVS1
hybrid-izing band which was upshifted with respect to the bands
ob-served in untransfected cells and which also cohybridized with
the
neo
probe (30).
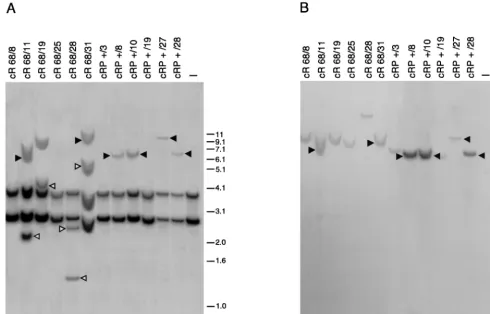
[image:8.612.54.294.92.164.2]According to this criterion, site-specific integration was
ob-served in 7 of 28 clones (25% frequency of site-specific
inte-gration) derived from cells cotransfected with ITR/Hook-Neo
and the expression vector for Rep68. This integration efficiency
is in line with previously published results (30). Also, Rep1–
491/P was able to mediate integration at the AAVS1 site, and
the frequency of site-specific integration was higher in the
presence of RU486 treatment (25%; 7 positive clones of 28
analyzed) than in its absence (3.2%; 1 positive clone of 31),
confirming that the activity of the fusion was in large part
under hormonal control. Of the clones derived from
Rep68-transfected cells, 40% showed additional AAVS1 bands not
cohybridizing with the
neo
probe. As illustrated in Fig. 6, where
the integration pattern of some representative clones is shown,
these
neo
-negative bands were evident both in clones scored as
positive for site-specific integration (Fig. 6, lanes 2 and 6) and
in those scored as negative (lanes 3 and 5). Interestingly,
AAVS1-positive and
neo
-negative bands were not observed in
any of the clones derived from cells transfected with Rep1–
491/P and treated or not treated with RU486 (lanes 7 to 12 and
data not shown). These results suggest that short-term (24-h)
treatment with RU486 enables Rep1–491/P to promote
site-specific integration as efficiently as wt Rep68 but with much
TABLE 2. Intracellular distribution of Rep C-terminal
deletion mutants
Protein Intracellular localization (%)
a
N N⫽C C
Rep1–520
95
5
0
Rep1–502
57
42
1
Rep1–491
16
42
42
Rep1–484
100
aStained Hep3B cells were classified as described in the legend to Fig. 1C. A
total of 600 stained cells, obtained from at least two different experiments, were analyzed for each protein, and the results are expressed as the percentage of cells included in each category.
TABLE 3. Activity of Rep68 C-terminal deletion mutants
in transfected cells
Protein p5/LUC activity (%)Repression of a replicationRescue-b Integrationc
Rep78
98
⫾
1
⫹
⫹
Rep68
98
⫾
1
⫹
⫹
Rep1–520
97
⫾
2
⫹
⫹
Rep1–502
98
⫾
1
⫹
⫹
Rep1–491
95
⫾
4
⫹
⫹
Rep1–484
40
⫾
3
⫺
⫺
aRepression of p5/LUC activity was analyzed as described in the legend to Fig.
2A. 293 or HeLa cells were cotransfected with plasmid p5/LUC and the expres-sion vectors for the various Rep derivatives. Represexpres-sion was calculated with respect to the luciferase activity observed in extracts from cells transfected with p5/LUC plasmid alone.
bRescue-replication of the Hook-neocassette in Ad2-infected HeLa, 293, or
Hep3B cells was monitored as described in the legend to Fig. 2B.⫹, detection of a rescue-replicated Hook-Neo cassette in the Hirt supernatant of transfected cells (the intensity of the signal was identical for all the active mutants);⫺, no replicated cassette could be detected. Similar results were obtained in all three cell lines.
cHeLa cells were cotransfected with the expression vectors for the various
mutants and plasmid ITR/Hook-Neo and then serially passaged for 14 days in the absence of selection. Genomic DNA was extracted and used as a template for two consecutive rounds of PCR with nested primers specific for the AAV ITRs and the AAVS1 site in human chromosome 19 to selectively amplify the ITR/ AAVS1 junctions (30). Amplification products were then detected by Southern blot analysis with an AAVS1-derived probe (30, 52, 55).⫹and⫺indicate that AAVS1-positive signals were observed or not, respectively.
288
RINAUDO ET AL.
J. V
IROL.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:8.612.311.548.490.574.2]less propensity to generate additional and undesired
rear-rangements at the AAVS1 site.
We finally checked whether the stable transformants scored
as positive for site-specific integration also expressed the other
[image:9.612.125.479.79.605.2]gene, Hook, contained between the AAV ITRs in plasmid
ITR/Hook-Neo. As shown in Fig. 7, Western blot analysis
revealed that all seven stable transformants derived from cells
transfected with Rep1–491/P also expressed the Hook gene
FIG. 4. Structure and intracellular distribution of chimeras made up of Rep68 C-terminal deletion mutants fused with PR891-HBD. (A) Diagram of Rep1–491/P and Rep1–484/Pn chimeric constructs. The region of the human PR spanning from aa 635 to 891 is shown at the top of the figure: the NLS (aa 635 to 642), which is maintained in the Rep1–484/Pn fusion but absent in the Rep1–491/P chimera, is indicated in black. Numbers above the hybrid proteins refer to the amino acid position in the parental Rep protein (see also the legend to Fig. 1A). (B) RU486 affects the intracellular distribution of Rep1–491/P and Rep1–484/Pn. Hep3B cells were transfected with 10g of the expression vectors pCMV/Rep1–491/P and pCMV/Rep1–484/Pn and treated with 100 nM RU486 or left untreated. The cells were stained with an anti-Rep polyclonal serum and classified as described in the legend to Fig. 1C. At least 1,000 stained cells, obtained from a minimum of three experiments, were scored for each fusion. The numbers below the micrographs represent the percentage of cells falling into each category.
on November 9, 2019 by guest
http://jvi.asm.org/
protein product (sFv/PDGFR) (6). Conversely, the Hook gene
protein product was detectable in only one of the seven
AAVS1 integrants isolated from cells cotransfected with wt
Rep68 (results not shown). This finding further suggests that
Rep1–491/P promotes a more precise integration of an
ITR-flanked cassette at the AAVS1 site.
DISCUSSION
In this paper we describe the construction and
characteriza-tion of a ligand-dependent AAV Rep protein. Among the two
ligand-dependent Rep/PR fusions generated, Rep1–491/P has
the most interesting properties in that in various assays it
displays a low basal activity which is highly stimulated by
RU486 and, more importantly, promotes integration into the
AAVS1 site in a ligand-dependent manner. The partial
dis-crepancy in the results that emerged from the PCR-based
integration assay (no junctions amplified from pooled and
un-selected cells in the absence of RU486 treatment [Fig. 5C, lane
4]) and the Southern blot analysis of individual selected clones
(site-specific integration detected in 1 of 31 clones derived
from untreated cells) might simply reflect different sensitivities
of the two assays. Nevertheless, further work is required to
clarify whether selection can increase the integration
fre-quency even in the absence of hormone treatment.
Rep1–491/P has several advantages over wt Rep78 or Rep68
for gene therapy purposes. This chimera triggers site-specific
integration in the presence of RU486 as efficiently as wt Rep68
does, and it does so without generating major unwanted
rear-rangements at the AAVS1 site, thus overcoming one of the
major limitations of the AAV-based integration strategy (31).
FIG. 5. Hormone-dependent activity of Rep1–491/P and Rep1–484. (A) Rep1–491/P and Rep1–484/Pn repress the p5 promoter in a ligand-dependent manner. 293 cells were transfected with 5g of plasmid p5/LUC and 50 ng of the expression plasmids pCMV/Rep68, pCMV/Rep1–491/P, and pCMV/Rep1–484/Pn. Luciferase activity was calculated as described in the legend to Fig. 2A. White and black columns represent the activities measured in the absence and in the presence of 100 nM RU486, respectively. Each column represents the mean and standard deviation for at least three different experiments, performed in duplicate with different plasmid preparations. (B) RU486 stimulates the activity of Rep1–491/P and Rep1–484/Pn in a rescue-replication assay. Ad2-infected HeLa cells were cotransfected with 10g of the ITR/Hook-Neo plasmid and 10g of the expression plasmid pCMV/Rep68 (lane 6), pCMV/Rep1–491/P (lanes 4 and 5), or pCMV/Rep1–484/Pn (lanes 2 and 3). Cell treatment and analysis of low-molecular-weight DNA was performed as described in the legend to Fig. 2B. Monomeric (about 3.7-kb) and dimeric (about 7.5-kb) forms of the rescued ITR-flanked cassette are visible: higher-order multimeric forms were detectable after longer exposures (data not shown). In control experiments, the ITR/Hook-Neo plasmid was cotransfected with the empty expression vector pcDNAIII (lane 1). Molecular sizes are shown in kilobases. The autoradiogram shown is representative of four different experiments which all gave similar results. (C) RU486-dependent site-specific integration mediated by Rep1–491/P. HeLa cells were transfected with 10g of plasmid ITR/Hook-Neo alone (lane 2) or together with 10g of the expression vector pCMV/Rep68 (lane 3), pCMV/Rep1–491/P (lanes 4 and 5), or pCMV/Rep1–484/Pn (lanes 6 and 7). At 15 h later, the cells were washed and incubated for 24 h with 100 nM RU486 or left untreated. ITR/AAVS1 junctions were amplified from the genomic DNA extracted from cells subcultured for 14 days and detected with an AAVS1-derived probe as described in the footnote to Table 3. Lane 1 contains untransfected cells. Molecular sizes are shown in base pairs. (D) Sequence analysis of ITR/AAVS1 junctions. The letters D and A refer to the accepted nomenclature for AAV/ITR sequences (4, 30, 46). The numbers above the diagrams refer to the last identifiable viral and AAVS1 nucleotides. Insertions between AAV/ITR and AAVS1 are boxed. AAVS1 breakpoints are based on published AAVS1 sequence (27). Nucleotide numbering of the AAV/ITR is relative to the right end of the AAV genome (51).
290
RINAUDO ET AL.
J. V
IROL.
on November 9, 2019 by guest
http://jvi.asm.org/
It is not yet clear how rearrangements are generated and
whether they occur at the same time as or after the initial
integration event (33), but our results with Rep1–491/P
indi-cate that it is possible to prevent or at least reduce them by
simply establishing a time limit to Rep activity in target cells.
These results tempt us to speculate that rearrangements at
AAVS1 are not a direct consequence of the integration process
itself but, rather, might be ascribed to unrestrained activity of
Rep at the integration locus. This is further corroborated by
the recent report that AAVS1 rearrangements are also
de-tected in cells transfected with a plasmid containing the entire
rep
ORF only and no ITR-flanked sequences (52; S. Lamartina
and C. Toniatti, unpublished results).
We might hence envision the following scenario: after the
initial integration event, rearrangements occur when
constitu-tively active Rep78/68 binds and nicks its target sequence
present not only at AAVS1 but also in the ITRs flanking the
integrated transgene. This leads to partial replication,
rear-rangements, and, possibly, translocation of the region (2, 31,
32, 52). In relation to this last point, it is of interest that the
Rep78 or Rep68 nicking sites located on the ITRs are
main-tained in all the AAVS1-ITR junctions sequenced so far and
that in the majority of cases, the ITRs flanking the integrated
DNA still retain their Rep binding site (30, 38, 42, 43, 46, 65).
In contrast, Rep1–491/P is capable of triggering site-specific
integration during the initial 24 h of RU486 treatment of
transfected cells; following withdrawal of the hormone, it no
longer binds and nicks DNA, thus reducing the instability of
the region.
[image:11.612.56.547.76.390.2]The two genes, Hook and
neo
, contained between the AAV
FIG. 6. Southern blot analysis of HeLaneo-resistant clones derived from cells cotransfected with plasmid ITR/Hook-Neo and expression vectors for either wt Rep68 or Rep1–491/P. Transfection and selection of Neorclones were carried out as described in Materials and Methods. Genomic DNA of isolated clones was digested with BamHI and blotted onto a nylon membrane. (A) Hybridization to an AAVS1-specific probe. (B) The same membrane after rehybridization to aneo-specific probe. Solid triangles mark upshifted bands which cohybridize with both probes and are therefore indicative of site-specific integration (panels A and B, lanes 2, 6, 8, 9, 11, and 12). Open triangles show nonspecific rearrangements (AAVS1-positive/neo-negative bands) observed in clones derived from cells cotransfected with wt Rep68 (panel A, lanes 2, 3, 5, and 6). cR68, clones derived from cells transfected with wt Rep68 (lanes 1 to 6); cRP⫹, clones derived from cells transfected with Rep1–491/P and treated for 12 h with 100 nM RU486 (lanes 7 to 12). Lane 13 contains untransfected cells. Molecular sizes are shown in kilobases.
FIG. 7. Hook gene expression in site-specific integrants derived from HeLa cells cotransfected with ITR/Hook-Neo and Rep1–491/P and treated with RU486. Whole-cell extracts were prepared from cRP⫹clones and run on an SDS-polyacrylamide gel. Fractionated proteins were transferred to a nitrocellu-lose membrane, which was probed with anti-mycepitope tag antibody (13) as described in Materials and Methods. The arrow marks the band, of the expected size, corresponding to the single-chain antibody (sFv/PDGFR) encoded by the Hook gene (6). Asterisks indicate nonspecific product recognized by the anti-myc monoclonal antibody 9E10.2 in untransfected cells. Lane 8 contains untrans-fected HeLa cells.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:11.612.80.266.528.645.2]ITRs were both expressed in all site-specific integrants derived
from hormone-treated cells transfected with Rep1–491/P (Fig.
7). In addition, in six of seven clones the size of the single
upshifted AAVS1 band are about 6.5 kb long (Fig. 6A, lanes 8,
9, and 12, and data not shown), a size which is consistent with
the integration of one single copy of the full-length
ITR/Hook-Neo cassette (3.7 kb). These findings are in striking contrast to
what was observed, in this and previous studies, in site-specific
stable integrants derived from cells cotransfected with wt
Rep68 (Fig. 6A) (2, 47, 52). We should mention that
integra-tion of the Hook gene at AAVS1 in our selected clones could
not be unequivocally demonstrated by Southern blotting,
be-cause the Hook-derived probe reveals a complex pattern of
multiple bands on genomic DNA (Hook codes for a
single-chain antibody [6]). The use of different ITR-flanked cassettes
will therefore be required for a careful study of the fine
struc-ture of the integrated DNA. This will be the object of fustruc-ture
work to clearly establish whether Rep1–491/P not only reduces
nonspecific rearrangements but also can promote a more
pre-cise integration of the ITR-flanked cassette.
Shorter variants of Rep (Rep1–491 and Rep1–484) fused
with PR891-HBD proved hormone dependent, while fusions
with full-length Rep78 or Rep68 did not. The observation that
tighter hormonal control can be achieved by reducing the
dis-tance between HBD and the active site of the heterologous
moiety is not unprecedented (35, 40, 50). Nevertheless, the
precise mechanisms by which heterologous proteins fused with
HBDs are regulated by the cognate ligand have not yet been
elucidated and are likely to vary according to the particular
steroid receptor used (41, 54). A point to note is that in our
case, the more stringent control achieved with the shorter
fusions did not parallel a concomitant tighter regulation of
their subcellular distribution. In fact, both fusions with Rep
deletions and fusions with full-length Rep78/68 were
predom-inantly cytoplasmic, presumably complexed with heat shock
protein 90 (Hsp90) (49, 54), in the absence of RU486 and were
localized into nuclei following hormone treatment. It is
con-ceivable that the constitutive activity of full-length Rep
pro-teins fused with PR891-HBD results from the highly sensitive
assays we used to monitor Rep activity in transfected cells.
Furthermore, immunofluorescence experiments gave a static
representation of what is probably a dynamic situation in
which, similarly to sex steroids such as progesterone, estrogen,
and glucocorticoid receptors (54), a specific Rep/PR fusion
continuously shuttles between the nucleus and the cytoplasm
with, at any given time, a major fraction of the protein being
localized in one of the two compartments (28, 54).
Neverthe-less, our results suggest that regulation of intranuclear
local-ization is probably neither the only nor the most important
mechanism responsible for the ligand-dependent activity of
Rep1–491/P and Rep1–484/Pn. Although we have no data to
support this hypothesis, it is tempting to speculate that the
major difference between full-length Rep/PR and deleted
Rep/PR fusions is that only the latter require RU486 to
as-sume the proper conformation, thus acquiring full activity
and/or the ability to interact with specific intracellular proteins.
In relation to this point, it has to be remembered that the
progesterone receptor is predominantly nuclear, regardless of
the presence of its ligand, but interacts with appropriate
co-factors and stimulates transcription only in the presence of the
hormone (28, 36, 64).
Rep78 and Rep68 repress the AAV p5 promoter: this
re-pression is postulated to be mediated in part by direct binding
to the p5 RBS and in part by interaction with as yet
unidenti-fied cellular factors that might facilitate repression (29, 39).
Rep1–491/P efficiently down-regulated the p5 promoter in a
hormone-dependent manner, although background activity
(20% repression [Fig. 5A]) was also observed in the absence of
RU486 treatment. It is worth noting that p5 promoter
repres-sion is an extremely sensitive test, as demonstrated by the
behavior of mutant Rep1–484, which is unable to promote
rescue-replication and to drive integration at AAVS1 but is
still active in this assay (21). Further investigation is required
to assess the activity of Rep1–491/P on heterologous
promot-ers, but it is reasonable to expect that the chimeric protein,
which is localized mainly in the cytoplasm in the absence of
hormone treatment, should prove less capable than Rep78/68
of interfering with the expression of cellular genes and,
ulti-mately, with cellular physiology. This hypothesis is further
sup-ported by the evidence that when transfected in 293 cells,
Rep78 reduced their cloning efficiency by 80% while Rep1–
491/P had only a modest effect in the absence of RU486
treat-ment (D. Rinaudo and C. Toniatti, unpublished results). These
results complement previous reports indicating that the growth
rate of 293 cells stably expressing an inducible Rep is altered
and reduced in the presence of the inducer (68).
An interesting property of Rep1–491/P is that it is not
re-sponsive to endogenous steroids such as progesterone, thus
rendering the use of this protein feasible not only for ex vivo
but also for in vivo gene therapy. It has been recently reported
that Rep-mediated site-specific integration at AAVS1 takes
place in transgenic rodents (mice and rats) carrying the human
AAVS1 site (43), and it would be of interest to test the
inte-gration competence of Rep1–491/P in this animal model. The
ideal vector for in vivo utilization of Rep1–491/P and, in
gen-eral, of the AAV integration machinery, has yet to be
con-structed, but one possible approach is that of introducing both
an ITR-flanked transgene and a Rep1–491/P expression
cas-sette into an Ad vector. This Ad/AAV chimeric virus would
borrow properties from both Ad vectors (i.e., infectivity, high
titer, and large capacity) and AAV (integration competence).
We have recently constructed a helper-dependent Ad vector
containing the Rep78 gene and demonstrated that this
chi-meric Ad/AAV vector is indeed capable of triggering
site-specific integration of a codelivered ITR-flanked cassette in
cultured cells (42). A further step in this direction will be to use
the ligand-dependent Rep1–491/P for the generation of
addi-tional Ad/AAV hybrid vectors to be tested in AAVS1
trans-genic rodents.
ACKNOWLEDGMENTS
We are grateful to B. O’Malley and S. Tsai for PR891-HBd cDNA.
We also thank Janet Clench for editing the manuscript and M. Emili
for contributing graphical work.
REFERENCES
1.Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.).1995. Current protocols in molecular biology. John Wiley & Sons, Inc., New York, N.Y.
2.Balague´, C., M. Kalla, and W.-W. Zhang.1997. Adeno-associated virus Rep78 protein and terminal repeats enhance integration of DNA sequences into the cellular genome. J. Virol.71:3299–3306.
3.Baulieu, E. E.1985. RU486: an antiprogestin steroid with contragestive activity in women, p. 1–25.InE. E. Baulieu and J. Segal (ed.), The antipro-gestin steroid RU486 and human fertility control. Plenum Press, New York, N.Y.
4.Berns, K. I., and R. M. Linden.1995. The cryptic life stile of adeno-associ-ated virus. Bioessays17:237–245.
5.Cadepond, F., A. Ulmann, and E.-E. Baulieu.1997. RU486 (mifepristone): mechanisms of action and clinical use. Annu. Rev. Med.48:129–156. 6.Chesnut, J. D., A. R. Baytan, M. Russell, M-P. Chang, A. Bernard, I. H.
Maxwell, and J. P. Hoeffler.1996. Selective isolation of transiently trans-fected cells from a mammalian cell population with vectors expressing a membrane anchored single-chain antibody. J. Immunol. Methods193:17–27. 7.Chiorini, J. A., S. M. Wiener, R. A. Owens, S. R. M. Kyo¨stio¨, R. M. Kotin,
292
RINAUDO ET AL.
J. V
IROL.
on November 9, 2019 by guest
http://jvi.asm.org/
and B. Safer.1994. Sequence requirements for stable binding and function of Rep68 on the adeno-associated virus type 2 inverted terminal repeats. J. Virol.68:7448–7457.
8.Chiorini, J. A., L. Yang, R. M. Kotin, and B. Safer.1995. Determination of adeno-associated virus Rep68 and Rep78 binding sites by random sequence oligonucleotide selection. J. Virol.69:7334–7338.
9.Crystal, R. G.1995. Transfer of genes to humans: early lessons and obstacles to success. J. Biol. Chem.270:404–410.
10. Cunliffe, V., D. Thatcher, and R. Craig.1995. Innovative approach to gene therapy. Curr. Opin. Biotechnol.6:709–713.
11. Davis, M. D., R. S. Wonderling, S. L. Walker, and R. A. Owens.1999. Analysis of the effects of charge cluster mutations in adeno-associated virus Rep68 protein in vitro. J. Virol.73:2084–2093.
12. Di Pasquale, G., and S. N. Stacey.1998. Adeno-associated virus Rep78 protein interacts with protein kinase A and its homologue PRKX and inhib-its CREB-dependent transcriptional activation. J. Virol.72:7916–7925. 13. Evan, G. I., and G. K. Lewis.1985. Isolation of monolonal antibodies specific
for c-mycproto-oncogene product. Mol. Cell. Biol.5:3610–3616. 14. Flotte, T. R., and B. J. Carter.1995. Adeno-associated virus vectors for gene
therapy. Gene Ther.2:357–362.
15. Giraud, C., E. Winocour, and K. I. Berns.1994. Site-specific integration by adeno-associated virus is directed by a cellular DNA sequence. Proc. Natl. Acad. Sci. USA91:10039–10043.
16. Guiochon-Mantel, A., H. Loosfelt, P. Lescop, S. Sar, M. Atger, M. Perrot-Applanat, and E. Milgrom.1989. Mechanisms of nuclear localization of the progesterone receptor: evidence for interaction between monomers. Cell 57:1147–1154.
17. Heilbronn, R., A. Bu¨rkle, S. Stephan, and H. zur Hausen.1990. The adeno-associated virusrep gene suppresses herpes simplex virus-induced DNA amplification. J. Virol.64:3012–3018.
18. Hermonat, P. L.1991. Inhibition of H-ras expression by the adeno-associated virus Rep78 transformation suppressor gene product. Cancer Res.51:3373– 3377.
19. Hermonat, P. L.1994. Down-regulation of the human c-fos and c-myc proto oncogene promoters by adeno-associated virus Rep78. Cancer Lett.81:129– 136.
20. Hirt, B.1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol.26:365–369.
21. Ho¨rer, M., S. Weger, K. Butz, F. Hoppe-Seyler, C. Geisen, and J. A. Klein-schmidt.1995. Mutational analysis of adeno-associated virus Rep protein-mediated inhibition of heterologous and homologous promoters. J. Virol. 69:5485–5496.
22. Im, D.-S., and N. Muzyczka.1990. The AAV origin-binding protein Rep68 is an ATP dependent site-specific endonuclease with DNA helicase activity. Cell61:447–457.
23. Im, D.-S., and N. Muzyczka.1992. Partial purification of adeno-associated virus Rep78, Rep68, Rep52, and Rep40 and their biochemical characteriza-tion. J. Virol.66:1119–1128.
24. Kearns, W. G., S. A. Afione, S. B. Fulmer, M. G. Pang, D. Erikson, M. Egan, M. J. Landrum, T. R. Flotte, and G. R. Cutting.1996. Recombinant adeno-associated (AAV-CTFR) vectors do not integrate in a site-specific fashion in an immortalized epithelial cell line. Gene Ther.3:748–755.
25. Kellendonk, C., F. Tronche, A.-P. Monaghan, P.-O. Angrand, F. Stewart, and G. Schu¨tz.1996. Regulation of Cre recombinase activity by the synthetic steroid RU486. Nucleic Acids Res.24:1404–1414.
26. Kotin, R. M., M. Siniscalco, R. J. Samulski, X. D. Zhu, L. Hunter, C. A. Laughlin, S. M. Laughlin, N. Muzyczka, M. Rocchi, and K. I. Berns.1990. Site-specific integration by adeno-associated virus. Proc. Natl. Acad. Sci. USA87:2211–2215.
27. Kotin, R. M., R. M. Linden, and K. I. Berns.1992. Characterization of a preferred site on human chromosome 19q for integration of adeno-associ-ated virus DNA by non-homologous recombination. EMBO J.11:5071–5078. 28. Kunar Tyagi, R., L. Amazit, P. Lescop, E. Milgrom, and A. Guiochon-Mantel.1998. Mechanisms of progesterone receptor export from nuclei: role of