Hijacking of Host Calreticulin Is Required for the White Spot
Syndrome Virus Replication Cycle
Apiruck Watthanasurorot,aEnen Guo,aSirinit Tharntada,bChu-Fang Lo,cKenneth Söderhäll,a,dIrene Söderhälla
Department of Comparative Physiology, Uppsala University, Uppsala, Swedena; Department of Veterinary Technology, Faculty of Veterinary Technology, Kasetsart University, Bangkok, Thailandb; College of Bioscience and Biotechnology, National Cheng Kung University, Tainan, Taiwan, Republic of Chinac; Science for Life Laboratory, Uppsala University, Uppsala, Swedend
ABSTRACT
We have previously shown that multifunctional calreticulin (CRT), which resides in the endoplasmic reticulum (ER) and is
involved in ER-associated protein processing, responds to infection with white spot syndrome virus (WSSV) by increasing
mRNA and protein expression and by forming a complex with gC1qR and thereby delaying apoptosis. Here, we show that
CRT can directly interact with WSSV structural proteins, including VP15 and VP28, during an early stage of virus
infec-tion. The binding of VP28 with CRT does not promote WSSV entry, and CRT-VP15 interaction was detected in the viral
genome in virally infected host cells and thus may have an effect on WSSV replication. Moreover, CRT was detected in the
viral envelope of purified WSSV virions. CRT was also found to be of high importance for proper oligomerization of the
viral structural proteins VP26 and VP28, and when CRT glycosylation was blocked with tunicamycin, a significant decrease
in both viral replication and assembly was detected. Together, these findings suggest that CRT confers several advantages
to WSSV, from the initial steps of WSSV infection to the assembly of virions. Therefore, CRT is required as a “vital factor”
and is hijacked by WSSV for its replication cycle.
IMPORTANCE
White spot syndrome virus (WSSV) is a double-stranded DNA virus and the cause of a serious disease in a wide range of
crusta-ceans that often leads to high mortality rates. We have previously shown that the protein calreticulin (CRT), which resides in the
endoplasmic reticulum (ER) of the cell, is important in the host response to the virus. In this report, we show that the virus uses
this host protein to enter the cell and to make the host produce new viral structural proteins. Through its interaction with two
viral proteins, the virus “hijacks” host calreticulin and uses it for its own needs. These findings provide new insight into the
in-teraction between a large DNA virus and the host protein CRT and may help in understanding the viral infection process in
gen-eral.
W
hite spot syndrome virus
(WSSV) is a double-stranded DNA
virus of a new virus family, the
Nimaviridae
(
1
). WSSV is the
cause of a serious disease in a wide range of crustaceans that often
leads to high mortality rates (
2
). Substantial progress in
under-standing the molecular biology of WSSV has been made during
the last decade (
2
,
3
). The emergence of genomic, transcriptomic,
and proteomic tools has resulted in important insight into WSSV
biology and into some of the host responses to the virus infection.
Using proteomic approaches, it has been discovered that the
vi-rion contains more than 40 structural proteins, and at least 5
ma-jor structural proteins without glycosylation have been identified:
VP15, VP19, VP24, VP26, and VP28 (
1
,
4
,
5
). Of these, VP26 and
VP28 are the most abundant proteins; VP28 is an envelope
pro-tein, and VP26 acts as a linker between the envelope and the
nu-cleocapsid (
1
,
6
,
7
). Homo-oligomers, mainly trimers of VP28,
have been detected in WSSV virions by X-ray crystallography, and
this protein functions in envelope assembly, cell attachment, and
host penetration (
8
). Formation of homo-oligomers has also been
observed to occur with the WSSV tegument proteins VP24 and
VP26, which play essential roles in envelope assembly (
8–10
).
However, no oligomerization of nucleocapsid VP15 has been
re-ported. The nucleocapsid protein VP15 is a DNA-binding protein
that resembles histone proteins and may function in viral
replica-tion (
11
,
12
).
In previous studies, both transcriptomic and proteomic
evi-dence has indicated an enhancement of calreticulin (CRT)
expres-sion in response to WSSV infection (
13–15
). Important roles of
host CRT have been detected in responses to several other
patho-genic viral infections, such as dengue virus (
16
), rubella virus (
17
),
hepatitis C virus (
18
), and influenza virus (
19
) infections. The
viral induction of host CRT is likely to confer several advantages to
these viruses. Although a CRT/gC1qR complex, which prevents
apoptosis, has recently been detected during WSSV infection (
15
),
little is known concerning possible direct functions of CRT in
WSSV infection. Because of the multiple functions of CRT, this
conserved endoplasmic reticulum (ER) chaperone has emerged as
a frequent target of viral exploitation (
20
).
CRT functions as a chaperone and is involved in the folding
of several viral and nonviral glycoproteins (
21
). CRT facilitates
the correct formation of disulfide bonds, for example, in the
viral glycoproteins influenza virus hemagglutinin (HA) and
Received10 April 2014Accepted3 May 2014
Published ahead of print7 May 2014
Editor:G. McFadden
Address correspondence to Irene Söderhäll, Irene.Soderhä[email protected]. Copyright © 2014, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.01014-14
RETRACTED
on November 7, 2019 by guest
http://jvi.asm.org/
Downloaded from
on November 7, 2019 by guest
http://jvi.asm.org/
Downloaded from
on November 7, 2019 by guest
http://jvi.asm.org/
Semliki forest virus spike protein p62 (SFV p62) (
18
,
22
). The
carbohydrates on the N-terminal residues of both HA and SFV
p62 play a central role in their recognition by CRT (
22
,
23
).
Direct binding of CRT to these viral structural and
nonstruc-tural proteins occurs in the ER. The proper protein folding
mediated by ER chaperones is required for proper production
of virions (
24
). In addition to the ER, the CRT is also
translo-cated to other intracellular compartments, as well as to the cell
surface and extracellular compartments, during a virus
infec-tion (
20
,
25
). The ability of CRT to bind to the RNA of rubella
virus and dengue 4 virus has been shown, suggesting a possible
role in viral replication (
17
,
26
). In addition, CRT signaling
when Ca
2⫹storage is depleted can activate ER stress, which also
supports a high level of viral replication (
27
).
Here, we focus on the host CRT molecule, which is hijacked
and exploited by WSSV. The aim of our study was to define
pos-sible roles for the ER in the entry process and to determine the
effect of CRT on WSSV, thereby contributing to a more detailed
understanding of the interactions between WSSV virus proteins
and the host cell CRT molecule.
MATERIALS AND METHODS
Animals, cell culture, virus, antibodies, and materials.Healthy inter-molt freshwater crayfish (Pacifastacus leniusculus) were obtained from Lake Vättern, Sweden, and maintained in aerated tap water at 10°C. Cray-fish hematopoietic tissue (HPT) cells were prepared and cultured as pre-viously described (28), and 1/3 of the medium was changed at 48-h inter-vals. WSSV was purified as previously described (29) and resuspended in sterile crayfish saline buffer at a concentration of 2⫻107copies/ml. The
viral titer was determined by real-time PCR (qPCR). A standard curve for WSSV was constructed as previously described (30). Quantitation of WSSV amplicons was accomplished by measuring the cycle threshold (CT) value. Since the plot of the log of the initial target copy number for
this assay was identical to that of WSSV DNA, it is considered thatCT
values obtained with infected crayfish DNA extracts can be converted to the numbers of viral genomic DNA targets by using the standard curve, and they are referred to here as viral titers. Polyclonal antibodies against CRT were a kind gift from Sirawut Klinbunga and Virak Visudtiphole (Aquatic Molecular Genetics and Biotechnology Laboratory, National Center for Genetic Engineering and Biotechnology, National Science and Technology Development Agency), and polyclonal antibodies against
VP15, VP26, and VP28 WSSV structural proteins were made as previously described (7). A mouse monoclonal antibody to human CRT (A-9; cata-log number SC-166837) used for the proximity ligation assay (PLA) and a goat polyclonal antibody to actin (C-11; catalog number SC-1615) were obtained from Santa Cruz Biotechnology. Dithiothreitol (DTT) was from Sigma, and thapsigargin was from Calbiochem.
Infection studies.All WSSV infection experimentsin vitroandin vivo and expression analysis by quantitative PCR (qPCR) were performed as described previously (31,32). In our study of ER processes associated with WSSV infection, pharmacological ER inhibitors consisting of 20M MG-132, 1M thapsigargin, and 5 mM DTT (final concentrations) were added either 30 min before or 6 h after the addition of WSSV.
dsRNA-mediated gene silencing.Gene-specific primers forP. lenius-culusCRT and green fluorescent protein (GFP) were incorporated into the T7 promoter (Table 1) at the 5=end and used to amplify PCR products as a template for double-stranded RNA (dsRNA) synthesis. A GFP tran-script was amplified in a pd2EGFP-1 vector (Clontech) as a template and used as the control. The amplified products were then purified using a GenElute gel extraction kit (Sigma), followed byin vitrotranscription using a MegaScript kit (Ambion). The RNA interference (RNAi) effi-ciency was estimated by PCR using the oligonucleotide primers shown in
Table 1. The dsRNA transfection and WSSV infection into HPT cell cul-tures were performed as described by Liu et al. (33).
Production and purification of recombinant protein.The coding sequences of crayfish CRT without signal peptide (the first 17 amino acids [aa] at the N terminus) were amplified (using the primers listed inTable 1), followed by cloning into a pGEX-4T-1 vector (GE Health-care) at the BamHI and XhoI cleavage sites. The viral structural pro-teins VP15 and VP28 were amplified (using the primers listed inTable 1), followed by cloning into a pGEX-4T-1 vector at the BamHI and NotI cleavage sites and a pET-17b vector at the HindIII and SacI cleav-age sites, respectively. These constructed vectors were then trans-formed intoEscherichia coliBL21. Single positive colonies were picked and cultured in LB medium containing 100 mg/ml ampicillin until the optical density at 600 nm (OD600) reached 0.6, and then the cultures were
induced with 1M isopropyl--D-thiogalactopyranoside (IPTG) for 5 h at 37°C. The proteins were expressed as fusion products with glutathione S-transferase (GST; pGEX-4T-1) or His tags (pET-17b). The GST and His6 fusion proteins were purified using a GST-trap FF column (GE Healthcare) and HisPur cobalt resin (Thermo Scientific), and the purity of the recombinant proteins was confirmed by SDS-PAGE and Western blotting (WB).
TABLE 1Primer pairs used in this articlea
Method
Primer sequence
Forward Reverse
qPCR
CRT GSP ATTGGGAGTCTCGTTGGGTTC TCAAGTATCCACCGCCACAGT
40S ribosomal protein GSP CCAGGACCCCCAAACTTCTTA GAAAACTGCCACAGCCGTTG
Ie1 GSP TCAATTTTATGTGGCTAATGGAGA CTTGAGTGGAGAGAGAGCTAGTTATAA
WSSV VP28 GSP TCACTCTTTCGGTCGTGTCG CCACACACAAAGGTGCCAAC
Recombinant protein production
Recombinant CRT TTGAATTCAAGGTGTTTTTCGAAGAGAAGTTC TTTCTCGAGTTACAGTTCATCGTGATCTCTCTCAT
Recombinant VP15 TTTCGGGATCCATGGTTGCCCGAAGCTCC TTTTTGCGGCCGCTTAACGCCTTGACTTGC
RNA interference
dsCRT taatacgactcactatagggTCTCTTGTTGAATCTAAGGTGTT taatacgactcactatagggTAGGACCTCGTAAGTGTTATCAG
dsGFP taatacgactcactatagggCGACGTAAACGGCCACAAGT taatacgactcactatagggTTCTTGTACAGCTCGTCCATG
CRT GSP for estimating RNAi efficiency
ATGAAGATCTTTTTGTTCGCCG TTACAGTTCATCGTGATCTCTCTCATTC
aGSP, gene-specific primers. Primer sequences are given in 5=-to-3=format. Lowercase letters indicate the sequence of the T7 promoter.
RETRACTED
on November 7, 2019 by guest
http://jvi.asm.org/
Watthanasurorot et al.
RETRACTED
on November 7, 2019 by guest
http://jvi.asm.org/
GST pulldown and far-Western overlay assays.To identify proteins in WSSV that are able to bind to CRT, WSSV envelope proteins were solubilized by incubation with Triton X-100 (0.4% Triton X-100 for 50l of purified WSSV) for 1 h at room temperature with gentle shaking. The envelope fraction was collected by centrifugation at 30,000⫻gfor 20 min at 4°C and suspended in phosphate-buffered saline (PBS) buffer.
In the GST pulldown assay, the interaction of WSSV envelope protein and CRT was examined by incubating 5g of purified GST-CRT, 5g of isolated WSSV envelope proteins, and glutathione-Sepharose 4B resin (50
l of 50/50 bed slurry) for 2 h at 4°C. In the control reaction, GST-CRT was replaced with GST. After incubation, the samples were washed 10 times with PBS, and then bound proteins were eluted by adding PBS containing 10 mM reduced glutathione. The eluted proteins were de-tected by 12% SDS-PAGE and stained with Coomassie blue. The presence of GST and GST-PlgCRT was confirmed by Western blot analysis.
Using far-Western overlay assays, the protein virus envelope fraction was subjected to 12.5% SDS-PAGE, transferred to polyvinylidene difluo-ride (PVDF) membranes and blocked with 10% skim milk in Tris-buff-ered saline containing Tween 20 (TBST) for 1 h at room temperature. After three washes with TBST, the membranes were incubated with 25 nM GST-CRT or GST (as a control) in 10 ml TBST for 1 h at room tempera-ture. The blots were washed twice and incubated for 1 h with a 1:1,000 dilution of an anti-GST antibody and then washed twice, incubated for 1 h with a 1:2,000 dilution of an anti-mouse IgG peroxidase-linked species-specific whole antibody for 1 h and, finally, washed three times before detection was performed using an enhanced chemiluminescence (ECL) WB reagent kit.
In situPLAs.Normal and WSSV-infected HPT cells were attached to Superfrost slides. The attached HPT cells were fixed in fresh 4% parafor-maldehyde (PFA) and carefully washed with 1⫻PBS. The slides were incubated with 70% ethanol, placed on ice for 60 min, and air dried. These slides were stored at⫺20°C until the proximity ligation assay (PLA). The PLA assay was performed using reagents and instructions from the Duolink II kit. Briefly, 40l of the blocking solution was added to each sample, and the slides were incubated in a humidity chamber for 1 h at 37°C. The primary antibodies (against CRT and either VP15 or VP28) were diluted to 1g/ml in a volume of 350l and added to all slides, including controls, after the blocking solution had been tapped off. The slides were incubated in a humidity chamber overnight (22 h) at 4°C. Detection was performed using PLA secondary probes and ligation ac-cording to the instructions from the Duolink II kit; finally, the slides were covered with 10l SlowFade Gold Antifade Reagent (Invitrogen catalog no. S36936), sealed, and stored at 4°C before analysis on an epifluores-cence microscope using⫻40 magnification. All images were run using a Duolink Image Tool (Olink Bioscience), and the drawing tool was used to exclude background signals and clumped nuclei. The nuclei were counted manually.
DNA-binding assay: EMSA.In vitroelectrophoretic mobility shift as-says (EMSAs) were performed according to the method described by Wit-teveldt et al. (11). Briefly, purified plasmid DNA (pET28a) was mixed with either recombinant VP15 protein (0, 0.06, or 0.24g) or recombinant CRT (0 or 1g) and two recombinant proteins, rgC1qR at a concentra-tion of either 0.06 or 0.24g and 1g of rCRT, in a final volume of 20l containing 300 mM MgCl2. This mixture was incubated at 37°C for 30
min and then mixed with 6⫻loading buffer (1 mM EDTA [pH 8.0]), followed by examination in 1% agarose gels in boric acid buffer (45 mM
boric acid, 45 mM Tris-HCl [pH 8.0]). For thein vivomobility shift assays, both normal and WSSV-infected cells at 24 h postinfection were lysed in Tris-HCl buffer (pH 7.4). These lysates were incubated with antibodies against VP15, CRT, or both antibodies together. Preimmune serum was used as a control. This mixture was incubated on ice for 1 h and then mixed with 6⫻loading buffer, followed by examination in 1% agarose gels in boric acid buffer.
In vitroandin vivoneutralization experiments.In vitroexperiments with either recombinant protein or antibody were performed using three groups with three replicates in each group. These experimental groups received different treatments as follows: group 1, WSSV (2⫻104copies/
well; negative control); group 2, WSSV with either recombinant GST or preimmune antibody (positive control); and group 3, either recombinant CRT and anti-CRT antibody with WSSV. Thereafter, the cells were har-vested 24 post-WSSV infection for extraction of total RNA. This experi-ment was repeated twice.
In vivoneutralization experiments with antibodies and crayfish (10 g fresh weight) were divided into three groups with three replicates. The crayfish were injected as follows: group 1 with WSSV (4⫻104copies/
crayfish; negative control), group 2 with WSSV plus preimmunized anti-body (positive control), and group 3 with WSSV plus antianti-body against CRT (10g/crayfish). For neutralization experiments with recombinant proteins, WSSV was preincubated with saline (negative control), recom-binant GST (positive control), or recomrecom-binant CRT at room temperature for 1 h. These mixtures were then injected into crayfish, as described above. The hemolymph of three crayfish from each group was extracted 24 h postinjection, and the hemocytes were separately preserved for RNA extraction. This experiment was repeated twice.
The cDNA from all samples was synthesized using ThermoScript (In-vitrogen). The transcription level of CRTin vitroandin vivowas detected using qPCR (using the primers listed inTable 1).
Fractionation of virion proteins by detergent treatment.A purified virus suspension was treated with 1% Triton X-100 at room temperature for 30 min in 1 M NaCl. The Triton X-100-treated samples were put on top of a 35% sucrose cushion and separated into two fractions, superna-tant and pellet, by centrifugation at 30,000⫻gfor 1 h. The insoluble pellets were dissolved in an equal volume of Tris buffer (envelope frac-tion), and the proteins in solution (nucleocapsid fraction) and in the intact purified virion control were separated by 12.5% SDS-PAGE. After separation, the SDS-PAGE gels were transferred to PVDF membranes, followed by Western blotting using antibody against CRT.
Tunicamycin treatment.HPT cells were incubated with 2⫻106
cop-ies of WSSV/well for 6 h, followed by treatment with 0.5g/ml tunicamy-cin (TUN). Twenty-four hours after the addition of TUN, the cells were washed three times with PBS and then harvested for RNA and protein extractions. Cells that were collected immediately after WSSV addition were used as a negative control. The relative expression level of VP28 was analyzed by qPCR and Western blotting.
Analysis of viral assembly measured as oligomerization of VP26 and VP28.Proteins isolated from purified WSSV or WSSV-infected cells with or without dsCRT, in the presence or absence of DTT, were subjected to nonreducing 12% SDS-PAGE. The separated proteins were transferred to PDVF membranes, followed by Western blotting using antibodies against VP26 and VP28.
Statistical analysis.The relative expression levels of different time groups were examined by one-way analysis of variance (ANOVA)
fol-FIG 1ER-associated CRT-mediated WSSV infection. (A) HPT cells were pretreated with inhibitors for ER processes, including 5 mM DTT, 1M thapsigargin, and 20M MG-132, for 30 min, and then WSSV was added and incubated for 24 h, followed by detection of WSSV-encoded VP28 mRNA expression by qPCR. (B) HPT cells were infected with WSSV, and at 6 h postinfection (h.p.i) the cells were treated with inhibitors for ER processes, including 5 mM DTT, 1M thapsigargin, and 20M MG-132, as for panel A. At 24 h.p.i. WSSV-encoded VP28 mRNA expression was detected by qPCR. (C and D) CRT mRNA expression was analyzed by qPCR (C), and the CRT protein level by Western blotting (D) in cells treated as in panel A. (E to J) CRT silencing was achieved in HPT cells by using dsRNAi (and dsRNAi for GFP as a control), followed by treatment of the cells as in panel A. The CRT knockdown samples with or without inhibitors, DTT (E and F), thapsigargin (G and H), and MG-132 (I and J), were analyzed for VP28 mRNA (E, G, and I) or CRT (F, H, and J) mRNA expression by qPCR. Significant differences are indicated by asterisks: *,P⬍0.05; **,P⬍0.01.
RETRACTED
on November 7, 2019 by guest
http://jvi.asm.org/
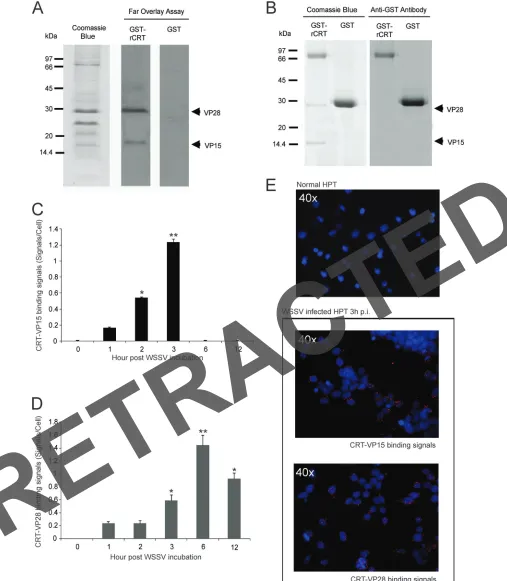
FIG 2Interaction of host CRT with viral structure proteins. (A) Binding of CRT to viral structure proteins was detected by far overlay assay. A WSSV envelope protein fraction was subjected to 12.5% SDS-PAGE, transferred to PDVF membranes, and blocked and washed; finally, the membranes were incubated with 25 nM GST-CRT or GST (as a control) and binding was detected with anti-GST antibodies. A recombinant CRT protein interacted with VP15 and VP28 of WSSV, whereas binding did not occur in the control using GST. (B) The interaction of WSSV envelope protein and CRT was examined by a GST pulldown assay. Purified GST-CRT (or GST as a control) was incubated together with isolated WSSV envelope proteins on a glutathione-Sepharose 4B resin. After washing, bound proteins were eluted and detected by 12% SDS-PAGE and stained with Coomassie blue. The presence of GST and GST-CRT was confirmed by Western blot analysis. (C and D)In situproximity ligation assay (PLA) of WSSV-infected HPT cells at different time intervals was performed using antibodies against CRT and VP15 (C) or CRT and VP28 (D). (E) Three hours postinfection, the complex formations between CRT and WSSV structure proteins are indicated by red signals; the control cells showed similar results when analyzed using both antibodies.
RETRACTED
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.44.551.35.616.2]lowed by Duncan’s new multiple range test and Tukey’s test. Differences were considered statistically significant at aPvalue of⬍0.05. The results are expressed as means⫾standard errors (SE).
RESULTS
CRT-mediated ER processes are associated with WSSV
infec-tion.
We have recently shown that calreticulin (CRT) is required
for the replication and survival of WSSV and is highly upregulated
at the mRNA and protein level 3 to 6 h after infection (
15
). To
determine whether CRT deficiency can alter WSSV replication, we
performed CRT RNAi knockdown, and as shown in
Fig. 1A
, this
resulted in a 50% decrease in the expression of VP28 mRNA.
Because CRT is known as a Ca
2⫹-binding ER chaperone and is
important for protein folding, we then decided to test whether
inhibitors affecting ER-associated processes could influence viral
entry and replication. We added inhibitors of ER Ca
2⫹homeosta-sis (thapsigargin), disulfide bond formation (DTT), and
protea-somal degradation (MG-132) to the HPT cells, which were then
followed by challenge of the cultures with WSSV. Significant
in-hibition of WSSV replication was detected 24 h postinfection
(h.p.i.) after 5 mM DTT treatment only, whereas 5
M
thapsi-gargin was able to significantly increase virus replication (
Fig. 1A
).
In contrast, no significant difference in virus replication was
found after 20
M MG-132 treatment. Interestingly, no effect of
the pharmacological inhibitors on WSSV replication was detected
when they were added 6 h.p.i. (
Fig. 1B
), suggesting that DTT and
thapsigargin may function only in the early events of virus
infec-tion. We were able to further show that there was an effect of these
inhibitors at the level of CRT mRNA and protein, which may
partly explain the role of these inhibitors on virus replication. The
CRT expression level was significantly decreased by DTT and
in-creased by thapsigargin treatment, but there was no effect after
MG-132 treatment (
Fig. 1C
and
D
).
Next, we used dsRNAi to knock down CRT in HPT cells,
fol-lowed by treatment with inhibitors and virus, to determine if the
inhibitor effect was due to an effect at the CRT protein level. We
found that a reduction in viral replication was observed for the
dsCRT groups (
Fig. 1E
,
G
, and
I
). As shown in
Fig. 1E
, the dsCRT
knockdown of HPT cells with 5 mM DTT resulted in the highest
reduction in WSSV replication. There was no difference in viral
replication between knockdown of CRT with or without either
thapsigargin or MG-132 treatment (
Fig. 1G
and
I
). These results
imply that CRT-mediated ER processes are involved in early virus
infection and replication.
Calreticulin interacts with WSSV envelope and nucleocapsid
proteins.
Although CRT is thought to play an important role in
many processes during virus infection, there are few data to show
a direct interaction between this protein and WSSV proteins. To
investigate if any viral protein has the potential to bind to CRT, far
overlay and GST pulldown assays were performed. We obtained
the same results in both of these assays, namely, that CRT can
interact with WSSV VP15 and VP28 (
Fig. 2A
and
B
). Binding
between CRT and these two virus proteins was also detected in
FIG 3Surface-located CRT is not required for WSSV entry. WSSV was preincubated with either recombinant CRT protein (A and B) or antibody (AB) against CRT (C and D). Preincubated viruses were added or injected into HPT cells (in vitro, panels A and C) or crayfish (in vivo, panels B and D), and WSSV replication was assayed as the level of VP28 mRNA expression by qPCR. Recombinant GST and preimmunized antibody were used as positive controls. No significant differences in viral replication were observed between the experimental treatments.
RETRACTED
on November 7, 2019 by guest
http://jvi.asm.org/
Watthanasurorot et al.
RETRACTED
on November 7, 2019 by guest
http://jvi.asm.org/
WSSV-infected HPT cells by a proximity ligation assay (PLA). The
CRT-VP15 interaction was found to increase from 1 to 3 h.p.i. and
then disappear (
Fig. 2C
and
E
), whereas the binding between CRT
and VP28 was detected at 3 to 12 h.p.i. (
Fig. 2D
and
E
). These
interactions confirmed the observations above that CRT may play
a role in the initial steps of WSSV infection.
Binding to cell surface CRT is not required to establish a
vi-rus infection.
Envelope protein VP28, which we have now
iden-tified as a CRT-binding protein, plays an essential role in WSSV
infection (
34
). Our previous report observed CRT on the cell
sur-face of HPT cells (
15
), where it could be suggested to serve as a
receptor for interaction with VP28 during the early stage of virus
infection. To evaluate a role for CRT in this process, we first
ana-lyzed viral replication in HPT cells that were infected with WSSV
(positive control), WSSV preincubated with recombinant CRT
(rCRT), and WSSV preincubated with recombinant GST (rGST;
positive control). These data showed that there were no significant
differences among these three treatments (
Fig. 3A
). We also
con-firmed this finding
in vivo
by injecting control WSSV and WSSV
coated with rCRT or rGST into crayfish, and this assay showed
results similar to the
in vitro
results (
Fig. 3B
).
Furthermore, we performed neutralization experiments in
which the HPT cells or crayfish were incubated or injected with
WSSV in the presence or absence of CRT antibody or preimmune
IgG as a control. The CRT antibodies were not able to inhibit
WSSV infection
in vitro
or
in vivo
(
Fig. 3C
and
D
). Taken together,
these results demonstrated that interference with binding of
WSSV VP28 to cell surface CRT via either recombinant CRT or
CRT antibody does not influence viral replication.
CRT-VP15 interaction is detected in virus DNA at an early
stage of infection.
Based on the function of VP15 as a
histone-like protein present in the WSSV nucleocapsid (
11
) and its
ability to bind CRT, and because CRT has been localized to the
nuclear compartment (
15
), we next asked whether CRT was
associated with the packaging of virus DNA. A purified plasmid
DNA (pET28a) was incubated with various amounts of
recom-binant CRT (rCRT) and VP15 (rVP15). The pET28a plasmid
can be separated into three major DNA topologies, a nicked
circle, linear, or supercoiled DNA, in an agarose gel. All these
topologies could be detected when 0 to 0.06
g of rVP15 was
incubated with or without 1
g of rCRT (
Fig. 4A
, lanes 1 to 4).
However, the supercoiled topology of plasmid DNA
disap-peared after the addition of 0.24
g of rVP15 (lane 5), and only
nicked circular DNA appeared when 1
g of rCRT was added to
plasmid DNA together with 0.24
g of rVP15 (lane 6). This
result suggests that incubation with a combination of rCRT
and rVP15 is involved in binding DNA.
To determine if binding of both CRT and VP15 to WSSV DNA
occurs in virus-infected HPT cells, cells were harvested at 12 h
after WSSV infection and homogenized in a Tris-based buffer (pH
7.5), followed by incubation with antibodies against CRT or VP15
(and preimmune serum as a control) on ice for 1 h. These
mix-tures were then subjected to electrophoresis for a gel mobility shift
assay. We found that a clear DNA band of approximately 300 kb
was detected exclusively in WSSV-infected cells and was not
de-tected in normal uninfected cells (
Fig. 4B
). After addition of either
CRT or VP15 antibodies, a band shift appeared, and the
incuba-tion of these two antibodies together resulted in a supershift (
Fig.
4B
). In contrast, preimmune serum did not affect DNA mobility
(
Fig. 4B
). When treated with dsCRT, all shifted bands disappeared
as expected, because dsCRT clearly affects viral replication.
Fur-thermore, the absence of CRT dramatically decreased viral DNA
replication (
Fig. 4B
) and also delayed the expression of virus
im-mediate early gene (
ie1
) in WSSV-infected HPT cells (
Fig. 4E
). We
also confirmed that all detected DNA in the band at 300 kb in
Fig.
4B
was from the WSSV genome, and this was confirmed using
virus gene-specific primers. The specific PCR products of
WSSV genes could be detected, whereas no crayfish-specific
PCR products could be found (
Fig. 4C
). Furthermore, the DNA
bands of the WSSV genome were cut out of the gel in
Fig. 4B
and crushed in PBS buffer, and a Western blot was performed.
As expected, we found that both CRT and VP15 proteins were
observed in the virus DNA bands (
Fig. 4D
). However, the CRT
protein could not be detected in a nucleocapsid fraction of
mature WSSV virions (
Fig. 4F
). These data suggest a possible
role for the CRT-VP15 interaction in viral replication.
Absence of CRT affected the assembly of viral structure
pro-teins.
It has been suggested that both VP26 and VP28 in the WSSV
envelope are present as trimers (
8
). We showed above that CRT
was detected in the envelope fraction of purified WSSV virions,
but not in their nucleocapsid (
Fig. 4F
). Thus, it is possible that
CRT may affect assembly of the viral envelope structure. To
ex-plore this possibility, oligomerization of these viral structure
pro-teins was detected in HPT cells after knockdown of CRT or after
DTT treatment. All samples (24 h.p.i.) were analyzed by
nonre-ducing SDS-PAGE, followed by Western blotting. In the WSSV
purified from infected crayfish, all three oligomers, including
monomers, dimers, and trimers of VP26 and VP28, could be
de-tected (
Fig. 5A
and
B
). However, in WSSV-infected cells treated
with dsGFP, no trimers could be detected (
Fig. 5A
and
B
), and
only monomers of either VP26 or VP28 were found when the
virus-infected cell was treated with DTT or dsCRT (
Fig. 5A
and
B
).
Interestingly, dimers of both viral structure proteins appeared
af-ter DTT or dsCRT treatment afaf-ter incubation for longer periods of
time (
Fig. 5C
and
D
). As shown in
Fig. 5E
, the protein level of CRT
is increased at 48 h.p.i. This indicates that an absence of CRT
delays oligomerization of the structural proteins in the WSSV
en-velope.
FIG 4A CRT-VP15 complex correlates with viral replication. (A) In anin vitroDNA-binding assay, plasmid DNA (pET28a) was mixed with either recombinant VP15 protein (0, 0.06 or 0.24g) or recombinant CRT (0 or 1g) and two recombinant proteins, rgC1qR at a concentration of either 0.06 or 0.24g and 1g rCRT, in a final volume of 20l containing 300 mM MgCl2, and this was followed by examination by agarose gel electrophoresis. The different plasmid topologies
are indicated to the left. (B) Electrophoretic mobility shift assay using normal (left), WSSV-infected cells (middle), or WSSV-infected cells after CRT RNAi (right) at 24 h.p.i. incubated with antibodies against VP15, CRT or both antibodies together. Preimmune serum was used as a control. (C) The WSSV DNA bands I-III were extracted from WSSV-infected cell samples in the gel shown in (B) and analyzed for the presence of VP15 DNA and host 40S by PCR. All detected bands were shown to contain viral VP15 DNA, but no bands were detected with primers for crayfish 40S ribosomal DNA. (D) The bands in (C) were analyzed for the presence of VP15 protein and host CRT protein by Western blotting, using antibodies against host actin as a control. (E) The expression of WSSVie1mRNA was analyzed at different time intervals after infection in normal or CRT knocked down HPT cells, by qPCR (*,P⬍0.05; **,P⬍0.01). (F) Detection by using Western blot of host CRT present in purified WSSV virions, isolated envelope proteins or isolated WSSV nucleocapsid.
RETRACTED
on November 7, 2019 by guest
http://jvi.asm.org/
Watthanasurorot et al.
RETRACTED
on November 7, 2019 by guest
http://jvi.asm.org/
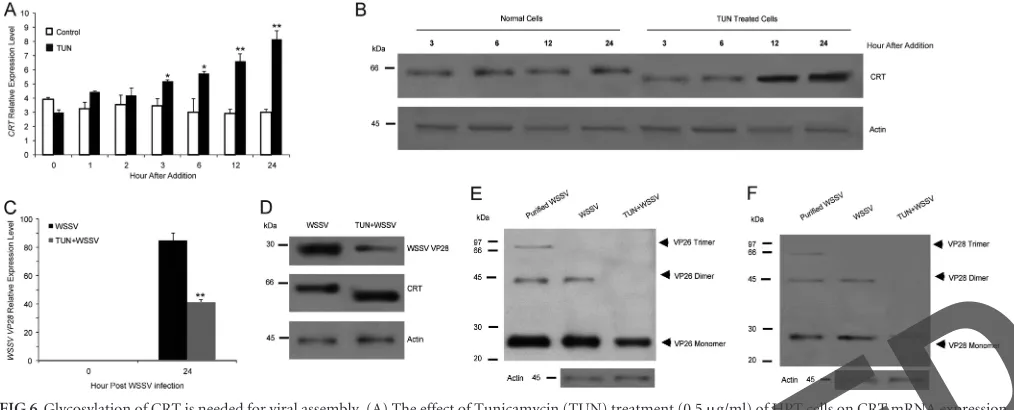
Tunicamycin mediated ER stress-inhibited viral replication
and assembly.
To further investigate if misfolded CRT has any
impact on viral replication and assembly, we treated
WSSV-in-fected cells with tunicamycin (TUN), a glycosylation inhibitor.
TUN inhibits the formation of N-linked glycoproteins and
thereby induces an unfolded protein response (UPR). As shown in
Fig. 6A
and
B
, TUN treatment resulted in increased mRNA
ex-pression of CRT, and at the same time, the CRT formed had a
lower apparent molecular mass by SDS-PAGE. Because CRT is a
glycoprotein, its structure is affected by the drug, as previously
shown (
35
). The WSSV structural proteins VP26 and VP28 are not
glycosylated (
5
), and thus their molecular sizes were not affected
by TUN treatment. However, TUN treatment significantly
de-creased WSSV replication (
Fig. 6C
and
D
), and oligomer
forma-tion of the viral structure proteins was clearly compromised (
Fig.
6E
and
F
, VP26 and VP28, respectively). Thus, the presence of
functional CRT appears to be necessary for WSSV replication and
assembly.
DISCUSSION
Our results show that host CRT is of high importance for WSSV to
complete its replication cycle because it is hijacked by the virus for
use in nucleocapsid and envelope formation. A significant
corre-lation was demonstrated between WSSV replication and the
ex-pression level of CRT in virus-infected cells after pretreatment
with some ER-associated inhibitors. This finding is supported by
several previous observations that CRT is associated with the
pro-duction of infective virions (
24
,
36
). The requirement for CRT has
been detected in the replication cycle of several viruses (
24
,
27
),
but the main function studied has been related to the role of CRT
as a chaperone and its involvement in proper folding and
assem-bly. Recently, the cell entry of simian virus 40 was shown to
de-pend upon ER proteins for correct folding, but the main proteins
responsible for this nonenveloped DNA virus were the two
oxi-doreductases, Erp57 and PDI (
37
). In our study, WSSV infectivity
was highly sensitive to DTT treatment at an early stage, before 6
h.p.i., which could be due partly to an effect of DTT on CRT
expression. However, there may be a dual effect by DTT, as
Schel-haas et al. (
37
) found an early inhibitory effect of DTT on simian
virus infection that was not related to the Erp57-linked ER process
and not linked to CRT.
VP28 is one of the major WSSV proteins, and it naturally forms
homotrimers in the virus envelope (
8
). Previous experiments have
demonstrated an essential role for VP28 during WSSV infection
(
34
), and this suggests a possible role of its envelope trimers in
virus-host interactions. Our results clearly show the ability of CRT
to bind to VP28. Many ER chaperone molecules can act as
core-ceptors for viral infection (
38
,
39
). Although the presence of CRT
on the cell surface of HPT was detected in our previous study (
15
),
we show here that CRT on the cell surface does not act as a
recep-tor for WSSV because no effect on virus replication was detected
between the control and preincubated virus with either the
re-combinant protein or a specific antibody to CRT. The interaction
between CRT and VP28 was detected at its highest point 6 h after
FIG 5Assembly of WSSV structural proteins is mediated through host CRT. (A to D) Western blot showing that the assembly of virions requires multimeric VP26 and VP28. Oligomerization of VP26 (A) and VP28 (B) was detected in WSSV-infected HPT cells after knockdown of CRT (lane 2), with GFP as a control (lane 1), or after DTT treatment (lane 4) and compared to results for purified virions (lane 3). All samples (24 h.p.i.) were analyzed by nonreducing SDS-PAGE, followed by Western blotting. (C and D) Western blot showing the effect of CRT knockdown by dsRNAi in HPT cells, or DTT treatment prior to infection with WSSV to delay the assembly of the viral structural proteins VP26 (C) and VP28 (D). All samples were analyzed by nonreducing SDS-PAGE, followed by Western blotting. (E) Western blot showing the translation level of CRT after different treatments and times.
FIG 6Glycosylation of CRT is needed for viral assembly. (A) The effect of Tunicamycin (TUN) treatment (0.5g/ml) of HPT cells on CRT mRNA expression was analyzed by qPCR. (B) The effect of TUN treatment (0.5g/ml) of HPT cells on CRT protein level and CRT size was analyzed by Western blotting using a CRT antibody. (C) The effect of TUN treatment (0.5g/ml) of HPT cells on VP28 mRNA expression was analyzed by qPCR. (D) The effect of TUN treatment (0.5g/ml) of HPT cells on VP 28 and CRT protein level as detected by Western blotting at 24 h.p.i. The level of actin was analyzed as a control. E and F) The effect of TUN treatment as detailed for panel A on viral structural protein oligomerization in HPT cells analyzed by Western blotting using VP26 antibody (E) or VP28 antibody (F). Significant differences are indicated by asterisks: *,P⬍0.05; **,P⬍0.01.
RETRACTED
on November 7, 2019 by guest
http://jvi.asm.org/
[image:10.585.41.548.67.272.2]WSSV infection, and this is when CRT protein and mRNA are at
their highest level according to our previous study (
15
). This is
also the time when a complex between CRT and gC1qR starts to
form in the cytoplasm. Therefore, it is possible that binding of
VP28 to CRT is important for initiation of this complex
forma-tion.
The most notable feature in WSSV envelope assembly is the
formation of multiprotein complexes (MPCs) at a late time point
postinfection. The majority of MPCs in the WSSV envelope
in-clude a homotrimer of VP26 and a homotetramer of VP28 (
9
).
However, the abundant natural form of VP28 in the viral envelope
is a homotrimer (
8
). The multimeric forms of these two viral
structure proteins became monomers in our DTT-treated WSSV.
The monomers in the trimeric forms of VP26 and the VP28 are
held together by an intramolecular disulfide bridge via cysteine
residues at their C terminus, which can be broken by DTT (
8
).
Oligomerization of these virus structural proteins is slower when
CRT is downregulated by either an individual dsRNAi or after
DTT treatment. Several observations suggest that ER chaperones
facilitate proper protein folding and assembly morphogenesis in
many viruses (
16
,
27
,
40
). The loss of CRT may result in a dramatic
impairment of virus protein folding, which affects and delays the
trimerization of VP26 and tetramerization of VP28. Our
observa-tion is consistent with influenza virus, where calnexin and CRT
clearly influence folding and trimerization of viral hemagglutinin
(HA) (
19
). When HA is prevented from binding to these
chaper-ones, it directly causes a decrease in the outcome of virion
produc-tion. Both castanospermine and tunicamycin (TUN) are
glycosy-lation inhibitors, and these inhibitors have been shown to cause
disassembly of influenza virus HA complex (
19
,
41
). In contrast,
the glycosylation of CRT has been shown to be decreased by TUN
treatment and various conditions associated with ER stress (
35
).
Because CRT is glycosylated by hyperthermic ER stress, CRT can
bind to misfolded nonglycosylated proteins and suppress their
irreversible aggregation (
21
). Here, we showed that enhancement
of misfolded CRT expression after TUN treatment will lead to
decreased WSSV replication. Indeed, Heal et al. (
35
) suggested
that CRT in normal cells is partially glycosylated, and it is
there-fore possible that only nonglycosylated CRT is required for virus
protein folding and assembly.
The CRT binds not only to VP28 of WSSV but also to VP15.
VP15 was identified in the nucleocapsid fraction of WSSV and was
proposed to function in virus replication based on its
DNA-bind-ing ability (
11
). Binding between CRT and VP15 inside the
in-fected cell occurred early during infection, and both CRT and
VP15 were detected in isolated WSSV genomic DNA by Western
blotting. As shown by the PLA assay (
Fig. 2A
), it is possible that
CRT may transiently work together with VP15 during viral
repli-cation or DNA packaging followed by a dissociation of this
CRT-VP15 interaction. The appearance of CRT-VP15 can be explained by its
histone-like ability to bind with WSSV DNA (
11
). Chromosomal
region maintenance 1 (Crm1) is indicated to be a general receptor
for nuclear protein export, and use of the Crm1 export pathway is
common among complex viruses, including DNA viruses (
42–
44
). CRT also has a nuclear export function for glucocorticoid
receptors (
45
), and thus it may be critical to carry either of the
receptors that are required for successful WSSV infection. With
the AIDS virus, Crm1 is required to export viral RNA from the
nucleus to the cytoplasm for HIV-1 replication (
46
,
47
). Similarly,
CRT is found to specifically bind to rubella virus RNA, which
suggests a possible role for such an interaction during viral
repli-cation (
17
).
Kobayashi et al. (
48
) have suggested that CRT plays a role as a
histone chaperone in chromatin dynamics of mitotic
chromo-somes. This is in line with our results showing that CRT and VP15
together interact with WSSV DNA and so may affect replication or
DNA packaging. A common feature of most DNA-binding or
hi-stone-like proteins is the ability to form DNA supercoils (
49
). In
this work, we showed that VP15 as well as CRT preferentially bind
to supercoiled DNA as previously shown (
11
,
48
). Further, a DNA
mobility shift assay confirmed the VP15 and CRT binding to the
WSSV genome by addition of anti-CRT and anti-VP15
antibod-ies, especially if they were incubated together. This DNA binding
also disappeared after CRT had been silenced by dsRNAi for CRT.
Moreover, the knockdown of CRT results in a significant decrease
in viral DNA duplication and viral gene transcription. Taken
to-gether, these observations show that an involvement of CRT with
VP15 is necessary for viral replication.
Although binding of CRT with VP15 can be observed for
WSSV-infected cells, no CRT was detected in the nucleocapsid
fraction of WSSV virions. In contrast, the CRT protein is found in
the virus envelope fraction and mature WSSV virions. Several
proteomic studies of human-pathogenic viruses have shown the
presence of many host proteins in their virions (
50–52
). However,
no host cellular proteins have yet been reported in WSSV, and
proteomic analyses so far report only 30 major viral proteins (
5
,
7
,
53
). These WSSV proteins are detected by SDS-PAGE and
two-dimensional (2D) gel electrophoresis because they are all highly
abundant proteins, whereas host proteins in viral particles are
present at very low levels. In the influenza virus, the host proteins
incorporated into its virion were only discovered using specific
antibodies (
54
,
55
). Also in our study, we were able to detect CRT
protein in the enveloped WSSV by using a specific antibody to
CRT. Enveloped viruses have considerable potential to
incorpo-rate numerous host proteins into their membranes as well as
in-side the envelope, and these can be present at low levels, thereby
making their detection difficult (
56
,
57
). We propose that CRT is
incorporated into WSSV virions through an interaction with
VP28, as demonstrated in this study.
In summary, this work provides the first evidence for a role of
CRT in the WSSV replication cycle. Although CRT binding with
gC1qR to prevent apoptosis has been reported during WSSV
in-fection (
15
), additional proof for a direct interaction between CRT
and virus proteins has been demonstrated in the present study.
Thus, our results may represent a common function for CRT in
other enveloped viruses during infection and viral assembly.
ACKNOWLEDGMENTS
This work was financed by Swedish Research Council VR (http://www.vr .se; grant 2011-4797 to I.S. and grants VR 319-2010-6250 and 621-2012-2418 to K.S.) and the Swedish Research Council Formas (http: //www.formas.se; grant 223-2011-606 to K.S.).
The funders had no role in the study design, data collection and anal-ysis, decision to publish, or preparation of the manuscript.
REFERENCES
1.van Hulten MC, Witteveldt J, Peters S, Kloosterboer N, Tarchini R, Fiers M, Sandbrink H, Lankhorst RK, Vlak JM.2001. The white spot syndrome virus DNA genome sequence. Virology286:7–22.http://dx.doi .org/10.1006/viro.2001.1002.
2.Liu H, Söderhäll K, Jiravanichpaisal P. 2009. Antiviral immunity in Watthanasurorot et al.
RETRACTED
on November 7, 2019 by guest
http://jvi.asm.org/
crustaceans. Fish Shellfish Immunol.27:79 – 88.http://dx.doi.org/10.1016 /j.fsi.2009.02.009.
3.Flegel TW, Sritunyalucksana K.2011. Shrimp molecular responses to viral pathogens. Mar. Biotechnol. (NY)13:587– 607.http://dx.doi.org/10 .1007/s10126-010-9287-x.
4.Xie X, Xu L, Yang F.2006. Proteomic analysis of the major envelope and nucleocapsid proteins of white spot syndrome virus. J. Virol.80:10615– 10623.http://dx.doi.org/10.1128/JVI.01452-06.
5.van Hulten MC, Goldbach RW, Vlak JM. 2000. Three functionally diverged major structural proteins of white spot syndrome virus evolved by gene duplication. J. Gen. Virol.81:2525–2529.
6.Wan Q, Xu L, Yang F.2008. VP26 of white spot syndrome virus functions as a linker protein between the envelope and nucleocapsid of virions by binding with VP51. J. Virol.82:12598 –12601.http://dx.doi.org/10.1128 /JVI.01732-08.
7.Tsai JM, Wang HC, Leu JH, Wang AH, Zhuang Y, Walker PJ, Kou GH, Lo CF.2006. Identification of the nucleocapsid, tegument, and envelope proteins of the shrimp white spot syndrome virus virion. J. Virol.80:3021– 3029.http://dx.doi.org/10.1128/JVI.80.6.3021-3029.2006.
8.Tang X, Wu J, Sivaraman J, Hew CL.2007. Crystal structures of major envelope proteins VP26 and VP28 from white spot syndrome virus shed light on their evolutionary relationship. J. Virol.81:6709 – 6717.http://dx .doi.org/10.1128/JVI.02505-06.
9.Li Z, Xu L, Li F, Zhou Q, Yang F.2011. Analysis of white spot syndrome virus envelope protein complexome by two-dimensional blue native/SDS PAGE combined with mass spectrometry. Arch. Virol.156:1125–1135.
http://dx.doi.org/10.1007/s00705-011-0954-7.
10. Chang YS, Liu WJ, Chou TL, Lee YT, Lee TL, Huang WT, Kou GH, Lo CF.2008. Characterization of white spot syndrome virus envelope protein VP51A and its interaction with viral tegument protein VP26. J. Virol. 82:12555–12564.http://dx.doi.org/10.1128/JVI.01238-08.
11. Witteveldt J, Vermeesch AM, Langenhof M, de Lang A, Vlak JM, van Hulten MC.2005. Nucleocapsid protein VP15 is the basic DNA binding protein of white spot syndrome virus of shrimp. Arch. Virol.150:1121– 1133.http://dx.doi.org/10.1007/s00705-004-0483-8.
12. van Hulten MC, Reijns M, Vermeesch AM, Zandbergen F, Vlak JM. 2002. Identification of VP19 and VP15 of white spot syndrome virus (WSSV) and glycosylation status of the WSSV major structural proteins. J. Gen. Virol.83:257–265.
13. Wang HC, Kou GH, Lo CF, Huang WP.2007. Identification of icp11, the most highly expressed gene of shrimp white spot syndrome virus (WSSV). Dis. Aquat. Organ.74:179 –189.http://dx.doi.org/10.3354/dao074179. 14. Wang HC, Leu JH, Kou GH, Wang AH, Lo CF.2007. Protein expression
profiling of the shrimp cellular response to white spot syndrome virus infection. Dev. Comp. Immunol.31:672– 686.http://dx.doi.org/10.1016/j .dci.2006.11.001.
15. Watthanasurorot A, Jiravanichpaisal P, Söderhäll K, Söderhäll I.2013. A calreticulin/gC1qR complex prevents cells from dying: a conserved mechanism from arthropods to humans. J. Mol. Cell Biol.5:120 –131.
http://dx.doi.org/10.1093/jmcb/mjt005.
16. Mondotte JA, Lozach PY, Amara A, Gamarnik AV.2007. Essential role of dengue virus envelope protein N glycosylation at asparagine-67 during viral propagation. J. Virol.81:7136 –7148.http://dx.doi.org/10.1128/JVI .00116-07.
17. Singh NK, Atreya CD, Nakhasi HL.1994. Identification of calreticulin as a rubella virus RNA binding protein. Proc. Natl. Acad. Sci. U. S. A.91: 12770 –12774.http://dx.doi.org/10.1073/pnas.91.26.12770.
18. Choukhi A, Ung S, Wychowski C, Dubuisson J.1998. Involvement of endoplasmic reticulum chaperones in the folding of hepatitis C virus gly-coproteins. J. Virol.72:3851–3858.
19. Hebert DN, Foellmer B, Helenius A.1996. Calnexin and calreticulin promote folding, delay oligomerization and suppress degradation of in-fluenza hemagglutinin in microsomes. EMBO J.15:2961–2968. 20. Johnson S, Michalak M, Opas M, Eggleton P.2001. The ins and outs of
calreticulin: from the ER lumen to the extracellular space. Trends Cell Biol.11:122–129.http://dx.doi.org/10.1016/S0962-8924(01)01926-2. 21. Saito Y, Ihara Y, Leach MR, Cohen-Doyle MF, Williams DB. 1999.
Calreticulin functions in vitro as a molecular chaperone for both glycosy-lated and non-glycosyglycosy-lated proteins. EMBO J.18:6718 – 6729.http://dx .doi.org/10.1093/emboj/18.23.6718.
22. Molinari M, Helenius A.2000. Chaperone selection during glycoprotein translocation into the endoplasmic reticulum. Science288:331–333.http: //dx.doi.org/10.1126/science.288.5464.331.
23. Peterson JR, Ora A, Van PN, Helenius A.1995. Transient, lectin-like association of calreticulin with folding intermediates of cellular and viral glycoproteins. Mol. Biol. Cell 6:1173–1184. http://dx.doi.org/10.1091 /mbc.6.9.1173.
24. Maruri-Avidal L, Lopez S, Arias CF.2008. Endoplasmic reticulum chap-erones are involved in the morphogenesis of rotavirus infectious particles. J. Virol.82:5368 –5380.http://dx.doi.org/10.1128/JVI.02751-07. 25. Galluzzi L, Kepp O, Morselli E, Vitale I, Senovilla L, Pinti M, Zitvogel L,
Kroemer G.2010. Viral strategies for the evasion of immunogenic cell death. J. Intern. Med. 267:526 –542. http://dx.doi.org/10.1111/j.1365-2796.2010 .02223.x.
26. Yocupicio-Monroy RM, Medina F, Reyes-del Valle J, del Angel RM. 2003. Cellular proteins from human monocytes bind to dengue 4 virus minus-strand 3=untranslated region RNA. J. Virol.77:3067–3076.http: //dx.doi.org/10.1128/JVI.77.5.3067-3076.2003.
27. Limjindaporn T, Wongwiwat W, Noisakran S, Srisawat C, Netsawang J, Puttikhunt C, Kasinrerk W, Avirutnan P, Thiemmeca S, Sriburi R, Sittisombut N, Malasit P, Yenchitsomanus PT. 2009. Interaction of dengue virus envelope protein with endoplasmic reticulum-resident chaperones facilitates dengue virus production. Biochem. Biophys. Res. Commun.379:196 –200.http://dx.doi.org/10.1016/j.bbrc.2008.12.070. 28. Söderhäll I, Kim YA, Jiravanichpaisal P, Lee SY, Söderhäll K.2005. An
ancient role for a prokineticin domain in invertebrate hematopoiesis. J. Im-munol.174:6153– 6160.http://dx.doi.org/10.4049/jimmunol.174.10.6153. 29. Xie X, Li H, Xu L, Yang F.2005. A simple and efficient method for
purification of intact white spot syndrome virus (WSSV) viral particles. Virus Res.108:63– 67.http://dx.doi.org/10.1016/j.virusres.2004.08.002. 30. Sritunyalucksana K, Utairungsee T, Sirikharin R, Srisala J.2012.
Virus-binding proteins and their roles in shrimp innate immunity. Fish Shellfish Immunol.33:1269 –1275.http://dx.doi.org/10.1016/j.fsi.2012.09.017. 31. Watthanasurorot A, Jiravanichpaisal P, Söderhäll I, Söderhäll K.2010.
A gC1qR prevents white spot syndrome virus replication in the freshwater crayfish Pacifastacus leniusculus. J. Virol.84:10844 –10851.http://dx.doi .org/10.1128/JVI.01045-10.
32. Watthanasurorot A, Jiravanichpaisal P, Liu H, Söderhäll I, Söderhäll K. 2011. Bacteria-Induced Dscam Isoforms of the Crustacean, Pacifastacus leniusculus. PLoS Pathog.7:e1002062.http://dx.doi.org/10.1371/journal .ppat.1002062.
33. Liu H, Jiravanichpaisal P, Söderhäll I, Cerenius L, Söderhäll K.2006. Antilipopolysaccharide factor interferes with white spot syndrome virus replication in vitro and in vivo in the crayfish Pacifastacus leniusculus. J. Virol.80:10365–10371.http://dx.doi.org/10.1128/JVI.01101-06. 34. van Hulten MC, Witteveldt J, Snippe M, Vlak JM.2001. White spot
syndrome virus envelope protein VP28 is involved in the systemic infec-tion of shrimp. Virology 285:228 –233. http://dx.doi.org/10.1006/viro .2001.0928.
35. Heal R, McGivan J.1998. Induction of calreticulin expression in response to amino acid deprivation in Chinese hamster ovary cells. Biochem. J. 329(Pt 2):389 –394.
36. Mehta A, Lu X, Block TM, Blumberg BS, Dwek RA.1997. Hepatitis B virus (HBV) envelope glycoproteins vary drastically in their sensitivity to glycan processing: evidence that alteration of a single N-linked glycosyla-tion site can regulate HBV secreglycosyla-tion. Proc. Natl. Acad. Sci. U. S. A.94: 1822–1827.http://dx.doi.org/10.1073/pnas.94.5.1822.
37. Schelhaas M, Malmstrom J, Pelkmans L, Haugstetter J, Ellgaard L, Grunewald K, Helenius A.2007. Simian Virus 40 depends on ER protein folding and quality control factors for entry into host cells. Cell131:516 – 529.http://dx.doi.org/10.1016/j.cell.2007.09.038.
38. Jindadamrongwech S, Thepparit C, Smith DR.2004. Identification of GRP 78 (BiP) as a liver cell expressed receptor element for dengue virus serotype 2. Arch. Virol.149:915–927.http://dx.doi.org/10.1007/s00705 -003-0263-x.
39. Triantafilou K, Fradelizi D, Wilson K, Triantafilou M.2002. GRP78, a coreceptor for coxsackievirus A9, interacts with major histocompatibility complex class I molecules which mediate virus internalization. J. Virol. 76:633– 643.http://dx.doi.org/10.1128/JVI.76.2.633-643.2002.
40. Courageot MP, Frenkiel MP, Dos Santos CD, Deubel V, Despres P. 2000. Alpha-glucosidase inhibitors reduce dengue virus production by affecting the initial steps of virion morphogenesis in the endoplasmic re-ticulum. J. Virol.74:564 –572.http://dx.doi.org/10.1128/JVI.74.1.564-572 .2000.
41. Molinari M, Eriksson KK, Calanca V, Galli C, Cresswell P, Michalak M, Helenius A.2004. Contrasting functions of calreticulin and calnexin in
RETRACTED
on November 7, 2019 by guest
http://jvi.asm.org/
glycoprotein folding and ER quality control. Mol. Cell13:125–135.http: //dx.doi.org/10.1016/S1097-2765(03)00494-5.
42. Fornerod M, Ohno M, Yoshida M, Mattaj IW.1997. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell90:1051–1060.http: //dx.doi.org/10.1016/S0092-8674(00)80371-2.
43. Soliman TM, Silverstein SJ.2000. Herpesvirus mRNAs are sorted for export via Crm1-dependent and -independent pathways. J. Virol.74: 2814 –2825.http://dx.doi.org/10.1128/JVI.74.6.2814-2825.2000. 44. Popa I, Harris ME, Donello JE, Hope TJ. 2002. CRM1-dependent
function of a cis-acting RNA export element. Mol. Cell. Biol.22:2057– 2067.http://dx.doi.org/10.1128/MCB.22.7.2057-2067.2002.
45. Holaska JM, Black BE, Love DC, Hanover JA, Leszyk J, Paschal BM. 2001. Calreticulin Is a receptor for nuclear export. J. Cell Biol.152:127– 140.http://dx.doi.org/10.1083/jcb.152.1.127.
46. Fischer U, Huber J, Boelens WC, Mattaj IW, Luhrmann R.1995. The HIV-1 Rev. activation domain is a nuclear export signal that accesses an export pathway used by specific cellular RNAs. Cell82:475– 483.http://dx .doi.org/10.1016/0092-8674(95)90436-0.
47. Malim MH, Hauber J, Le SY, Maizel JV, Cullen BR.1989. The HIV-1 rev trans-activator acts through a structured target sequence to activate nu-clear export of unspliced viral mRNA. Nature338:254 –257.http://dx.doi .org/10.1038/338254a0.
48. Kobayashi S, Uchiyama S, Sone T, Noda M, Lin L, Mizuno H, Matsu-naga S, Fukui K.2006. Calreticulin as a new histone binding protein in mitotic chromosomes. Cytogenet. Genome Res.115:10 –15.http://dx.doi .org/10.1159/000094795.
49. Huang CC, Chang KM, Cui H, Jayaram M. 2011. Histone H3-variant Cse4-induced positive DNA supercoiling in the yeast plasmid has
implica-tions for a plasmid origin of a chromosome centromere. Proc. Natl. Acad. Sci. U. S. A.108:13671–13676.http://dx.doi.org/10.1073/pnas.1101944108. 50. Bechtel JT, Winant RC, Ganem D.2005. Host and viral proteins in the
virion of Kaposi’s sarcoma-associated herpesvirus. J. Virol.79:4952– 4964.
http://dx.doi.org/10.1128/JVI.79.8.4952-4964.2005.
51. Johannsen E, Luftig M, Chase MR, Weicksel S, Cahir-McFarland E, Illanes D, Sarracino D, Kieff E.2004. Proteins of purified Epstein-Barr virus. Proc. Natl. Acad. Sci. U. S. A.101:16286 –16291.http://dx.doi.org /10.1073/pnas.0407320101.
52. Franke EK, Yuan HE, Luban J.1994. Specific incorporation of cyclophi-lin A into HIV-1 virions. Nature372:359 –362.http://dx.doi.org/10.1038 /372359a0.
53. Tsai JM, Wang HC, Leu JH, Hsiao HH, Wang AH, Kou GH, Lo CF. 2004. Genomic and proteomic analysis of thirty-nine structural proteins of shrimp white spot syndrome virus. J. Virol.78:11360 –11370.http://dx .doi.org/10.1128/JVI.78.20.11360-11370.2004.
54. Zebedee SL, Lamb RA.1988. Influenza A virus M2 protein: monoclonal antibody restriction of virus growth and detection of M2 in virions. J. Virol.62:2762–2772.
55. Richardson JC, Akkina RK.1991. NS2 protein of influenza virus is found in purified virus and phosphorylated in infected cells. Arch. Virol.116:69 – 80.http://dx.doi.org/10.1007/BF01319232.
56. Maxwell KL, Frappier L.2007. Viral proteomics. Microbiol. Mol. Biol. Rev.71:398 – 411.http://dx.doi.org/10.1128/MMBR.00042-06. 57. Cantin R, Methot S, Tremblay MJ.2005. Plunder and stowaways:
incor-poration of cellular proteins by enveloped viruses. J. Virol.79:6577– 6587.
http://dx.doi.org/10.1128/JVI.79.11.6577-6587.2005. Watthanasurorot et al.
RETRACTED
on November 7, 2019 by guest
http://jvi.asm.org/
Retraction for Watthanasurorot et al., Hijacking of Host Calreticulin
Is Required for the White Spot Syndrome Virus Replication Cycle
Apiruck Watthanasurorot,aEnen Guo,aSirinit Tharntada,bChu-Fang Lo,cKenneth Söderhäll,a,dIrene Söderhälla
Department of Comparative Physiology, Uppsala University, Uppsala, Swedena; Department of Veterinary Technology, Faculty of Veterinary Technology, Kasetsart University, Bangkok, Thailandb; College of Bioscience and Biotechnology, National Cheng Kung University, Tainan, Taiwan, Republic of Chinac; Science for Life Laboratory, Uppsala University, Uppsala, Swedend
Volume 88, no. 14, p.
8116 – 8128
, 2014. The authors and the journal hereby retract this article. After publication, this article was found
to have multiple images that were unacceptably manipulated by Apiruck Watthanasurorot, a clear violation of ASM’s ethical standards.
The figures in question are Fig. 1D, where the actin panels seem to be duplicates of the actin panels in Fig. 1G in Watthanasurorot et al.,
J Mol Cell Biol, 5:120 –131, 2013 (
http://dx.doi.org/10.1093/jmcb/mjt005
), Fig. 2A, where there are likely duplications of Coomassie
Blue and glutathione
S
-transferase (GST) lanes from Fig. 9A in Watthanasurorot et al., J Virol 84:10844 –10851, 2010 (
http://dx.doi
.org/10.1128/JVI.01045-10
), and Fig. 5 and 6, where the bands representing actin as well as VP26 dimer and trimer bands may have been
duplicated between the figures. Since the integrity of the data as presented was compromised and A. Watthanasurorot cannot provide
reliable original files for these figures, this publication is retracted in its entirety. We apologize to the readers of
Journal of Virology
and
regret any inconvenience this causes. A. Watthanasurorot could not be reached when asked to agree to the retraction.
CitationWatthanasurorot A, Guo E, Tharntada S, Lo C-F, Söderhäll K, Söderhäll I.
2016. Retraction for Watthanasurorot et al., Hijacking of host calreticulin is required for the white spot syndrome virus replication cycle. J Virol 90:1155.
doi:10.1128/JVI.02629-15.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.