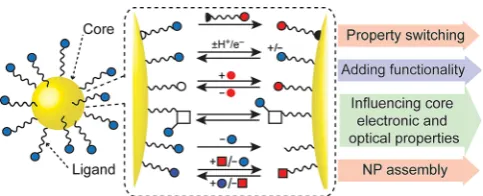
Stimuli-Responsive Nanoparticles
Manipulating the Monolayer: Responsive and Reversible Control
of Colloidal Inorganic Nanoparticle Properties
William Edwards and Euan R. Kay*
[a]Abstract:For a wide range of nanomaterials, surface-bound molecules play a central role in defining properties, and are key to integration with other components—be they mole-cules, surfaces, or other nanoparticles. Predictable and gen-eral methods for manipulating the surface monolayer are therefore crucial to exploiting this new region of chemical space. This review highlights limitations of the few estab-lished methods for controlling nanoparticle-bound molecular functionality, then focuses on emerging new strategies. In
particular, approaches that can achieve stimuli-responsive and reversible modification of surface-bound molecules in colloidal solution are examined, with an emphasis on using these methods to control nanoparticle properties such as solvent compatibility, catalytic activity and cytotoxicity. Final-ly, the outstanding challenges and future potential for pre-cisely controlled nanoparticle-bound monolayers are discussed.
1. Introduction
1.1. The role of the surface monolayer
Monolayer-stabilized inorganic nanoparticles (NPs) have been among the most intensively investigated nanomaterials to emerge over the past quarter of a century. These new chemical entities can often be manipulated as colloidal suspensions using existing molecular and macromolecular techniques and infrastructure, and have engendered considerable interest on account of unique properties that are tunable according to NP material, size and shape. Consequently, considerable efforts have been made to optimize synthetic control over these pa-rameters.[1] Yet, the core nanomaterial only partly determines NP properties; the surface-bound molecular species are equally important in determining overall behavior, and control over this aspect of NP structure remains comparatively under-developed.[2]
Irrespective of core material, the NP-bound surface ligands play a common role: chemical passivation of coordinatively un-saturated surface sites, and steric or electrostatic stabilization of the material in nanoparticle form. Beyond this, the surface molecular monolayer is also crucial for adding function and de-fining properties (Figure 1). For example, surface speciation of colloidal quantum dots (QDs) strongly influences the intrinsic semiconductor electronic and optical properties that are key to applications from photovoltaics to lighting technology and fluorescence bioimaging.[3]For noble metal NPs, the dielectric environment defined by the ligand shell affects the surface plasmon resonance features, and therefore their behavior as sensors.[4]Physicochemical properties that derive from interac-tions between a NP and the surrounding matrix are also de-fined, in large part, by the surface-bound species. Nowhere is this more important than in the large number of potential
ap-plications for monolayer-stabilized NPs in biomedical fields,[5] where cytotoxicity, immunogenicity, biodistribution, and phar-macokinetics are all determined by the NP surface details.[6] Fi-nally, NP-bound ligands present the opportunity to introduce entirely new functionality: from targeting ligands to therapeu-tics; catalysts to imaging probes. For virtually all applications, it is now imperative that synthetic nanochemists develop robust and generalizable strategies for manipulating the NP-bound monolayer.
The de novo synthesis of new NP entities with differing sur-face monolayers is tedious, and intrinsically limiting. Only a small subset of monolayer functionalities will be compatible with any given synthesis conditions, while even subtle varia-tions in ligand molecular structure can have a significant impact on the outcome of a NP synthetic protocol.[7]Far more attractive are postsynthetic methods, through which a range of systems can be accessed from a small number of optimized building blocks, ideally with the ability to switch and tune properties in response to specific triggers.
1.2. Ligand exchange for NP property control
Directly replacing one NP-bound species with another in a ‘ligand exchange’ process has long been established as a powerful method for postsynthetic NP modification.[2] First developed by Murray and co-workers on AuNPs,[8] ligand ex-change allows functionality to be incorporated independent of the NP synthesis conditions, including the combination of
sev-Figure 1.Cartoon representation of a monolayer-stabilized NP and postsyn-thetic methods for ligand modification. From top to bottom: ligand ex-change, charge switching, host–guest complexation, molecular switches, ir-reversible covalent reactions, ir-reversible covalent reactions. Monolayer mo-lecular structure plays a crucial role in determining a variety of important system features.
[a]Dr. W. Edwards, Dr. E. R. Kay EaStCHEM School of Chemistry University of St Andrews
North Haugh, St Andrews, Fife, KY16 9ST (UK) E-mail: [email protected]
Homepage: http://kaylab.wp.st-andrews.ac.uk
ORCID(s) from the author(s) for this article is/are available on the WWW under http://dx.doi.org/10.1002/cnma.201500146.
[image:2.595.306.548.225.323.2]eral different ligands to produce mixed monolayers,[8b]making it an attractive approach for modifying a host of NP properties. Despite widespread adoption, however, this strategy presents several significant challenges. Exchange can often be slow, and complete replacement of one ligand for another is difficult to achieve.[3, 9]For this reason, essentially irreversible replacement of a weakly bound species with stronger ligands is often fa-vored. Disrupting the NP–ligand bond can be a relatively harsh process, leading to unwanted changes in NP properties, size distribution, or even NP decomposition. Perhaps most signifi-cantly, the mechanistic details of ligand exchange are specific to each particular nanomaterial–ligand combination.[3, 9] Evi-dence points to a complex network of reversible and irreversi-ble processes, that often depend on sample history or other batch-to-batch differences. Despite continued development of innovative ligand exchange methodology on a variety of NP surfaces,[10]this lack of understanding, and intrinsic system-de-pendent variability, hinders progress towards predictable and generalizable methods that may be adopted irrespective of— or even blind to—the underlying nanomaterial details.
1.3. Scope of this review
In this Focus Review, we discuss emerging methods for tuning NP properties in colloidal suspension, through postsynthetic manipulation of the surface monolayer, without requiring dis-ruption of the NP-monolayer bond. Through selective exam-ples, we highlight the strengths and weaknesses of each strat-egy, with a focus on approaches that may achieve reversible and stimuli-responsive property control. Crucial to developing predictable methods, we examine evidence correlating phe-nomenologically observed property changes with underlying molecular changes, and we point to some of the outstanding challenges that must now be addressed. Although the scope here is restricted to colloidal suspensions of small inorganic nanoparticles stabilized by a surface monolayer (i.e., metal, metal oxide, and semiconductor NPs), it should be noted that many of the same strategies may be extended to (or in some cases were pioneered on) other types of nanomaterial (e.g., silica particles, carbon dots, fullerenes, polymer nanoparticles or liposomes), as well as for creating and controlling NP assem-blies, and properties in condensed phases.
2. Manipulating Monolayer Charge State for
Nanoparticle Property Tuning
Electrostatics are particularly important in determining interac-tions between a NP and the surrounding matrix, strongly influ-encing colloidal stability and NP adhesion to exogenous mole-cules or surfaces. Reversible charge switching is readily ach-ieved through ionization of acidic or basic groups in the mon-olayer, so for many applications careful consideration must be given to maintaining an appropriate charge under the expect-ed range of operating conditions. Overall neutral monolayers of zwitterionic ligands, for example, have been shown to ex-hibit excellent aqueous colloidal stability, resistance to nonspe-cific binding in biological media and low cytotoxicity.[11]
Re-cently, rational design of acylsulfonamide ligand 1 provided AuNPs displaying a sharp transition between neutral zwitter-ionic, and cationic states around pH 6.5 (Figure 2 a).[12] Congru-ent with the mildly acidic microenvironmCongru-ent surrounding many solid tumors, the transition to positive surface charge en-hanced NP endocytosis and cytotoxicity, suggesting promise for tumor-selective therapeutic or imaging applications.
William Edwards received his MChem degree from the University of York in 2009, staying there to complete a PhD studying multicom-ponent supramolecular gels under the super-vision of Prof. David K. Smith in 2013. Follow-ing this he joined the group of Dr Euan R. Kay as a Postdoctoral Research Fellow. His current research focuses on the controlled synthesis and functionalization of ligand stabilized nanoparticles.
[image:3.595.307.547.69.254.2]Euan Kay received his MChem from the Uni-versity of Edinburgh in 2002 and completed his PhD there in 2006 under the supervision of David A. Leigh. He was the recipient of a 2007 IUPAC Prize for Young Chemists. Fol-lowing postdoctoral work in Edinburgh, he joined Prof. Moungi Bawendi at MIT (2008– 2010). Since 2011, he has been a Royal Society of Edinburgh/Scottish Government Personal Research Fellow at the University of St An-drews. His research interests focus on translat-ing dynamic and stimuli-responsive (supra)-molecular systems into the nanoworld.
Figure 2.Manipulating NP surface charge by proton transfer. a) Homogene-ous monolayers of acylsulfonamide1allow switching between neutral (zwit-terionic) and cationic surface charges at mildly acidic pH values.[12]b) Mixed
Neutral surface charge may alternatively be attained by combining two oppositely charged ligands in a mixed mono-layer, allowing for switching between positive, neutral and negative charge states. Mixing permanently positively charged tetraalkyl ammonium ligand 2, with an excess of carboxylic acid 3, produced pH-responsive AuNP-2/3 that could switch between highly positive and negative surface charges (Fig-ure 2 b).[13]The NPs were colloidally stable in aqueous media at both low and high pH, but underwent reversible precipitation at an intermediate pH, corresponding to surface charge neu-tralization. Interestingly, the neutralization point could be tuned across a wide pH range (4 to7) according to both the ratio2:3and NP radius of curvature. This illustrates an im-portant point: molecular properties of monolayer constituents such as pKacan be strongly influenced by nanoscale features, including monolayer composition and NP radius of curva-ture.[14] The mixed-charge monolayer allowed AuNP-2/3, dis-playing an overall negative surface charge, to be internalized by cells—counter to the commonly held understanding that cellular uptake requires overall positive surface charge.[13] It must be noted here that accurate characterization ofabsolute
NP surface charge is challenging. Most commonly, light scatter-ing is used to measure NP electrophoretic mobility, which is then converted into z-potential. However this conversion is fraught with complications, while z-potential itself is only a proxy for the attached surface charge, and is highly sensitive to the measurement conditions.[15]Electrochemical charging of redox-active monolayer constituents provides a simple—but relatively under-exploited—means to reversibly modulate NP surface charge.[16] In a similar manner to pK
a, redox potentials are sensitive to both the nanoscopic and molecular environ-ment, presenting opportunities for sensing applications,[16a] or control of host–guest interactions (see Section 3.1).[16b]
3. Manipulating Noncovalent Interactions for
Nanoparticle Property Tuning
One of the most highly developed approaches for postsynthet-ic NP monolayer modifpostsynthet-ication exploits reversible and specifpostsynthet-ic hybridization to NP-bound single-stranded oligonucleotides. Introduced simultaneously by Alivisatos,[17] and Mirkin,[18] this has become one of the touchstone methods for colloidal NP functionalization, leading to diverse applications including sensing, gene delivery, and NP assembly.[19] Detailed physical-organic studies have revealed much about the parameters de-termining the remarkable behavior of these systems, allowing NP-bound oligonucleotide hybridization to be exploited in an increasingly predictable manner. Together with the rapid, auto-mated synthesis of oligonucleotides with any given sequence, DNA-coated NPs can genuinely now be considered as ‘Pro-grammable Atom Equivalents’.[20] Yet, the chemical and struc-tural stability of double-stranded DNA is only maintained within a relatively narrow window of conditions. Furthermore, incorporation of non-nucleotide molecular functionality and in situ molecular-level characterization of these complex, oligo-meric chemical structures is extremely challenging. Here, we
will concentrate on newly emerging nonbiomolecular approaches.
3.1. Intermolecular noncovalent monolayer modification
Encapsulating semiconductor QDs in micelles,[21]or amphiphilic polymers,[22]was one of the earliest solutions for converting as-synthesized hydrophobic nanocrystals into biocompatible fluo-rophores, while preserving their attractive optical features. Driven by hydrophobic interactions between the surfactant and the lipophilic NP-bound ligands, bilayer-protected NPs can similarly be prepared from any core nanomaterial bearing a hy-drophobic surface monolayer.[2, 23] Polymeric amphiphiles have proven particularly convenient for incorporating a wide range of functional groups on the bilayer surface through this mild, one-step approach.[24] However, encapsulation is essentially ir-reversible, the challenges of balancing long-term stability while avoiding NP cross-linking can be significant, and the attendant increase in NP solvodynamic size is a drawback for many applications.
Small-molecule host–guest systems present diverse possibili-ties for addressable noncovalent NP monolayer modification.[25] For example, colloidal NP phase transfer has been achieved by masking hydrophobic NP-bound ligands with water-soluble cy-clodextrin (CD) macrocycles.[26] Conversely, incorporating CDs as one of the surface-bound ligands on QDs has been used to create a fluorescence turn-on sensor for various hydrophobic small molecules, operating through the displacement of a quencher from the CD cavity.[27]Yet, these early systems illus-trate the major challenges faced by nonbiomolecular host– guest systems: optimizing complex stability and reversibility, while avoiding kinetic traps; and achieving sufficient selectivity for application in complicated real-world environments.
Recently, Rotello and co-workers have successfully exploited the strong and selective recognition properties of
cucurbit-Figure 3.Competitive host–guest complexation between ADA and CB[7] for selective NP property switching. a) Intracellular decomplexation of AuNP-4·CB[7] reveals cytotoxic AuNP-4.[28]b) Intracellular removal of CB[7] provides
[image:4.595.307.545.507.700.2][n]uril macrocycles to achieve remarkable control over AuNP properties in live cells (Figure 3).[28] Diaminohexane-functional-ized AuNPs were shown to form strong host–guest interactions with cucurbit[7]uril (CB[7]) ; clear evidence for a NP-bound threaded host–guest complex was provided by 1H NMR spec-troscopy. The resulting AuNP-4·CB[7] complex (Figure 3 a) is water soluble and readily taken up into the endosomes of human breast cancer cells, with no apparent cytotoxicity at concentrations 50mm. Subsequent incubation with
1-ada-mantylamine (ADA)—a bioorthogonal competitive guest for CB[7]—resulted in endosomal escape of the NPs and a cytotox-ic response very similar to that elcytotox-icited by AuNP-4alone; pre-sumably the result of intracellular disruption of the NP-bound host–guest complex by ADA.[28] This approach has now been extended to achieve intracellular control of a bioorthogonal catalytic reaction (Figure 3 b).[29]Complex formation in a similar manner to before was used to prevent substrate access to-wards an organometallic catalyst intercalated within the hydro-phobic region of the monolayer; subsequently, catalytic activi-ty could be restored by competitive binding of CB[7] to ADA. Kinetic analysis revealed selective and reversible catalyst inhibi-tion, and excellent agreement to CB[7] binding affinities mea-sured independently by isothermal titration calorimetry. With this approach, intracellular activation of the anticancer drug 5-fluorouracil (5FU) was achieved on co-localization of a 5FU pro-drug with a NP-bound, CB-protected catalyst and ADA.[29]
This elegant strategy suggests possibilities for tuning a bio-logical response according to local concentrations of NP, com-petitive binder and (where relevant) catalytic substrate. There is exciting long-term potential for smart delivery of drugs or imaging agents with spatiotemporal dose control, while more immediate applications include stimuli-responsive tools for chemical biology. However, in these proof-of-concept studies relatively high concentrations of competitive binder are incu-bated over several hours, and translation to in vivo settings still presents a significant challenge.
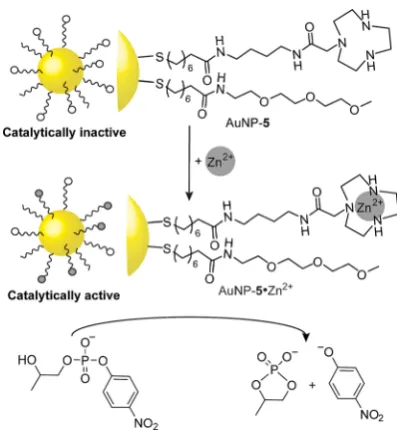
Scrimin, Prins and co-workers have developed a series of AuNPs functionalized with azacrown macrocycles, such as AuNP-5 (Figure 4).[30] Coordination of Zn2+ to the NP-bound
macrocycles affords active transphosphorylation catalysts that mimic phosphodiester cleavage by ribonucleases. A sigmoidal increase in observed reaction rates with increasing metal ion loading indicates a cooperative multinuclear catalytic site, which is facilitated by the multivalent nature of the NP-bound monolayer, and leads to catalytic activities over 600-fold great-er than mononuclear molecular analogues.[30b] The monolayer composition was characterized by a combination of NMR anal-yses and spectrophotometry. Yet, the structure of the active catalyst can still only be inferred on the basis of kinetic meas-urements. Although the evidence strongly suggests a catalytic site involving two Zn2+ centers within the same monolayer,
the prospect of several different catalytically active structures operating simultaneously, or a non-random distribution of li-gands in mixed NP monolayers, cannot be ruled out. Detailed structural characterization of active sites—such as can be pro-vided by crystallography for small molecule catalysts or
en-zymes—remains an important long-term challenge for NP-bound catalysts.
The observation that AuNP-5·Zn2+ also acts as a high-affinity
host for small oligoanions has provided a powerful, and syn-thetically adaptable, system with which fundamental features of host–guest recognition at responsive multivalent nanoscale surfaces can be investigated.[31]Reversible tuning of NP valency can be achieved by adding and removing (using a competitive ligand) Zn2+ from AuNP-6.[31b] The change in binding capacity (i.e., valency) and complexation kinetics has been monitored via emission quenching of fluorescent anionic guests when in close proximity to the metal surface, affording data consistent with at least two distinct anion binding modes. It was further-more observed that AuNP-6·Zn2+ exhibits a marked selectivity
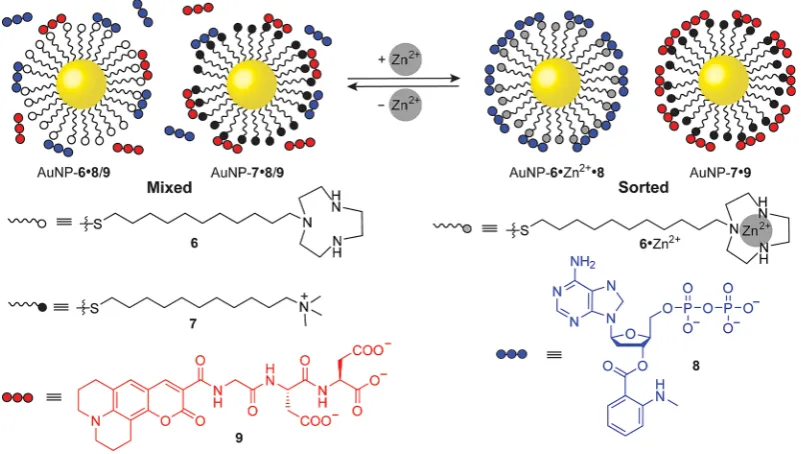
for binding diphosphates over dicarboxylates,[31a] while anion binding to nonmetalated AuNP-6 is pH-dependent.[31b] This multiparameter responsiveness has allowed increasingly com-plex behaviors to be achieved, including self-sorting of diphos-phate (8) and tricarboxylate (9) guests onto two different NP monolayers: phosphate-selective AuNP-6·Zn2+ and
nonselec-tive cationic AuNP-7 (Figure 5).[32] Although a comprehensive series of experiments strongly supports self-sorting, it is still not possible to directly distinguish binding of each fluorescent guest to the different NP surfaces. Switching between the sorted and unsorted states was achieved by removal and addi-tion of Zn2+, while specific stimuli could trigger sequential
re-lease of the probes from their respective NP-hosts. In related work, complexation to AuNP-6·Zn2+ has been used for the
[image:5.595.326.525.62.278.2]dy-namic combinatorial identification of metal-linked bis-nucleo-tide dimers from a pool of lower-affinity monomers, which could be developed either as a metal sensing platform, or for identifying optimal metal-binding nucleotide ligands.[33]
Nonbiomolecular systems offer several advantages over oli-gonucleotides in terms of structural simplicity, adaptability of molecular design and synthetic routes, and applicability under a wider range of environmental conditions. These pioneering designs are slowly closing the gap on the exquisite specificity, selectivity and kinetic tunability exhibited by DNA-functional-ized NPs, but in only a handful of cases is binding strong enough that property changes can be achieved and main-tained independent of an unbound excess of reagent. Signifi-cant challenges also remain in directly linking structural charac-terization of dynamic noncovalent complexes within NP-bound monolayers to observed property changes.
3.2. Intramolecular monolayer conformational and configu-rational changes
Monolayers that respond to a specific stimulus by undergoing a conformational change present a self-contained strategy to tune NP properties without requiring addition of exogenous species. Stimuli-responsive polymers—such as those displaying lower critical solution temperature (LCST) behavior—are attrac-tive ready-made systems for such strategies.[34] Phase transfer across oil–water interfaces has been achieved for AuNPs func-tionalized with random copolymers of 2-(2-methoxyethoxy)eth-yl methacr2-(2-methoxyethoxy)eth-ylate and oligo(eth2-(2-methoxyethoxy)eth-ylene glycol) meth2-(2-methoxyethoxy)eth-yl methacry-late.[35]However, the polymer switching behavior is significant-ly affected by attachment to the NP surface, and bidirectional switching required careful adjustment of both temperature and aqueous phase ionic strength.[35b] Theoretical rationaliza-tion of these subtle effects based on thermodynamic consider-ations underlines the importance of understanding the influ-ence of the nanosurface-bound environment on molecular behavior.
Cross-linked thermosensitive polymers display volume phase transitions between expanded and collapsed states, and have been used to modulate NP catalytic activity and selectivity by controlling substrate diffusion towards the NP surface. For AuNPs coated with cross-linked poly(N-isopropyl acrylamide), a decrease in NP hydrodynamic diameter on increasing tem-perature indicated polymer collapse, and was correlated with a sharp reduction in the rate of NP-catalyzed ferricyanide re-duction by borohydride.[36] In a similar system, substrate selec-tivity could be altered on polymer phase transition (Figure 6 a): reduction of hydrophilic nitro-aromatic substrates being pre-ferred at T<LCST (swollen polymer shell), and hydrophobic substrates reacting faster at T>LCST (collapsed polymer shell).[37]
The same strategy has been used to reversibly control the approach of Raman-active and fluorescent analytes towards a AuNP surface (Figure 6 b). For a probe molecule that does not interact strongly with the NP surface, intercalation into the swollen polymer monolayer results in strong surface-enhanced fluorescence (SEF), but weak surface-enhanced Raman scatter-ing (SERS); conversely, polymer collapse results in a strong SERS signal but quenching of the fluorescence, presumably a result of confining the analyte closer to the NP surface.[38] The ability to capture probes that have little or no affinity with the NP surface, and to switch between two spectroscopic mo-dalities, with an easily addressable, colloidally stable platform, suggests several opportunities for quantitative sensing in com-plex environments. A recent exciting extension exploits pho-tonic heating of hollow gold nanoshells to simultaneously trig-ger the reversible polymer phase-transition and monitor the concomitant SERS response using a near-IR light source.[39]
The rich set of behaviors attained through nonspecific con-formational changes suggest that an even greater level of sophistication may be reached by harnessing the more
precise-Figure 5.Switchable noncovalent self-sorting on NP-bound monolayers. Anionic guests8and9bind nonselectively to AuNP-6and AuNP-7, producing a mixed guest-monolayer state. Addition of Zn2+
produces AuNP-6·Zn2+
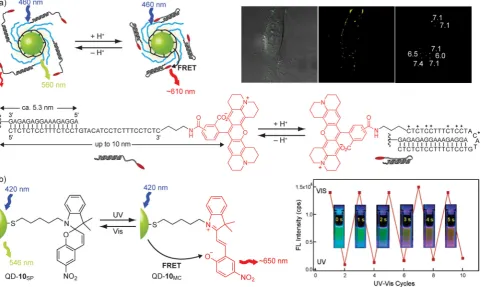
[image:6.595.98.499.67.294.2]ly defined stimuli-induced conformational, co-conformational or configurational changes achieved by modern-day artificial molecular machines.[40]In one of the first examples of a molecu-lar-machine-controlled NP device, a pH-responsive oligonucleo-tide conformational switch was combined with fluorescent QDs to create a biocompatible pH sensor (Figure 7 a).[41] A ra-tiometric (self-referencing) optical response to changing pH was generated by modulating Fçrster resonance energy trans-fer (FRET) between the QD and a pH-insensitive molecular fluo-rophore through large-amplitude proton-triggered folding/un-folding of the molecular machine. Excellent agreement be-tween energy transfer efficiency and the physical dimensions of the sensor construct as pH is varied confirmed the molecu-lar-mechanical transduction mechanism. This defines a universal fluorescent sensor design strategy whereby the analyte-re-sponsive, and the optical reporting components may be opti-mized independently, allowing the unique—but otherwise en-vironment-insensitive—optical properties of QD fluorophores to be harnessed for tracking pH within individual endosomes of living cells (Figure 7 a).[41] However, high-resolution imaging
of very small QD populations (in theory down to individual particles) requires highly homogeneous probe behavior, and so methods for accessing such devices with tightly controlled NP:machine stoichiometry represent the next challenge.
Stimuli-responsive changes in configuration of isomerizable photoswitches also provide a powerful strategy for controlling NP behavior. This area has recently been reviewed extensively elsewhere,[42] and a large proportion of these studies have been directed towards controlling NP aggregation and NP properties in condensed phases. In colloidal suspension, the optical properties of photochromic switches have been applied in a number of instances to control emission from fluorescent NPs. For example, fluorescence from CdSe–ZnS core–shell QDs functionalized with spiropyrans could be reversibly switched on and off by photoirradiation (Figure 7 b).[43]This effect can be attributed to photoconversion between spiropyran (10SP) and merocyanine (10MC) isomers in the NP-bound monolayer; the latter form absorbs strongly in the visible region (lmax= 588 nm), and can therefore quench QD emission (lFL=546 nm) by FRET. Consistent with this mechanism, weak red emission, characteristic of the merocyanine dye, is observed in the ‘off’ state, and quenching efficiency depends on both the number of photoswitches in the monolayer and the spectral overlap between QD emission and meorcyanine absorption bands.[43] Similar results have been achieved with fluorescent gold nano-crystals,[44]and rare-earth upconverting nanophosphors.[45]
These relatively simple examples only hint at the functionali-ty that might be achieved by interfacing more sophisticated artificial molecular machines with NPs. Large-amplitude co-conformational motions of mechanically interlocked molecular shuttles have already been achieved within NP monolayers, creating some of the most structurally advanced switchable NP systems to date.[46]It now remains to properly understand the influence of the surface-confined environment on machine op-eration, and define the most appropriate properties and practi-cal uses to be addressed with these remarkable systems.
4. Manipulating Covalent Connectivity for
Nanoparticle Property Tuning
4.1. Kinetically controlled covalent monolayer modification
[image:7.595.47.289.62.422.2]With the potential for accessing a huge range of kinetically stable, well-defined structures, through a wide array of reac-tions, modifying the covalent connectivity of NP-bound ligands in situ is a highly attractive strategy. Early work established that simple reactions such as ester or amide coupling can di-rectly modify reactive NP-bound ligands,[47] and subsequently a wide range of protocols—many inspired by bioconjugation methods—have been adapted for functionalization of NP-bound monolayers.[48] These established techniques, however, each exploit essentially irreversible reactions, providing a “one-shot” opportunity to modify the monolayer, with no degree of error checking, and resulting in a statistical product distribu-tion that can be difficult to control. Incorporating orthogonally reactive ligands in the ligand shell can allow stepwise modifi-cation,[49] but this in turn requires precise control of the initial
Figure 6.Nanoparticle property switching using thermosensitive polymer ligand shells. a) Switching substrate selectivity for a catalytically active ‘yolk– shell’ AuNP coated with poly(N-isopropyl acrylamide).[37]b) Temperature
monolayer composition. Stimuli responsiveness in these sys-tems is limited to controlling reagent stoichiometry, or reaction conditions to kinetically promote or inhibit reaction progress. A few systems have used bond-breaking reactions to ach-ieve property changes in response to a particular stimulus. Emrick and co-workers reported the inversion of NP-stabilized Pickering emulsions, induced by the acid-promoted cleavage of ether protecting groups (Figure 8 a).[50]AuNP-11
0.9120.1, bear-ing a mixture of hydrophobic (11) and hydrophilic (12) ligands, was found to stabilize a water-in-oil emulsion. Acid initiated deprotection of 11 increases NP hydrophilicity, disrupting the emulsion and leading to phase separation. Adding more water, with agitation, produced an oil-in-water emulsion, stabilized by the more hydrophilic AuNP-12. Appealingly, this approach could also be photonically triggered by including a photoacid generator in the organic phase.[50]
Incorporating directly photocleavable units within the ligand design has been used to produce a number of irreversibly switchable NP systems.[51] For example, ortho-nitrobenzyl ligand13was used to create ‘caged’ CdTe/CdS core/shell
QD-130.25140.75 exhibiting highly quenched photoluminescence on excitation at 405 nm. Irradiation at 365 nm caused the photo-luminescence quantum yield to increase by over 400-fold, a consequence of removing the aromatic substituent.[51a] Simi-lar approaches have exploited enzymatic cleavage of peptidic linkers,[52] or redox modification[52b, 53] of energy- or electron-transfer quenchers on QD surfaces to irreversibly modify fluo-rescence, thereby providing detection and sensing functions.
4.2. Thermodynamically controlled covalent monolayer modification
Approaches that combine the reversibility and stimuli respon-siveness displayed by noncovalent systems or configurational switches, with the stability and structural diversity of covalent chemistry, would afford access to a diverse range of self-con-tained NP systems with precisely controlled and reconfigurable monolayer features, and therefore properties. Dynamic cova-lent reactions have the potential to meet all these require-ments.[54]Applied to NP-bound ligands, this would allow cova-lent bonds to be formed and broken reversibly, with product distributions that are predictably determined by thermody-namic stabilities, and responsive to reaction conditions. How-ever, a change in conditions (e.g. , removal of a catalyst), can ki-netically lock the products, allowing isolation, purification, and application without fear of further changes.
Largely exploited for their ability to operate under relatively mild conditions, there have been a number of examples of conjugation to NPs in ostensibly irreversible processes that in fact exploit potentially reversible covalent bonds.[48]In particu-lar, hydrazone formation under pseudo-irreversible conditions has been developed as a convenient bioconjugation method for incorporating a small number of modifications on both AuNP and QD monolayers.[55]Conversely, acid-catalyzed hydra-zone hydrolysis has been used to selectively release NP-bound cargoes.[56]Conceptually, however, these approaches are
essen-Figure 7.Nanoparticle properties controlled by molecular switches. a) Proton-triggered conformational switching modulates FRET efficiency between a QD donor (green) and molecular acceptor (red), producing a ratiometric response that can be exploited for intracellular pH sensing.[41]
b) Light-triggered spiropyr-an-merocyanine interconversion reversibly modulates FRET from a QD donor over several cycles.[43]
[image:8.595.58.541.67.354.2]tially indistinguishable from the covalent bond forming and breaking reactions discussed in Section 4.1.
[image:9.595.49.289.62.451.2]Recently, Otto and co-workers reported the first example of template-driven dynamic covalent NP functionalization, creat-ing optimal DNA-bindcreat-ing monolayers via selection from a dy-namic combinatorial library of AuNP-bound imines (Figure 9).[57] A mixed monolayer of aldehyde ligand 15 and cationic ligand 16 was treated in aqueous solution with an excess of three aromatic amines (17–19), chosen for their dif-fering potential to form noncovalent interactions with DNA. Using HPLC to quantify the population of unbound amines, it could be inferred that negligible concentrations of imine form in the absence of a template. However, on adding short oligo-nucleotides, selective uptake of different amines was observed, with imine product distributions dependent on the base-pair sequence of the DNA template. The multivalent presentation
of functionality on a NP surface, combined with rapid dynamic covalent exchange of monolayer components opens the way to combinatorial selection of binding surfaces for probing a va-riety of large-area noncovalent biomolecular interactions, which have proven particularly challenging to address using traditional host–guest systems.
In the same journal issue, we reported the first example of dynamic covalent exchange used to reversibly switch NP-bound dynamic covalent functionality between multiple kineti-cally stable states (Figure 10).[58]AuNPs coated with hydrazone terminated ligands could be modified at will by reaction with
Figure 8.Covalent cleavage reactions for nanoparticle property control. a) Acid-triggered cleavage of a hydrophobic tetrahydropyran group disrupts water-in-oil Pickering emulsions stabilized by AuNP-110.9120.1; subsequent
addition of more water and agitation leads to an inverted an oil-in-water emulsion stabilized by AuNP-12. Confocal microscopy images show emul-sions with FITC-dextran dye labelling the aqueous phase.[50]Adapted with
[image:9.595.304.548.64.221.2]permission from reference [50], copyright 2013 Wiley-VCH Verlag GmbH &Co. KGaA, Weinheim. b) Photocleavage ofortho-nitrobenzyl ligand13 re-leases a fluorescence quencher, restoring QD emission.[51a]
Figure 9.DNA-templated formation of dynamic covalent imine AuNP mono-layers. Monolayer composition depends on the specific base pair sequence of the DNA template introduced; negligible imine formation is observed in the absence of DNA.[57]
Figure 10.Reversible dynamic covalent modification of a NP ligand shell by hydrazone exchange, and concomitant solvent compatibility switching. Sol-vents: A=hexane, B=chloroform, C=tetrahydrofuran, D=methanol, E=N,N-dimethylformamide, F=water.[58]
[image:9.595.317.539.431.680.2]an aldehyde exchange unit under acid-catalyzed conditions. The structural change could be harnessed to produce a con-comitant change in NP solvent compatibility: AuNP-20, colloi-dally stable only in polar aprotic solvents, was converted into AuNP-21, which exhibits colloidal stability in less polar organic solvents, by reaction with hydrophobic aldehyde22. Likewise, exchange with sulfonated aldehyde 23 produced water-com-patible AuNP-24. The reversible exchange process allowed each state to be accessed from either of the other two, simply by selecting the appropriate aldehyde exchange moiety (22,
23, or25), while the acid catalyst and all other unbound mo-lecular species could be easily removed at any stage. By incor-porating appropriate fluorine labels, and ensuring complete purification from unbound species, the exchange process could be monitored in real time by 19F NMR, providing direct quantitative assessment of reactivity in the NP-bound mono-layer.[58]
Although dynamic covalent NP monolayer functionalization is clearly still in its infancy, these first two examples illustrate a key advantage of this approach. One system relies on kineti-cally and thermodynamically unstable dynamic covalent bonds, the other exploits reversible covalent exchange under specific conditions, to produce kinetically stable products that may be isolated, purified, characterized and stored. The ability to access such a wide range of reactivity, combined with the inherent structural diversity afforded by modern organic chemistry, suggests that dynamic covalent approaches can provide an extremely powerful addition to the range of strat-egies for the reversible and responsive control of NP-bound functionality and properties.
5. Conclusions and Prospects
Defining the interface between a colloidal NP and the outside world, the surface-bound molecules are crucial to controlling a host of NP properties ; precise control over monolayer struc-tural details at the molecular, supramolecular and nanoscale levels is therefore essential for virtually any potential applica-tion. While well-established direct synthetic routes,[1]ligand ex-change,[8–10]oligonucleotide hybridization,[17–19]and amphiphile encapsulation[21–24]approaches will continue to provide essen-tial solutions, methods for reversible and stimuli-responsive postsynthetic manipulation of NP properties using nonbiomo-lecular systems can lead to an as-yet unimagined array of smart, functional NPs.
A proper understanding of the multifarious parameters af-fecting molecular behavior within the NP-bound monolayer will be essential to arriving at predictable and generalizable strategies. Notwithstanding the significant challenges facing molecular-level characterization within dilute, heterogeneous and polydisperse systems, many of the examples discussed here illustrate how advances in synthetic strategy, purification protocols, analytical methods and theoretical understanding are now combining to achieve unprecedented levels of infor-mation, and therefore link phenomenological behavior to structural details.
At the same time, complexity is one of the most exciting as-pects of these multicomponent systems, presenting possibili-ties that just do not exist in the solution-phase molecular world. Protonation state of surface-bound species is an ostensi-bly simple parameter that can have a profound effect on NP properties (Section 2), yet the nanoscale environment provides unique opportunities for controlling NP surface charge.[14a]For example, exploiting the effect of local surface curvature on pKa,[14b]distinct charge ‘patches’ can be created on nonspheri-cal NPs bearing an otherwise compositionally uniform mono-layer.[59] In the complex environment of a NP-bound monolay-er, several inter-related factors can modify molecular behav-ior,[60] and therefore have the potential to influence system-level properties. Analytical methods that can better probe mo-lecular states with nanoscale resolution will be essential in fully understanding these effects and exploiting the potential that they offer for NP property control. Mixed monolayers introduce further important challenges, including improved control and analysis of ligand composition and distribution (which can vary from intimately mixed to fully phase-separated). From this will come truly multifunctional NP systems where two or more sur-face-bound ligands each play an active role in defining stimuli-responsive system properties.
Advances in supramolecular design have allowed NP-bound nonbiomolecular host–guest systems to begin to rival oligonu-cleotide hybridization, to the extent that they can control re-markable stimuli-responsive NP systems in living cells (Sec-tion 3.1). Developing analytical methods capable of directly probing supramolecular structure and dynamics within the monolayer, of distinguishing different noncovalent binding modes, and even different nano-locations will be key to driving future advances here. Having independently enjoyed similarly rapid development trajectories, recent examples suggest that the interface between artificial molecular machines and func-tional NPs is now ripe for exploiting (Section 3.2). Understand-ing the interdependent relationships between NP features, ma-chine performance and system properties presents formidable challenges, but there are undoubtedly great rewards to be had by combining the remarkable achievements of these two fields of modern synthetic chemical technology.
While kinetically controlled covalent chemistry has been a mainstay of postsynthetic monolayer functionalization,[2, 48] the recent introduction of stimuli-responsive and dynamic co-valent systems that combine the stability and diversity of cova-lent structures, with the tunable and adaptable behavior of equilibrium systems, promises a step-change in capabilities (Section 4). These methods will afford unprecedented control over monolayer composition, in response to either physical or chemical inputs, using only well-defined covalent structures and without disrupting the NP-ligand interaction. Nondestruc-tive methods for monitoring reaction processes in situ will be crucial to developing predictable methodology here; just as molecular synthesis is underpinned by detailed mechanistic understanding, so too must synthetic methods on the nano-scale.
NP—strategies that manipulate the monolayer in place are more readily generalizable to a wide range of nanomaterials than those that attempt to adapt NP synthetic routes, or to re-place the surface-bound species in their entirety. The ultimate goal to which synthetic chemists must aspire is a set of univer-sal strategies, and a predictable structure–property under-standing, applicable irrespective of NP material, size, or shape. When considered in totality, the remarkable and unique prop-erties possible for the core nanomaterial, combined with the vast diversity of modern-day molecular and supramolecular chemistry, and the multitude of parameters that can be called upon to manipulate surface-bound molecular behavior (vide supra), suggest we are only just beginning to scratch the sur-face in terms of what can be achieved by postsynthetic manip-ulation of NP-bound monolayers. The exciting possibilities for controlling NP properties and designing smart NP devices will play an important role in fully realizing the technological po-tential of monolayer-stabilized nanomaterials.
Acknowledgements
We thank the EPSRC (EP/K016342/1) and the Leverhulme Trust (RPG-2015-042) for funding support. E.R.K. is a Royal Society of Edinburgh/Scottish Government Personal Research Fellow.
Keywords: molecular devices · monolayers · nanoparticles ·
property control·supramolecular chemistry
[1] For leading reviews on the synthesis of monolayer-stabilized NPs, see: a) J. Park, J. Joo, S. G. Kwon, Y. Jang, T. Hyeon,Angew. Chem. Int. Ed.
2007,46, 4630 – 4660;Angew. Chem.2007,119, 4714 – 4745; b) Y. Xia, Y. Xiong, B. Lim, S. E. Skrabalak,Angew. Chem. Int. Ed.2009,48, 60 – 103;
Angew. Chem.2009,121, 62 – 108; c) N. T. K. Thanh, N. Maclean, S. Ma-hiddine,Chem. Rev.2014,114, 7610 – 7630.
[2] For an introduction to the role of the monolayer on colloidally stable inorganic nanoparticles, and traditional routes to monolayer modifica-tion via ligand exchange, encapsulamodifica-tion, or bioconjugamodifica-tion, see: R. A. Sperling, W. J. Parak, Philos. Trans. R. Soc. London Ser. A 2010, 368, 1333 – 1383.
[3] J. Owen,Science2015,347, 615 – 616.
[4] H. Jans, Q. Huo,Chem. Soc. Rev.2012,41, 2849 – 2866.
[5] a) F. Caruso, T. Hyeon, V. M. Rotello,Chem. Soc. Rev.2012,41, 2537 – 2538; b) E.-K. Lim, T. Kim, S. Paik, S. Haam, Y.-M. Huh, K. Lee,Chem. Rev.
2015,115, 327 – 394.
[6] a) M. P. Monopoli, C. berg, A. Salvati, K. A. Dawson,Nat. Nanotechnol.
2012,7, 779 – 786; b) S. T. Kim, K. Saha, C. Kim, V. M. Rotello,Acc. Chem. Res.2013,46, 681 – 691.
[7] Subtle variations in stabilizing ligand structure (not directly involving the surface-coordinating group) can lead to significant changes in NP synthesis outcomes. For example, AuNP size distribution: a) R. Sardar, J. S. Shumaker-Parry,Chem. Mater.2009,21, 1167 – 1169; or morphology of branched CdSe nanocrystals: b) J. Huang, M. V. Kovalenko, D. V. Tala-pin,J. Am. Chem. Soc.2010,132, 15866 – 15868.
[8] a) M. J. Hostetler, S. J. Green, J. J. Stokes, R. W. Murray,J. Am. Chem. Soc.
1996,118, 4212 – 4213 ; b) R. S. Ingram, M. J. Hostetler, R. W. Murray,J. Am. Chem. Soc.1997,119, 9175 – 9178.
[9] A. Caragheorgheopol, V. Chechik, Phys. Chem. Chem. Phys.2008, 10, 5029 – 5041.
[10] a) J. S. Owen, J. Park, P.-E. Trudeau, A. P. Alivisatos,J. Am. Chem. Soc.
2008,130, 12279 – 12281; b) M. V. Kovalenko, M. Scheele, D. V. Talapin,
Science2009,324, 1417 – 1420 ; c) A. Dong, X. Ye, J. Chen, Y. J. Kang, T. Gordon, J. M. Kikkawa, C. B. Murray,J. Am. Chem. Soc.2011,133, 998 – 1006; d) X. Zhang, M. R. Servos, J. W. Liu,J. Am. Chem. Soc.2012,134,
7266 – 7269; e) E. L. Rosen, R. Buonsanti, A. Llordes, A. M. Sawvel, D. J. Milliron, B. A. Helms,Angew. Chem. Int. Ed.2012,51, 684 – 689;Angew. Chem.2012,124, 708 – 713; f) J. Hassinen, V. Liljestrçm, M. A. Kostiainen, R. H. A. Ras,Angew. Chem. Int. Ed.2015,54, 7990 – 7993 ;Angew. Chem.
2015,127, 8101 – 8104.
[11] K. P. Garca, K. Zarschler, L. Barbaro, J. A. Barreto, W. O’Malley, L. Spiccia, H. Stephan, B. Graham,Small2014,10, 2516 – 2529.
[12] T. Mizuhara, K. Saha, D. F. Moyano, C. S. Kim, B. Yan, Y.-K. Kim, V. M. Ro-tello,Angew. Chem. Int. Ed.2015,54, 6567 – 6570;Angew. Chem.2015,
127, 6667 – 6670.
[13] P. P. Pillai, S. Huda, B. Kowalczyk, B. A. Grzybowski, J. Am. Chem. Soc.
2013,135, 6392 – 6395.
[14] a) K. P. Browne, B. A. Grzybowski,Langmuir2011,27, 1246 – 1250; b) D. Wang, R. J. Nap, I. Lagzi, B. Kowalczyk, S. Han, B. A. Grzybowski, I. Szlei-fer,J. Am. Chem. Soc.2011,133, 2192 – 2197.
[15] T. L. Doane, C.-H. Chuang, R. J. Hill, C. Burda,Acc. Chem. Res.2012,45, 317 – 326.
[16] a) D. Astruc, M.-C. Daniel, J. Ruiz, Chem. Commun.2004, 2637 – 2649; b) R. Klajn, L. Fang, A. Coskun, M. A. Olson, P. J. Wesson, J. F. Stoddart, B. A. Grzybowski,J. Am. Chem. Soc.2009,131, 4233 – 4235.
[17] A. P. Alivisatos, K. P. Johnsson, X. Peng, T. E. Wilson, C. J. Loweth, M. P. Bruchez Jr., P. G. Schultz,Nature1996,382, 609 – 611.
[18] C. A. Mirkin, R. L. Letsinger, R. C. Mucic, J. J. Storhoff,Nature1996,382, 607 – 609.
[19] J. I. Cutler, E. Auyeung, C. A. Mirkin,J. Am. Chem. Soc.2012,134, 1376 – 1391.
[20] R. J. Macfarlane, M. N. O’Brien, S. H. Petrosko, C. A. Mirkin,Angew. Chem. Int. Ed.2013,52, 5688 – 5698;Angew. Chem.2013,125, 5798 – 5809. [21] B. Dubertret, P. Skourides, D. J. Norris, V. Noireaux, A. H. Brivanlou, A.
Libchaber,Science2002,298, 1759 – 1762.
[22] X. Y. Wu, H. J. Liu, J. Q. Liu, K. N. Haley, J. A. Treadway, J. P. Larson, N. F. Ge, F. Peale, M. P. Bruchez,Nat. Biotechnol.2002,21, 41 – 46.
[23] a) H. Fan, K. Yang, D. M. Boye, T. Sigmon, K. J. Malloy, H. Xu, G. P. Lpez, C. J. Brinker,Science2004,304, 567 – 571; b) T. Pellegrino, L. Manna, S. Kudera, T. Liedl, D. Koktysh, A. L. Rogach, S. Keller, J. Rdler, G. Natile, W. J. Parak,Nano Lett.2004,4, 703 – 707; c) Y. Kang, T. A. Taton,Angew. Chem. Int. Ed.2005,44, 409 – 412;Angew. Chem.2005,117, 413 – 416. [24] D. Jan´czewski, N. Tomczak, M.-Y. Han, G. J. Vancso,Nat. Protoc.2011,6,
1546 – 1553.
[25] For reviews focusing on specific host–guest systems applied to metal NP surfaces, see: a) U. Drechsler, B. Erdogan, V. M. Rotello,Chem. Eur. J.
2004,10, 5570 – 5579; b) V. Montes-Garca, J. Prez-Juste, I. Pastoriza-Santos, L. M. Liz-Marzn,Chem. Eur. J.2014,20, 10874 – 10883; c) Y.-W. Yang, Y.-L. Sun, N. Song,Acc. Chem. Res.2014,47, 1950 – 1960. [26] a) N. Lala, S. P. Lalbegi, S. D. Adyanthaya, M. Sastry,Langmuir2001,17,
3766 – 3768; b) Y. Wang, J. F. Wong, X. Teng, X. Z. Lin, H. Yang,Nano Lett.
2003,3, 1555 – 1559 ; c) D. Dorokhin, N. Tomczak, M. Han, D. N. Rein-houdt, A. H. Velders, G. J. Vancso,ACS Nano2009,3, 661 – 667. [27] R. Freeman, T. Finder, L. Bahshi, I. Willner,Nano Lett. 2009, 9, 2073 –
2076.
[28] C. Kim, S. S. Agasti, Z. Zhu, L. Isaacs, V. M. Rotello,Nat. Chem.2010,2, 962 – 966.
[29] G. Y. Tonga, Y. Jeong, B. Duncan, T. Mizuhara, R. Mout, R. Das, S. T. Kim, Y.-C. Yeh, B. Yan, S. Hou, V. M. Rotello,Nat. Chem.2015,7, 597 – 603. [30] a) F. Manea, F. B. Houillon, L. Pasquato, P. Scrimin,Angew. Chem. Int. Ed.
2004, 43, 6165 – 6169 ; Angew. Chem. 2004, 116, 6291 – 6295; b) G. Zaupa, C. Mora, R. Bonomi, L. J. Prins, P. Scrimin,Chem. Eur. J.2011,17, 4879 – 4889.
[31] a) G. Pieters, A. Cazzolaro, R. Bonomi, L. J. Prins,Chem. Commun.2012,
48, 1916 – 1918; b) G. Pieters, C. Pezzato, L. J. Prins, J. Am. Chem. Soc.
2012,134, 15289 – 15292; c) R. Bonomi, A. Cazzolaro, A. Sansone, P. Scri-min, L. J. Prins, Angew. Chem. Int. Ed. 2011, 50, 2307 – 2312 ; Angew. Chem.2011,123, 2355 – 2360; d) C. Pezzato, B. Lee, K. Severin, L. J. Prins,
Chem. Commun.2013,49, 469 – 471.
[32] C. Pezzato, P. Scrimin, L. J. Prins,Angew. Chem. Int. Ed.2014,53, 2104 – 2109;Angew. Chem.2014,126, 2136 – 2141.
[33] a) S. Maiti, C. Pezzato, S. G. Martin, L. J. Prins,J. Am. Chem. Soc.2014,
[34] M. A. Cohen Stuart, W. T. S. Huck, J. Genzer, M. M ller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov, S. Minko,Nat. Mater.2010,9, 101 – 113.
[35] a) E. W. Edwards, M. Chanana, D. Wang, H. Mçhwald,Angew. Chem. Int. Ed.2008,47, 320 – 323;Angew. Chem.2008,120, 326 – 329; b) A. Stocco, M. Chanana, G. Su, P. Cernoch, B. P. Binks, D. Wang,Angew. Chem. Int. Ed.2012,51, 9647 – 9651;Angew. Chem.2012,124, 9785 – 9789. [36] S. Carregal-Romero, N. J. Buurma, J. Prez-Juste, L. M. Liz-Marzn, P.
Hervs,Chem. Mater.2010,22, 3051 – 3059.
[37] S. Wu, J. Dzubiella, J. Kaiser, M. Drechsler, X. Guo, M. Ballauff, Y. Lu,
Angew. Chem. Int. Ed.2012,51, 2229 – 2233;Angew. Chem.2012,124, 2272 – 2276.
[38] R. A. lvarez-Puebla, R. Contreras-Cceres, I. Pastoriza-Santos, J. Prez-Juste, L. M. Liz-Marzn, Angew. Chem. Int. Ed. 2009, 48, 138 – 143;
Angew. Chem.2009,121, 144 – 149.
[39] H. Kearns, N. C. Shand, K. Faulds, D. Graham,Chem. Commun.2015,51, 8138 – 8141.
[40] a) V. Balzani, A. Credi, M. Venturi,Molecular Devices and Machines: Con-cepts and Perspectives for the Nanoworld, 2nd ed., Wiley-VCH, Weinheim, 2008; b) J. M. Abendroth, O. S. Bushuyev, P. S. Weiss, C. J. Barrett,ACS Nano2015,9, 7746 – 7768 ; c) E. R. Kay, D. A. Leigh,Angew. Chem. Int. Ed.
2015,54, 10080 – 10088;Angew. Chem.2013,125, 10080 – 10088. [41] E. R. Kay, J. Lee, D. G. Nocera, M. G. Bawendi, Angew. Chem. Int. Ed.
2013,52, 1165 – 1169;Angew. Chem.2013,125, 1203 – 1207.
[42] a) R. Klajn, J. F. Stoddart, B. A. Grzybowski, Chem. Soc. Rev.2010, 39, 2203 – 2237; b) A. C. Fahrenbach, S. C. Warren, J. T. Incorvati, A.-J. Aves-tro, J. C. Barnes, J. F. Stoddart, B. A. Grzybowski,Adv. Mater.2013,25, 331 – 348.
[43] L. Zhu, M.-Q. Zhu, J. K. Hurst, A. D. Q. Li,J. Am. Chem. Soc.2005,127, 8968 – 8970.
[44] B. Liao, J. Chen, H. Huang, X. Li, B. He,J. Mater. Chem.2011,21, 5867 – 5869.
[45] C. Zhang, C.-H. Xu, L.-D. Sun, C.-H. Yan,Chem. Asian J.2012,7, 2225 – 2229.
[46] A. Coskun, P. J. Wesson, R. Klajn, A. Trabolsi, L. Fang, M. A. Olson, S. K. Dey, B. A. Grzybowski, J. F. Stoddart,J. Am. Chem. Soc.2010,132, 4310 – 4320.
[47] a) H. Noglik, W. J. Pietro,Chem. Mater.1994,6, 1593 – 1595 ; b) A. C. Tem-pleton, M. J. Hostetler, C. T. Kraft, R. W. Murray,J. Am. Chem. Soc.1998,
120, 1906 – 1911; c) A. C. Templeton, M. J. Hostetler, E. K. Warmoth, S. Chen, C. M. Hartshorn, V. M. Krishnamurthy, M. D. E. Forbes, R. W. Murray,J. Am. Chem. Soc.1998,120, 4845 – 4849 ; d) K. J. Watson, J. Zhu, S. T. Nguyen, C. A. Mirkin,J. Am. Chem. Soc.1999,121, 462 – 463. [48] K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H.
Stewart, I. L. Medintz,Chem. Rev.2013,113, 1904 – 2074.
[49] X. Li, J. Guo, J. Asong, M. A. Wolfert, G.-J. Boons,J. Am. Chem. Soc.2011,
133, 11147 – 11153.
[50] I. Kosif, M. Cui, T. P. Russell, T. Emrick,Angew. Chem. Int. Ed.2013,52, 6620 – 6623;Angew. Chem.2013,125, 6752 – 6755.
[51] a) G. Han, T. Mokari, C. Ajo-Franklin, B. E. Cohen,J. Am. Chem. Soc.2008,
130, 15811 – 15813; b) S. Impellizzeri, B. McCaughan, J. F. Callan, F. M. Raymo,J. Am. Chem. Soc.2012,134, 2276 – 2283.
[52] a) L. F. Shi, V. De Paoli, N. Rosenzweig, Z. Rosenzweig,J. Am. Chem. Soc.
2006,128, 10378 – 10379 ; b) R. Gill, R. Freeman, J.-P. Xu, I. Willner, S. Wi-nograd, I. Shweky, U. Banin,J. Am. Chem. Soc.2006,128, 15376 – 15377; c) M. Suzuki, Y. Husimi, H. Komatsu, K. Suzuki, K. T. Douglas, J. Am. Chem. Soc.2008,130, 5720 – 5725.
[53] R. Freeman, R. Gill, I. Shweky, M. Kotler, U. Banin, I. Willner, Angew. Chem. Int. Ed.2009,48, 309 – 313;Angew. Chem. 2009,121, 315 – 319. [54] a) S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders, J. F. Stod-dart,Angew. Chem. Int. Ed.2002,41, 898 – 952;Angew. Chem.2002,114, 938 – 993; b) P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders, S. Otto,Chem. Rev.2006,106, 3652 – 3711.
[55] a) N. Nagahori, M. Abe, S.-I. Nishimura,Biochemistry2009,48, 583 – 594; b) M. B. Thygesen, K. K. Sørensen, E. Cl, K. J. Jensen, Chem. Commun.
2009, 6367 – 6369; c) J. B. Blanco-Canosa, I. L. Medintz, D. Farrell, H. Mat-toussi, P. E. Dawson, J. Am. Chem. Soc.2010,132, 10027 – 10033; d) G. Iyer, F. Pinaud, J. Xu, Y. Ebenstein, J. Li, J. Chang, M. Dahan, S. Weiss, Bio-conjugate Chem.2011,22, 1006 – 1011; e) T. L. Jennings, S. G. Becker-Cat-ania, R. C. Triulzi, G. Tao, B. Scott, K. E. Sapsford, S. Spindel, E. Oh, V. Jain, J. B. Delehanty, D. E. Prasuhn, K. Boeneman, W. R. Algar, I. L. Medintz,
ACS Nano2011,5, 5579 – 5593.
[56] F. Wang, Y.-C. Wang, S. Dou, M.-H. Xiong, T.-M. Sun, J. Wang,ACS Nano
2011,5, 3679 – 3692.
[57] P. Nowak, V. Saggiomo, F. Salehian, M. Colomb-Delsuc, Y. Han, S. Otto,
Angew. Chem. Int. Ed.2015,54, 4192 – 4197;Angew. Chem.2015,127, 4266 – 4271.
[58] F. della Sala, E. R. Kay, Angew. Chem. Int. Ed. 2015, 54, 4187 – 4191;
Angew. Chem.2015,127, 4261 – 4265.
[59] D. A. Walker, E. K. Leitsch, R. J. Nap, I. Szleifer, B. A. Grzybowski, Nat. Nanotechnol.2013,8, 676 – 681.
[60] a) T. Zdobinsky, P. Sankar Maiti, R. Klajn,J. Am. Chem. Soc.2014, 136, 2711 – 2714 ; b) C. Raimondo, B. Kenens, F. Reinders, M. Mayor, H. Uji-i, P. Samor,Nanoscale2015,7, 13836 – 13839.
Manuscript received : September 14, 2015
REVIEW
Stimuli-Responsive Nanoparticles
William Edwards, Euan R. Kay*
&&–&&
Manipulating the Monolayer: Responsive and Reversible Control of Colloidal Inorganic Nanoparticle Properties
There and back again: Many properties
![Figure 2. Manipulating NP surface charge by proton transfer. a) Homogene-ous monolayers of acylsulfonamide 1 allow switching between neutral (zwit-terionic) and cationic surface charges at mildly acidic pH values.[12] b) Mixedmonolayers of permanently cationic (2) and switchable carboxylic acid (3) li-gands allow switching between positive, neutral and negative surface charg-es, accompanied by reversible modulation of colloidal stability.[13]](https://thumb-us.123doks.com/thumbv2/123dok_us/8928994.391844/3.595.307.547.69.254/manipulating-acylsulfonamide-mixedmonolayers-permanently-switchable-accompanied-reversible-modulation.webp)
![Figure 3. Competitive host–guest complexation between ADA and CB[7] forselective NP property switching](https://thumb-us.123doks.com/thumbv2/123dok_us/8928994.391844/4.595.307.545.507.700/figure-competitive-host-guest-complexation-forselective-property-switching.webp)
![Figure 6. Nanoparticle property switching using thermosensitive polymerligand shells. a) Switching substrate selectivity for a catalytically active ‘yolk–shell’ AuNP coated with poly(N-isopropyl acrylamide).[37] b) Temperature con-trolled swelling of a poly(N-isopropyl acrylamide) ligand shell simultaneouslyalters surface-enhanced Raman scattering (SERS) and surface enhanced fluo-rescence (SEF) effects for an intercalated dye.[38]](https://thumb-us.123doks.com/thumbv2/123dok_us/8928994.391844/7.595.47.289.62.422/nanoparticle-thermosensitive-polymerligand-selectivity-catalytically-temperature-simultaneouslyalters-intercalated.webp)
![Figure 9. DNA-templated formation of dynamic covalent imine AuNP mono-layers. Monolayer composition depends on the specific base pair sequenceof the DNA template introduced; negligible imine formation is observed inthe absence of DNA.[57]](https://thumb-us.123doks.com/thumbv2/123dok_us/8928994.391844/9.595.317.539.431.680/templated-formation-monolayer-composition-sequenceof-introduced-negligible-formation.webp)
