LEABHARLANN CHOLAISTE NA TRIONOIDE, BAILE ATHA CLIATH
TRINITY COLLEGE LIBRARY DUBLIN
OUscoil Atha Cliath
The University of Dublin
Terms and Conditions of Use of Digitised Theses from Trinity College Library Dublin
Copyright statement
All material supplied by Trinity College Library is protected by copyright (under the Copyright and
Related Rights Act, 2000 as amended) and other relevant Intellectual Property Rights. By accessing
and using a Digitised Thesis from Trinity College Library you acknowledge that all Intellectual Property
Rights in any Works supplied are the sole and exclusive property of the copyright and/or other I PR
holder. Specific copyright holders may not be explicitly identified. Use of materials from other sources
within a thesis should not be construed as a claim over them.
A non-exclusive, non-transferable licence is hereby granted to those using or reproducing, in whole or in
part, the material for valid purposes, providing the copyright owners are acknowledged using the normal
conventions. Where specific permission to use material is required, this is identified and such
permission must be sought from the copyright holder or agency cited.
Liability statement
By using a Digitised Thesis, I accept that Trinity College Dublin bears no legal responsibility for the
accuracy, legality or comprehensiveness of materials contained within the thesis, and that Trinity
College Dublin accepts no liability for indirect, consequential, or incidental, damages or losses arising
from use of the thesis for whatever reason. Information located in a thesis may be subject to specific
use constraints, details of which may not be explicitly described. It is the responsibility of potential and
actual users to be aware of such constraints and to abide by them. By making use of material from a
digitised thesis, you accept these copyright and disclaimer provisions. Where it is brought to the
attention of Trinity College Library that there may be a breach of copyright or other restraint, it is the
policy to withdraw or take down access to a thesis while the issue is being resolved.
Access Agreement
By using a Digitised Thesis from Trinity College Library you are bound by the following Terms &
Conditions. Please read them carefully.
E ffect o f g en e stru ctu re changes on th e rate of
p ro tein seq u en ce e v o lu tio n
by
Brian Cusack
B.Sc. M.Res.
A Thesis su b m itted to
The University of Dubhri
for the degree of
D octor of Philosophy
D epartm ent of Genetics
T rinity College
University of Dublin
^ T R IN IT Y C O L L E G E ^
0 5 J U L 20Q 7_
^
LIBRARY DUBLIN ^
D ecla ra tio n
This thesis has not been subm itted as an exercise for a degree a t any other University.
Except where otherwise stated, the work described herein has been carried out by the
author alone. This thesis may be borrowed or copied upon request w ith the permission of
the Librarian, University of Dubhn, Trinity College. The copyright belongs jointly to the
University of Dublin and B rian Cusack.
Signature of A uthor
A ck now ledgem en ts
Ken - thanks for your patient supervision and encouragement through the well-judged
application of b o th stick and carrot.
T hanks to all current and past members of the Wolfe Lab for providing a great working
environm ent. T hanks to Devin, Gavin, Jeff, Jonathan, Kevin, M arie, M att, Meg, N adia
and Nora for their good hum our and willingness to help. T hanks to Gavin for help w ith his
like-tri-test software, to Marie for help w ith
and to Meg for knocking my gram m ar
into shape. Many thanks to Andrew for inspiring the work in C hapter 2.
C o n ten ts
1
I n t r o d u c t io n
21
1.1
Preface ...
21
1.2
Causes of variation in the ra te of protein sequence
evolution
...
21
1.2.1
Early approaches to explaining protein rate v a r ia tio n ...
23
1.2.2
Codon-based models of protein evolution ...
25
1.2.3
The im pact of fm ictional and comi)arative g e n o m ic s ...
27
1.2.4
Pitfalls in interpreting genomic c o r r e la tio n s ...
27
1.2.5
The controversy surrounding g e n e -d is p e n s a b ility ...
28
1.2.6
Quantifying pleiotroj)y in yeast: protein interaction d a t a ...
29
1.2.7
Evolutionary ra te and protein structure: the “designability”
of proteins
...
31
1.2.8
Most variation in ra te of yeast j^rotein evolution is explained by a
single d e t e r m in a n t ...
32
1.2.9
Translational R obustness ...
33
1.2.10 Fitness density versus functional d e n s ity ...
34
1.2.11 D eterm inants of evolutionary rate of m am m alian p r o t e i n s ...
35
1.2.12 Heterogeneity of the m annnalian genome ...
35
1.2.13 The transition to tissue d iffe re n tia tio n ...
37
1.2.14 Im pact of bread th of expression on protein evolution in m am m als . .
37
1.2.15 Expression breadth versus tissu e -sp e c ific ity ...
39
1.2.16 Tissue-specificity and protein se c re tio n ...
40
CONTENTS
1.3 Im pact of gene duplication on rates of molecular
evolution ...
44
1.3.1 The broad spectrum of gene duplications ...
44
1.3.2 B irth and death of duplicate g e n e s ...
45
1.3.3 Mechanisms for duplicate gene p re s e rv a tio n ...
46
1.3.4 Gene duplicate preservation and its inijmct on evolutionary rate . .
48
1.4 Im pact of alternative splicing on rates of molccular
evolution
...
52
1.4.1 A lternative splicing is associated with gene stru ctu re changes . . . .
53
1.4.2 Differing selective pressures associated with alternative splicing . . .
55
1.4.3 Heterogeneity in intragenic seciueuce evolution due to alternative
s p l i c i n g ...
56
1.4.4 Com plem entarity of alternative si)liciug and gene duplication . . . .
57
2 N o t b o r n e q u a l:
I n c r e a s e d r a te a s y m m e t r y in r e lo c a te d a n d r e t r o tr a n s p o s e d
r o d e n t g e n e d u p lic a t e s
59
2.1 A b s t r a c t ...
59
2.2 In tro d u c tio n ...
60
2.3 M e th o d s ...
62
2.3.1 Recent rodent duplicates ...
62
2.3.2 Gene duplication c a te g o r ie s ...
62
2.3.3 Direction of (retro)transposition of distant duplicates ...
63
2.3.4 Measures of sequence e v o lu tio n ...
64
2.3.5 Prevalence of significantly asymmetric sequence d i v e r g e n c e ...
65
2.3.6 Gene expression inform ation ...
66
2.4 R e s u lts ...
67
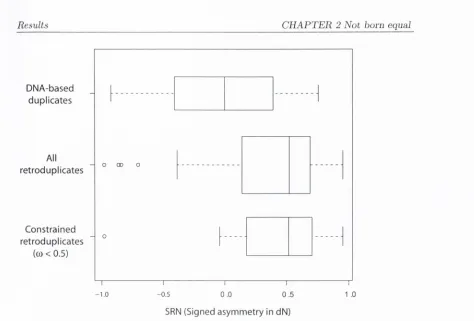
2.4.1
Asym m etry in
is greater among relocated duplicates and
duplicates created by retrotransposition...
67
2.4.2 Separating relocation from retrotransj^osition...
69
2.4.3 Directional sequence asymmetry: retrogenes accelerate relative to
their paralogs...
70
C O N T E N T S
2.6
A ck n o w led g em en ts...
76
3
C h a n g e s in a lt e r n a t iv e s p lic in g o f h u m a n a n d m o u s e g e n e s are
a c c o m p a n ie d b y fa s te r e v o lu t io n o f c o n s t it u t iv e e x o n s
7 7
3.1
A b s t r a c t ...
77
3.2
In tro d u c tio n ...
78
3.3
M e th o d s ...
80
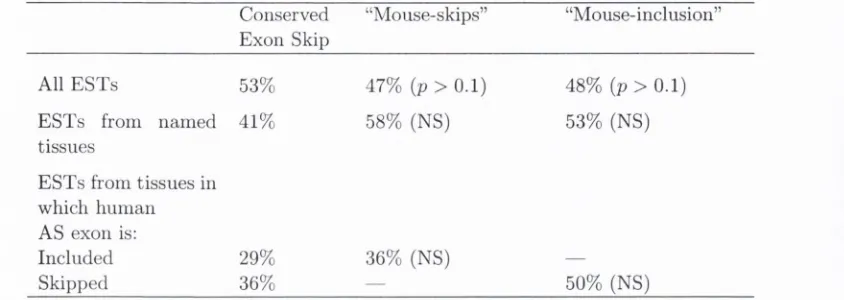
3.3.1 Hum an-mouse exon-skip c o n s e r v a ti o n ...
80
3.3.2 O rthology m a p p in g ...
80
3.3.3 Identification of “representative orthologs” in f i s h ...
81
3.3.4 Assessing levels of selective c o n s t r a i n t ...
82
3.3.5 D eterm ining alternatively spliced exon presence/absence in the
hum an-m ouse a n c e s to r ...
82
3.3.6 Influence of frequency of incorporation of alternatively spliced
se q u e n c e ...
83
3.3.7 Level and b re ad th of constitutive exon ex p re ssio n ...
83
3.3.8 E stim ating adecjuacy of mouse EST sam pling in genes with
putatively human-specific alternative s p l i c i n g ...
84
3.4
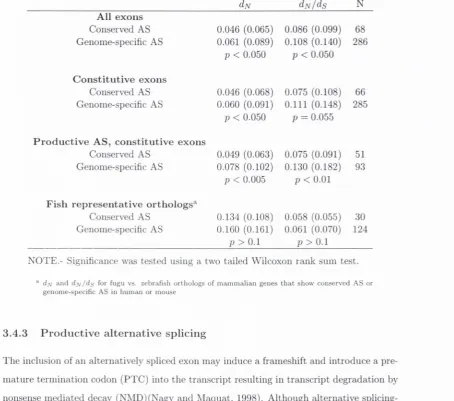
R e s u lts ...
84
3.4.1
Genes showing exon-skii)ping are more conserved th a n the genome
a v e r a g e ...
84
3.4.2 Genome-specific alternative splicing is associated w ith faster
protein evolution and weaker selective constraint in constitutive
re g io n s...
85
3.4.3 P roductive alternative s p l i c i n g ...
87
3.4.4 Differences in strength of selective constraint in m am m als are not a
reflection of inherent constraint differences...
89
3.4.5 Genes th a t have changed in alternative splicing p a tte rn have also
undergone changes in
dj\j/ds r a t i o ...
89
C O N T E N T S
3.4.8
Influence of frequency of incorporation of alternatively spliced
exons ...
94
3.4.9
Species-specific alternative splicing in genes w ith conserved
exon-intron s tr u c tu r e ...
95
3.5 D isc u ssio n ...
97
3.6 A c k n o w led g em en ts... 100
4 W h e n g e n e m a rria g es d o n ’t work out: d ivorce by s u b fu n c tio n a lisa tio n 101
4.1 A b s t r a c t ... 101
4.2 In tro d u c tio n ... 101
4.3 R esults and D isc u ssio n ... 102
4.4 A ck n o w led g em en ts... 107
4.5 Sources of nucleotide sequence d a t a ... 108
5 C o n clu sio n s
110
List o f Figures
1-1
R ates of amino acid substitu tio n in fibrinopeptides, haemoglobin, and
cytochrom e c...
22
2-1
D eterm ining the direction of transj)osition for distantly separated duplicates. 64
2-2
Signed nonsynonymous sequence asym m etry among d istan t duplicates . . .
71
3-1
Categories of alternative sjiliciug conservation retrieved from the ASAP
d a ta b a se ...
81
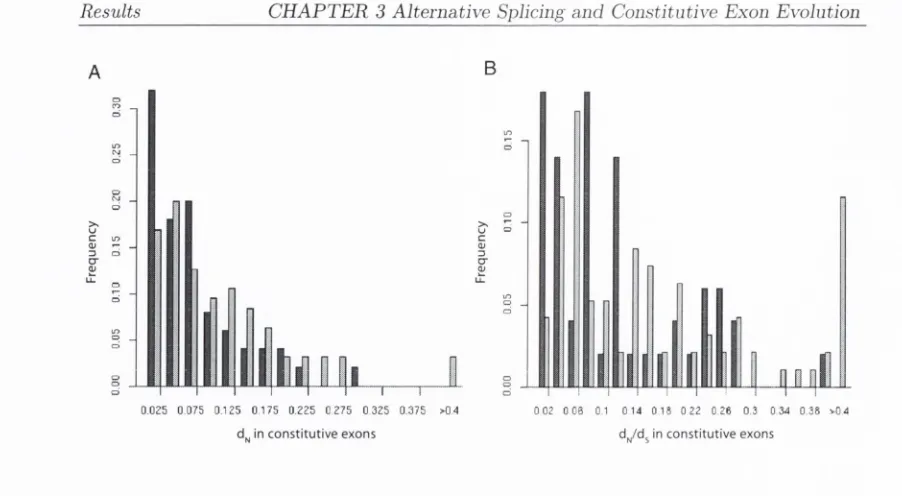
3-2
D istributions
ot djv and
d ^ / d s for constitutive exons...
88
3-3
Incorporation frequency of Inunan genome-specific alternative exons and
d ^
in constitutive exons...
95
4-1
O rganisation of SODcp, R P L32 and chimeric genes...
103
4-2
Amino acid sequence alignm ents of SODcp, RPL32
and chimeric genes. . . 106
List o f T ables
2.1 M agnitude of relative sequence asynnnetry in rodent duplicates categorised
by location and mechanism of duplication...
68
2.2 Prevalence of statistically significant sequence asym m etry in rodent
duplicates categorised by location and mechanism of duplication...
73
3.1 Evolutionary rates of alternatively spliced and non-alternatively spliced
hum an/m ouse orthologs...
85
3.2 Evolutionary rates of hum an/m ouse orthologs with conserved or
genome-specific alternative splicing...
87
3.3 D etection of chicken honiologs of hum an alternatively spliced exons...
92
3.4 Mouse EST coverage for genes with putatively human-specific alternative
splicing...
97
A b b r e v ia tio n s
BLAST
Basic Local Alignment Search Tool
bp
base pairs
CAI
Codon A daptation Index
cDNA
com plem entary DNA
D PE
D ownstream P rom oter Elem ent
E-value
E xpectation value
ESE
Exon Splicing Enhancer
ESS
Exon Splicing Silencer
EST
Expressed Sequence Tag
kb
kilobase
Mb
megabase
Mya
Million years ago
Myr
Million years
NMD
N onsense-m ediated decay
ORE
O pen R eading Frame
P T C
P rem atu re Term ination Codon
rRNA
ribosom al RNA
“T he race is not always to the swift, uor the b attle to the strong...
b u t tim e and chance happen to them all.”
Sum m ary
T he elaborate architecture of the genes of niulticellular eukaryotes is likely to underpin the
unique complexity of eukaryotic gene functions. The structure of eukaryotic genes differs
from th a t of prokaryotes and represents an assemblage of coding exons, iutrons th a t are
spliced out of precursor mRNAs, extended UTRs and complex regulatory regions. It is
likely th a t these features provided a platform for the evolution of the complex tra its th a t
typify m etazoans including alternative splicing and complex gene regulation.
Here I performed genome-wide studies of the association between the ra te of protein se
quence evolution and the modification of gene structures th a t can result from the processes
of gene duplication and alternative si)liciug. By considering recent gene duplicates in ro
dents I investigated genomic relocation following duplication and gene stru ctu re alteration
by retrotransposition as possible determ inants of evolutionary ra te differences between du
plicates. I found evidence th a t retrotranspositioii frequently results in asym m etric evolution
of gene duplicates and th a t functional retrogenes consistently accelerate relative to their
paralogs. A lthough the act of relocating a gene duplicate by transposition explains p a rt of
this effect my results show th a t the mechanism of retrotransposition makes an independent
contribution to this acceleration. This is likely to reflect the fact th a t duplicates created
by retrotransposition violate the assum ption connnon to most theoretical models th a t gene
duplicates are born equal. My results further suggest th a t the rate acceleration of functional
retrogenes is likely to be m ediated by changes in their expression.
these gains have resuhed in an acceleration in the rate of sequence evolution of constant
regions of th e encoded protein. Moreover, this effect is shown to strongly correlate w ith
the frequency of incorporation of these new exons. I argue th a t this correlation reflects a
causative relationship between these variables and dem onstrates the im pact on constitutive
parts of proteins of the acquisition of functional alternative s])lice forms.
Finally I present evidence from a single gene study supporting the intuition th a t al
ternative splicing and gene duplication can be jiarailel and complem entary routes to the
generation of functional diversity. I describe a gene fusion event th a t created a bifunctional
gene coding for two proteins by alternative splicing. This chimeric gene persists in the m an
grove genome b u t has duplicated in poplar and undergone subfunctionalisation to re-form
its constituent genes through the com plementary degeneration of its exons. T his example
is a clear illustration of the partitioning of alternative splice forms by subfunctionalisation
at the level of gene structure. I also discuss evidence th a t accelerated protein sequence
evolution occurred sim ultaneously w ith the gene structure changes corresponding to the
initial gene fusion and the subsequent gene fission following duplication.
C h ap ter 1
In tro d u ctio n
1.1
P reface
In the first p a rt of this introduction I describe the state of the field in the stu d y of protein
sequence evolution and the ongoing quest for the determ inants of the evolutionary rate
of proteins. In the second p a rt I address the im pact on the ra te of protein evolution of
the processes of gene duplication and alternative splicing. This section also outlines the
research chapters th a t investigate the im pact on evolutionary ra te of the changes in gene
stru ctu re th a t are frequently associated w ith both of these phenomena.
1.2
C au ses o f variation in th e rate o f p ro tein seq u en ce
e v o lu tio n
Causes o f variation in the rate o f protein sequence
evolution
Introduction
2 2
0-abcd«
?
180-160
Evolution of
ttie
g l o b i n s
140-2
120
-
100-S ep o ral io n of
o n c e s t o fs of
p l a n t s an d
a n im o ls
20-* T--20-*--- — r — — *--- --- T --- --- ---
1---200 300 400
500 500
TOO
800
900
1000
MOO 1200
1500 1400
Millions
of y e a r s since diver gen ce
cli
F i g u r e 1 -1 : R a t e s o f a m i n o a c i d s u b s t i t u t i o n in. f i b r i n o p e p t i d e s , h a e m o g l o b i n , a n d c y t o c h r o m e c.
C o m p a r i s o n s f o r w h i c h n o adeq^l,ate t i m e c o o r d i n a t e i s a v a i l a b l e a r e i n d i c a t e d b y n u m b e r e d c r o s s e s .
P o i n t 1 r e p r e s e n t s a d a t e o f 1 , 2 0 0 ±
7 5
M y r f o r th e s e p a r a t i o n o f p l a n t s a n d a n i m a l s , b a s e d o n a
l i n e a r e x t r a p o l a t i o n o f t h e c y t o c h r o m e c c u r v e . P o i n t s 2 - 1 0 r e f e r to e v e n t s i n t h e e v o l u t i o n o f t h e
g l o b i n f a m i l y . T h e 6 / ( 3 s e p a r a t i o n i s a t p o i n t 3, 'y/(3 is a t
4
. a n d o / f ) i s a t 5 0 0 M y r ( c a r p / l a m p r e y ) .
R e p r o d u c e d f r o m D i c k e r s o n ( 1 9 7 1 ) .
[image:22.523.7.477.65.484.2]Causes o f variation in the rate o f protein sequence
evolution
Introduction
(where m utations are deleterious) and sites at which changes are neutral (w ith no effect on
fitness) (Dickerson, 1971). Under the neutral theory (Kimura, 1983) the substitution rate
per site (fc) simply equals the neutral m utation rate per site (i^o)- Furtherm ore, if a certain
fraction (/o) of m utations are neutral or nearly neutral and the rest are deleterious, then
k — V o = V T f o
(
1
-1
)
where v t is the total ra te of m utation. Under this model /o is a m easurem ent of selective
constraint on a sequence. G reater values of /o indicate th a t m utations a t m ost sites are not
selected against and are fixed a t a faster rate. This predicts th a t less im p o rtan t proteins
should evolve at faster rates (have greater values of
k) because /o should be greater for
less im portant proteins. This model explains the observation th a t pseudogenes, which are
assum ed to have no function, show the highest rates of nucleotide su b stitu tio n because they
are free of selective constraint (/o = 1) (G raur and Li, 2000).
Therefore, proteins th a t are fmictionally less im portant are assum ed to evolve a t faster
rates reflecting the low level of selective constraint operating on them . It would appear
reasonable to tu rn this statem ent around and use observed rates of sequence substitution
to infer the intensity of selective constraint operating on a gene and therefore infer its func
tional im portance. Despite the circularity of this logic (G raur and Li, 2000) the application
of this principle has become connnon practice in molecular biology where sequence conser
vation is routinely used as a m easure of functional im portance. It has been suggested, for
example, th a t the fast evolution of proteins such as fibrinopeptides may be due to the ‘ac
ceptability’ of virtually any amino acid change in the protein sequence (K im ura and O hta,
1974).
1.2.1
E arly approaches to ex p la in in g p ro tein rate variation
Causes o f variation in the rate o f protein sequence
evolution
Introduction
F = Us/N
(
1.2
)
where N
is the to tal num ber of sites in the protein.
Intuitively this quantity should reflect the ratio of constrained to neutral amino acids
for a given protein which should be directly proportional to its rate of sequence evolution.
More recent work has led to an extension of this concept and the proposal of the term
“fitness density” (see section 1.2.10, page 34).
In a pioneering study Dickerson (1971) suggested th a t the surface residues of a protein
should be constrained by the p ro tein ’s interactions with its partners. There are potentially
many surface residues th a t could engage in such interactions relative to the handful of sites
concerned w ith an enzym e’s catalytic activity. Therefore these “contact functions” were
proposed to make a relatively large contribution to the functional density of a protein. This
assum ption finds a contem porary echo in the proposal th a t proteins with high connectivity
in protein-protein interaction (P P I) networks (i.e. high densities of contact functions)
should evolve slowly (see section 1.2.6, page 29).
Tests of the im pact of functional density on protein evolution are hindered by the absence
of direct m easurem ents of F
(such as those provided by saturation mutagenesis). For those
proteins for which functional density has been (ixperinientally determ ined there is a rough
negative correlation between
F
and the rate of protein evolution,
k (G raur and Li, 2000).
However, most work has attem p ted to explain variation in evohitionary rate using variables
th a t are assumed to be adequate surrogates of functional density, such as expression level,
pleiotropy, gene essentiality and gene dispensability.
One of the im plications of K im ura’s neutral theory of evolution is the prediction th a t
im portant genes (those m aking the largest contributions to organismal fitness) should be
subject to the strongest purifying selection. Wilson et al. (1977) therefore proposed th a t
in addition to “functional density” the other m ajor determ inant of protein evolution is
“dispensability” as form ulated in the expression
where
P
is the probability th a t a substitution is compatible with the function of the
protein and
Q is the probability th a t the organism can survive and reproduce w ithout the
Causes o f variation in the rate of protein sequence
evolution
Introduction
protein, reflecting protein dispensability. In other words, P is a m easure of the change in
function of the m utant protein relative to the w ild-type and
Q
scales this functional im pact
by the overall im portance of the protein (i.e., its dispensability).
Therefore, predicting the effect of selection on the protein as a whole requires knowledge
not only of th e fraction of sites engaged in protein function b u t also of th e im pact of
deleterious m utations of those sites on organism al survival. In m odern biology (at least for
unicellular organisms) a gene’s dispensability is quantified using the reduction of growth
ra te relative to th e w ild-type to approxim ate the fitness effect associated w ith deletion of the
gene. An alternative discrete classification distinguishes between essential and non-essential
genes depending on w hether deletion of the gene is lethal or not.
1 .2 .2
C o d o n -b a s e d m o d e ls o f p r o te in e v o lu tio n
Genome projects have allowed the evolution of proteins to be studied from th e perspective
of the nucleotide sequences th a t encode them . Codon-based analyses of protein-coding
sequences tre a t th e codon as the unit of evolution and distinguish between synonymous
and nonsynonymous rates of evolution. Synonymous nm tations yield a different codon
w ithout changing the encoded amino-acid and therefore do not affect the protein sequence.
Nonsynonymous m utations, on the other hand, result in replacem ent of one aniino-acid with
another. This distinction enables the calculation of two substitu tio n rates:
d s,
the number
of synonymous su b stitutions per synonymous site and djv, the num ber of nonsynonymous
substitutions per nonsynonymous site (Goldm an and Yang, 1994; Muse and G aut, 1994).
By distinguishing between synonymous
(ds)
and nonsynonymous substitu tio n rates
{df^)
it is possible to draw inferences regarding the natu re of the selection operating on the
protein-coding sequence. In particular, the ratio of these rates
{ d ^ / d s )
is commonly used
to estim ate a; (the am ino acid selection pressure) corrected for tt (the background nucleotide
m utation rate). This follows from the fact th a t, because synonymous changes are silent at
the protein level, synonymous sites are typically regarded as neutrally evolving (ignoring
selection on codon usage). Therefore, the synonymous rate is dependent on the nucleotide
m utation rate, tt and not on amino acid selection pressure,
u).
Nonsynonymous sites, on
the o ther hand, evolve a t a rate determ ined by both these processes.
Causes o f variation in the rate of protein sequence
evolution
Introduction
has led to the common use of the ratio
dpj / ds to estim ate the nature and m agnitude of
different types of am ino acid selection pressure. Values ot
< 1 indicate the operation
of purifying selection in causing a reduction in the fixation ra te of amino acid changes th a t
are deleterious relative to th e silent synonymous rate. Positive selection for beneficial amino
acid changes is frequently inferred when
d ^ / d s >
1-Estim ates of these rates are commonly derived in a m axinnmi likelihood framework
th a t sta rts w ith an explicit model of codon substitution and searches for the com bination
of param eter values th a t best describes the observed data. This approach accounts for
unequal substitution rates for nucleotide transitions compared to transversions (the tra n
sition/transversion ra te ratio,
k) as well as differences in codon frequencies. T he model
param eters estim ated from the d ata include
k.
the time
t and the
d ^ / d s
ratio w. This
allows subsequent derivation of the rates
d ^
and
dg- The procedure sinm ltaneously cor
rects for the occurrence of multijjle substitutions at the same site and i)erforms a realistic
weighting of alternative pathw ays of change between codons (Yang and Bielawski, 2000).
C auses o f va ria tio n in the rate o f p rotein sequence
evolution
In tro d u ctio n
1 .2 .3
T h e im p a c t o f fu n c tio n a l an d c o m p a r a tiv e g e n o m ic s
T h e d ev elo p m en t o f h ig h -th ro u g h p u t fu n ctio n al genom ics m e th o d s in th e recen t p a s t has
en a b le d th e re -a p p ra isal of som e early p red ictio n s in m olecular evolution th a t w ere for
m u la te d largely from an ecd o tal exam jjles. T h is has h ad a p a rtic u la rly significant im p act
on stu d ies of th e d e te rm in a n ts of p ro te in evolution, ex p an d in g on th e ea rly w ork of Zuck-
erk an d l, D ickerson a n d W ilson. T h e benefits of th is w ealth of genom ic d a ta are however
p a rtly offset by th e hid d en cost of ex p e rim e n ta l noise. For exam ple, m e asu rem en ts o f gene
expression are p a rtic u la rly noisy reflecting th e com bined effects of m easu rem en t in accu racy
a n d biological v a ria b ility across g ro w th conditions an d stra in s (C oghlan a n d W olfe, 2000;
D ru m m o n d e t al., 2006).
F u rth e rm o re , th e new w ealth of genom ics d a ta is n o t tax o n o m ically well spread.
E ven am ong m odel organism s th e unicellular b u d d in g y east
Saccharom yces cerevisiae has
am assed th e g re a te st v ariety and C[uantity of d a ta . Accordingly, before a tte m p tin g to ex
p lain th e h etero g en eity of p ro tein ra te s in higher eukaryotes it is in stru c tiv e to consider th e
e x te n t to w hich p ro te in ra te v ariatio n can be explained using genom ic ap p ro ach es in yeast.
1 .2 .4
P itfa lls in in te r p r e tin g g e n o m ic c o r r e la tio n s
Causes o f variation in the rate of protein sequence
evolution
Introduction
expression-m ediated selection on nucleotide substitutions).
A further m ajor source of error is th a t an observed strong pairwise correlation may
be induced as a trivial consequence of the m utual dependency of each variable on a third,
confounding, variable. In th is context, the deluge of genomics d ata has brought with it
the paradoxical side-effect th a t large num bers of d a ta points can suggest highly significant
associations between variables th a t are only weakly correlated. In such a situation the
task becomes one of disentangling the primary, evolutionarily relevant associations from
secondary, induced, correlations (Koonin and Wolf, 2006).
A recent, far-reaching, suggestion is th a t approaches th a t try to remove the confounding
effect of expression (e.g., partial correlation analysis) tail to do so when measurements
of expression level are noise-prone (Drum m ond et al., 2006). The authors argued th at
techniques such as partial correlation analysis and nudtiple linear regression are inapplicable
to situations where the variables under study intercorrelate (are '‘collinear”) and are further
underm ined by m easurem ent noise. Sinmlations showed th a t highly significant bu t entirely
spurious partial correlations can be detected between unrelated variables when analysing
noisy d a ta and crucially this might underlie the significant j)artial correlation between
the ra te of protein evolution and dispensability (Hirsh and Fraser, 2001; Pal et al., 2003;
Wall et al., 2005) th a t rem ains after attem pting to control for noise-prone measurem ents of
expression level. An alternative approach advocated by Druunnond
et al.
is th a t of principal
com ponent regression (PC R) (Drummond et al. (2006); see section 1.2.8, page 32).
1 .2 .5
T h e c o n tr o v e r sy su r r o u n d in g g e n e -d is p e n s a b ility
O f all th e potential candidates th a t might determ ine the rate of protein evolution, essen
tiality and dispensability would seem to come closest to capturing the essence of a gene’s
‘im portance’. The im pact of gene essentiality on protein evolution should therefore be un
equivocal: we would expect genes th a t are essential to organism survival (or fertility) to
evolve slowly, reflecting the strong selective constraints on their function. However, the pit
falls described above beset th e proposed association between the rate of protein evolution
and any candidate explanatory variable. This is clearly illustrated by the controversy th a t
has centred on the value of dispensability in explaining evolutionary rate, w ith the debate
foundering on several sources of error.
C auses o f variation in the rate o f p rotein sequence
evolution
Intro d u ctio n
surprising conclusion th a t, in mammals, there is no association between the fitness effect
of a gene’s deletion and its evolutionary rate once positively selected genes were excluded
(H urst and Sm ith, 1999). Although subsequent studies did claim to establish a connection,
the association was found to be surprisingly weak (Ilirsh and Fraser, 2001; Jordan et al.,
2002). In fact, even this marginal effect was diluted in the light of evidence th a t expres
sion level is a m ajor predictor of evolutionary rate in yeast (Pal et al., 2001) and following
use of partial correlation analysis to remove expression’s confounding influence (Pal et al.,
2003). More recent studies (Wall et al., 2005; Zhang and He, 2005; D rum m ond et al.,
2006) have attem p ted not only to account for the confounding effect of expression level bu t
also to address the problem of experim ental noise th a t causes observed m easurem ents to
deviate from real values of the underlying biological variables. Two of these studies con
cluded th a t gene dispensability, although weak, is a significant and independent correlate
of evolutionary ra te once expression level is controlled for (Wall et aL, 2005; Zhang and
He, 2005). Moreover, it was suggested th a t the tru e association between dispensability and
rate of protein evolution could only be uncovered when measuring sequence divergence on
short evolutionary time scales which b etter api)roximate the instantaneous ra te of protein
evolution (thus illustrating the problem of phylogenetic scale (Herbeck and W'all, 2005)).
However, the issue remains unresolved since by modelling the im pact of noise on expression
d a ta one of these studies concluded th a t the apparent correlation between gene dispens
ability and evolutionary rate is spurious and results purely from noise in the m easurem ent
of expression level (Drum m ond et al., 2006).
1 .2 .6
Q u a n tify in g p le io tr o p y in y ea st: p r o te in in te r a c tio n d a ta
Pleiotropic m utations are those having multiple phenotypic effects. By extension pleiotropic
genes are inferred to be nm ltifunctional since their rrmtation m ay affect m ultiple phenotypic
traits.
Causes o f va ria tio n in the rate o f p rotein sequence
evolution
Introduction
For m ulti-functional genes pleiotropic m utations will incurr a fitness cost amplified by the
num ber of affected tra its leading to stronger selective constraint on these m utations. Sec
ondly, pleiotropy is thought to impede the process of adaptive evolution by reducing the
likelihood th a t a m utation is advantageous (Fisher, 1930).
An interesting theoretical study implicates jsleiotropy as a possible determ inant of evo
lutionary rate. This study suggests th a t when many characters are affected by a m utation
this leads to the predom inance of a single optimal gene secjuence. This leads to a reduction
of w ithin-population variation w ith a resultant lowering in substitution ra te (W axman and
Peck, 1998).
A lthough pleiotropy is an im portant biological jihenomenon an adequate measurem ent
has proven elusive. There are several variables th a t might serve as proxies of pleiotropy
and for which large-scale genomics d ata is available in yeast. Among these, the number
of interactions in which a protein participates may be particularly informative. Therefore,
proteins w ith many interaction partners (“hubs”) might be considered to be m ultifunctional
and are expected to show high levels of pleiotropy. However, the search for an independent
correlation between protein evolutionary ra te and the number of interaction partners has
become mired in technical problems sim ilar to those encountered in studies of the role of
protein dispensability (Fraser et al., 2003; Jordan et al., 2003).
Despite these difficulties an appealing distinction has recently been draw n between
protein-interaction hubs engaging in multiple, simultaneous interactions (intram odule
“p arty ” hubs) and those th a t interact with different partners at different times (interm odule
“d ate” hubs). It
w eis
suggested th a t date hubs (having low coexpression w ith their interac
tors) are more pleiotropic th an p arty hubs (exhibiting high coexi)ression w ith their inter
actors) because of their transient interactions with many, functionally sem i-autonomous,
modules (Fraser, 2005). However, the observation th a t party hubs are, in fact, more con
served th an date hubs is contrary to expectation given the proposed difference in their
pleiotropic level. Moreover, recent work has cast doubt on the meaningfulness of this dis
tinction in hub types (B atada et al., 2006).
Causes o f variation in the rate o f protein sequence
evolution
Introduction
in m ultiple sim ultaneous interactions a large proportion of their surface residues is expected
to be involved in interactions (i.e. the density of contact functions is high) w ith a resultant
increase in the strength of purifying selection (Drum mond et al., 2006; Rocha, 2006). D ate
hubs on the other hand may interact w ith their many partners through repeated interaction
a t the same site and are therefore likely to be less conserved, by definition.
A lternative approaches to quantifying pleiotropy have used the num ber of biological
processes annotated for a gene to approxim ate the num ber of phenotypic tra its it affects.
However, less th a n 1% of the variation in selective constraint (m easured by
d ^ l d s )
of
yeast genes seems to be explained by this variable (Salathe et al., 2006). A sim ilar result
was obtained using the effects on growth of yeast m utants in 21 different conditions to
quantify pleiotropy (Salathe et al., 2006). A parallel study found a similarly weak, although
significant, association between a p rotein’s evolutionary ra te (m easured by dyv) and the
num ber of biological processes in which it participates. However, no correlation was found
between protein conservation and other potential m easurem ents of pleiotropy (e.g. num ber
of annotated molecular functions and num ber of protein domains) (He and Zhang, 2006).
It seems, therefore, th a t even in well-studied model organisms such as yeast, an adequate
description of pleiotropy remains tantalisingly out of reach.
1 .2 .7
E v o lu tio n a r y r a te a n d p r o te in str u c tu r e : th e “d e s ig n a b ility ”
o f p r o te in s
According to the conventional view of protein activity the existence of a correctly folded
three-dim ensional stru ctu re is a prerequisite for protein function. However, protein struc
tures differ w ith respect to their “designability”, i.e., the num ber of j)0ssible sequences th a t
can fold into th a t stru ctu re (Li et al., 1996; Koehl and Levitt, 2002). Highly designable
structures are determ ined by a large “neighbourhood” of such sequences and this reflects
their robustness to random m utations. It is therefore reasonable to expect th a t highly
designable proteins evolve at faster rates th an less designable proteins.
Causes o f variation in the rate o f protein sequence
evolution
Introduction
in protein designability. T his positive correlation could be considered as being at odds with
Zuckerkandl’s supposition th a t the contact density of proteins should correlate negatively
with their evolutionary rate. T he apparent contradiction may be explicable by the fact th a t
Bloom
et a l’s
stu d y only considered intram olecular contacts in calculating contact density.
Therefore, the possibility of a negative correlation between the density of intermolecular
contacts and ra te of protein evolution is not rejected by this result.
It is possible th a t protein stru ctu ra l constraints will b etter explain variation in evolu
tionary rates among sites w ithin a given protein, than rate differences between proteins.
This is suggested by the fact th a t non-synonymous rates correlate with the solvent acces
sibility of residues, and are twice as fast on the sm face of globular proteins th an in buried
regions (G oldm an et al., 1998).
1.2.8
M ost variation in rate o f yeast p rotein ev o lu tion is exp lained by a
sin gle d eterm in a n t
Expression level is frequently observed to be one of the strongest jiredictors of protein evo
lutionary rate. Techniques such as partial correlation analysis or nniltiple linear regression
have been used in an a tte m p t to reveal the ])riniary association between protein rate and
the focal variable by su b tractin g the secondary effect of exi)rossioii. However, until recently,
most studies did not seek to explain w hat underlies the recurrent association between ex
pression level and the ra te of protein evolution.
C auses o f va ria tio n in the rate o f protein sequence
evolution
Intro d u ctio n
role for p ro te in d isp en sab ility in p ro tein evolution.
T h is final resu lt is in strik in g c o n tra st to a second stu d y th a t used different m ethodology
to ad d ress th e sam e problem of m easu rem en t inaccuracy on p a rtia l co rrelatio n analysis
(W all e t al., 2005). Using a s tru c tu ra l eq u atio n m odel W all
et al.
p ro p o sed th a t gene
d isp e n sab ility m akes a sm all b u t significant co n trib u tio n to th e ra te of p ro te in evolution.
T h e fact th a t roughly h alf of th e v ariab ility in p ro te in ra te rem ain s to b e explained
suggests th a t o th e r, unconsidered, causative variables m ay acco u n t for a significant degree
o f p ro te in ra te v ariatio n . T h is possibility is largely d iscounted by D ru m m o n d a n d coworkers
o n th e g ro u n d s th a t th e co rrelatio n s th ey describe are necessarily u n d e re stim a te s d ue to th e
in h eren t sto c h a stic ity of th e ev o lu tio n ary process, atte rm a tio n by m e a su rem en t noise and
th e possible n o n -lin earity of th e relatio n sh ip s betw een th e p red icto rs a n d ev o lu tio n ary rate.
However, given b e tte r su rro g ates of functional d ensity and disp en sab ility , th e se variables
m ight b e found to account for som e fractio n of th e resid u al p ro te in r a te v a ria tio n yet to be
ex plained (R ocha, 2006).
1 .2 .9
T r a n sla tio n a l R o b u s tn e s s
T h e existence for each p ro te in s tru c tu re of a n eighbourhood of co m p a tib le p ro te in sequences
w£is discussed above in th e co n tex t of "p ro tein d esig n ab ility ” a t th e g en o ty p ic level. P a rts of
th is n eig h b o u rh o o d are also explored a t th e jjhenotypic level as a consequence of erro rs in
th e tra n s la tio n of th e genotype into th e p h enotype. T h e rib o so m e’s e rro r ra te is e stim a te d to
cause th e m istra n sla tio n of 20% of p roteins and in m any cases th ese m istra n sla te d p ro tein s
m ay m isfold (D ru m m o n d et al., 2005). However, som e p ro te in sequences reside in th e
m iddle of th e “n eig h b o u rh o o d ” of sequences th a t can each co rrectly d e te rm in e th e p ro te in ’s
n ativ e s tru c tu re . As a resu lt, w hen these “tra n sla tio n a lly ro b u s t” p ro te in sequences are
m istra n sla te d , m isfolding is avoided.
T h e cellular b u rd e n im posed by th e toxicity an d ag gregation o f m isfolded p ro tein s p ro
vides selective p ressu re for tra n sla tio n a l robustness. In fact th e fitness cost o f a m isfolded
p ro tein is p re d icted to be p ro p o rtio n a l to its frequency of tra n sla tio n . T h erefo re th e tra n s
latio n al ro b u stn ess hyp o th esis p red icts th a t highly expressed p ro te in s evolve slowly because
th ey are u n d er in ten se purifying selection to preserve those relativ ely ra re sequences th a t
are ro b u st to m istra n sla tio n (D rum m ond et al., 2005).
expression-Causes o f variation in the rate o f protein sequence
evolution
Introduction
related variables are th e most im portant determ inants of evolutionary rate in yeast. The
underlying phenom enon captured by these variables is likely to be the frequency of tran s
lation of each gene. Therefore the production rate of yeast proteins appears to determ ine
their evolutionary rate.
The paradoxical im plication of this hypothesis is th a t "translationally robust” protein
molecules are encoded by “m utationally fragile” genes. Thus while a considerable frac
tion of highly conserved sites in the prim ary sequence can be m utated (e.g. in site di
rected mutagenesis) w ith no inactivating effect on protein function, these m utations will
be selected against to preserve translational robustness. This may explain the observa
tion th a t genetic studies of the slowly evolving and highlj' abundant jjlant enzyme Rubisco
(ribulose-l,5-bisphosphate carboxylase/oxygenase) have revealed very few inactivating mu
tations (Drum mond et al., 2005). Therefore the sequence conservation of Rubisco to a large
extent reflects translational robustness and not functional fragility.
1 .2 .1 0
F itn e s s d e n s ity v e r su s fu n c tio n a l d e n sity
A fundam ental consequence of the translational rotnistness hypothesis is th a t selection not
directly related to protein function can also constrain the evolution of protein sequences.
Therefore, in addition to the selective constraint ojierating on specific residues to conserve
protein function (contributing to functional density) selection also operates on a sequence-
wide background of residues not directly constrained by function to conserve translational
robustness. Collectively these sites contribTite to the '“fitness density” of a protein i.e.,
the proportion of residues in a protein constrained by natural selection w ith each site
weighted by the fitness effect of m utation (Pal et al., 2006). According to the definition
of Pal
et a l, fitness density is a m easure of the change in fitness of the m utant
protein
(relative to the wildtype molecule). To determ ine the fitness difference of the individual
m utant
organism (relative to wildtype individuals) this measure nmst be scaled by the
overall im portance of the protein to the organism (Pal et al., 2006). Accordingly, the most
im portant determ inants of protein evolution should be fitness density and dispensability.
However, as highlighted earlier the role of dispensability remains the subject of vigorous
debate.
Causes o f variation in the rate of protein sequence
evolution
Introduction
URAlO
(orotate phosphoribosyltransferases 1 and 2) differ more th a n 60-fold in expression
level and six-fold in evolutionary rate w ith
URA5
being the m ore highly expressed and
slower-evolving. Given the similar functions of these proteins a sim ilar fraction of their
residues are expected to be constrained by function (i.e., their functional densities should
be equivalent). However, the higher expression level of
U RA5
should increase selective
constraint on the remaining residues to ensure correct folding in the event of m istranslation.
Selection for translational robustness, therefore, increases fitness density of
U RA5
compared
to
URAlO
while their functional densities should rem ain com parable (D rum m ond et al.,
2005).
1.2.11
D eterm in a n ts o f ev o lu tion ary rate o f m am m alian p rotein s
Studies of the causes of protein rate variation in yeast may provide only a limited first
approxim ation to explain the variability of rates in m ulticellular organism s such as m am
mals. T he difficulties inherent in any extrapolation over broad phylogenetic distances are
amplified by three sorts of evolutionary transition.
F irst, at a fundam ental level the transition from large effective population sizes in uni
cellular eukaryotes to smaller effective pojjulation sizes in m etazoans is expected to influence
the efficiency of selection against deleterious m utations. Second, com pared to unicellular
organisms, the m am m alian genome shows considerable heterogeneity w ith respect to both
m utation rate and fixation rate. Third, the emergence of tissue and organ differentiation is
likely to be associated with selective constraints unique to m etazoans (e.g., m am m als) com
pared to unicellular organisms (e.g., budding yeast). These last two evolutionary transitions
may contribute to intra-genomic variability in the rates of m am m alian protein evolution
and will be considered in turn.
1.2.12
H e te ro g en eity o f th e m am m alian gen o m e
In m am m als there is considerable genomic variability of both of the variables th a t dictate
the neutral ra te of protein evolution according to K im ura’s fornm lation (equation 1.1). In
other words different p arts of the genome can differ both in their ra te of m utation
{
v t
)
and
in their ra te of fixation of m utations (/o).
C auses o f variation in the rate o f protein sequence
evolution
Introduction
and this has been corroborated using other measures of the ra te of neutral substitution
(M atassi et al., 1999; Lercher et al., 2001; Sm ith et al., 2002). W h at causes this regional
variation of m utation rate? One possible explanation lies in the observation th a t GC content
varies considerably across the m am m alian genome, contributing to a genomic landscape of
long (> 300kb) regions of homogenous GC content (“isochores” (Eyre-W alker and Hurst.
2001)). Moreover, the neutral substitution ra te is likely to positively correlate w ith GC
content according to a non-equilibrium isochore model under which the
GC ^ AT ra te is higher th a n the AT ^ GC rate (and both rates are constant across the
genome) (Piganeau et al., 2002). Therefore, it has been suggested th a t the m utation rate of
genes located in GC-rich regions should be greater than th a t of genes in GC-poor regions
(Sm ith et al., 2002). A second possible explanation for intra-genomic variability in m utation
ra te is provided by variation in recom bination rate in the m annnalian genome (Kong et al..
2002). Because recom bination is m utagenic in mammals (Hellniann et al., 2003; Lercher
and H urst, 2002) genes in highly recombining regions should have higher m utation rates
th an those residing in regions of low recom bination rate.
Genomic heterogeneity is also seen in the fixation rate of nuitations. This is a conse
quence of genome-wide variation in the balance between the efficiency of selection on the
one hand and the power of genetic drift on the other. In fact the regional variation in re
com bination ra te described above also plays a role in this ty]>e of within-genome variability.
T he efficiency of selection is greatest in highly recombining regions because the disruption
of genetic linkage by recom bination allows selection to act on single alleles w ithout interfer
ence from alleles at neighbouring loci (i.e., Hill-R.obertson effects are reduced). Therefore,
purifying selection will be a t its most efficient in regions of high recombination. If most
m utations are deleterious this should mean th a t genes in highly-recombining regions should
evolve more slowly th a n those in regions of low recombination.
Causes o f variation in the rate of protein sequence
evolution
Introduction
diversity of protein rates (but see Wyckoff et al. (2005)).
1 .2 .1 3
T h e tr a n s itio n to tis s u e d iffe r e n tia tio n
A m ajor im plication of the emergence of tissue differentiation is th a t the expression of a
m am m alian gene m ust be described not only in term s of its level b u t also in term s of the
“bread th ” of its tissue distribution, i.e. the number of tissues in which it is expressed.
The m ultiplicity of m am m alian cell-types underlies an extraordinary diversity of highly
differentiated tissues th a t adds two additional dimensions to the concept of gene pleiotropy.
F irst, the developm ental tim ing of gene expression during tissue differentiation might corre
late w ith pleiotropy. According to the “hourglass model” interm ediate developmental stages
are highly conserved while earlier and later stages show greater evolutionary plasticity (Raff,
1996). M utations in proteins expressed at interm ediate stages in development are therefore
expected to have greater pleiotropic effects and these proteins should evolve more slowly as
a consequence. There is some support for this prediction in the case of mouse development
(Castillo-Davis et al., 2004). T he second potential correlate of pleiotropy in mamm als is
th e tissue breadth of a gene’s expression. Specifically, a situation of antagonistic pleiotropy
might result if a new allelic variant th a t benefits a gene’s function in one tissue is delete
rious to its function in a different tissue. These m utations are expected to be elim inated
efficiently by purifying selection leading to slower j)rotein evolution.
1 .2 .1 4
Im p a c t o f b r e a d th o f e x p r e ss io n o n p r o te in e v o lu tio n in m a m m a ls
Causes o f variation in the rate of protein sequence
evolution
Introduction
conditions where it encounters a wide range of niolecuhar interaction partners.
These early observations were extended by a genoine-wide study of the relationship
between the ra te of protein evolution of 2400 hunian-rodent orthologs and their breadth
of expression determ ined using expressed sequence tag (EST) d a ta from 19 tissues (Duret
and M ouchiroud, 2000). This study drew two m ajor conclusions regarding protein rate
variability. First, with regard to the effect of tissue Ijreadth, it was shown th a t tissue-specific
proteins evolve up to three times faster th an ubiquitously expressed proteins. Second, the
influence of tissue identity was reflected in the roughly 2.5 fold variation in the rate of
protein evolution among genes having similar breadths bu t different tissue-specificities.
D uret and M ouchiroud (2000) proposed th a t tlie hrst of these differences is of larger
m agnitude th an could be explained by H asting’s suggestion of increased functional con
straint on broadly expressed genes due to inter-tissue variation in cellular environment.
This led to the alternative explanation th a t the fitness effect of a m utation th a t is slightly
deleterious to a gene’s fimction is m ultiplied by the munber of tissues in which the gene
is expressed. Thus, D uret
et al.
attributeci the slower evolution of ubiquitously expressed
genes to an increase in the stren g th of selection pro])ortional to the number of tissues in
which a gene is expressed. This echoes the intuition th at, in multicellular organisms, the
breadth of expression of a gene should correlate with the gene’s ])leiotropic level. There
fore, the slower evolution of ubiquitously exjiressed compared to tissue-specific genes in
m am mals (D uret and Mouchiroud, 2000; Zhang and Li. 2004), should reflect an increase in
constraint associated w ith increased pleiotropy.
Strikingly, D uret and M ouchiroud’s second observation dem onstrating the influence of
tissue-identity implies th a t, for tissue-specific genes, inter-tissue differences account for
much variation in the rate of protein evolution. How'ever, they suggested th a t the slower
evolution of brain-specific com pared to liver-specific genes reflects the relatively peripheral
role of the liver com pared to the brain rather than reflecting inter-tissue variability in
cellular environm ent. Thus, the more central role of the brain is expected to manifest itself
in greater fitness effects of sequence changes among brain-specific proteins.
Causes o f variation in the rate of protein sequence
evolution
Introduction
b u t could be a simple consequence of the gene’s expression in a single rate-determ ining
tissue (e.g. brain). D uret and M ouchiroud’s two m ajor results were borne ou t by a more
recent study perform ed by Zhang and Li (2004). This study found a nearly two fold
increase in the ra te of non-synonymous divergence of tissue-specific genes com pared to
ubiquitously expressed “housekeeping” genes defined on the basis of m icroarray data. The
large effect of tissue identity was confirmed by the finding th a t lung-specific proteins evolve
on average nearly three times faster th an muscle-specific genes. However, Zhang and Li
also dem onstrated th a t tissue-specific genes in the slowest evolving categories (i.e. brain
and muscle) were significantly faster evolving th an broadly expressed genes thus negating
the possibility of a “rate-determ ining tissue”. This result therefore supports the concept of
an additive pleiotropic effect of expression breadth on the evolutionary ra te of m am m ahan
proteins.
1.2.15
E xp ression bread th versus tissu e-sp ec ificity
Previous studies have claimed th a t the level of a gene’s expression is highly correlated
w ith its expression b readth (Lercher et al., 2002; Subrarnanian and K um ar, 2004). This
is believed to reflect the assum ption th a t housekeeping genes tend to be highly expressed
(Vinogradov, 2004). However, the term "housekeeping gene” has occasionally been applied
loosely (Lercher et al., 2002) and in a m anner th a t has not always accorded w ith the strict
definition of housekeeping genes as those genes th a t are always expressed in every tissue
to m aintain cellular functions (W atson et al., 1965). A more recent working definition has
defined housekeeping genes as “those genes critical to the activities th a t m ust be carried out
for successful completion of the cell cycle” (W arrington et al., 2000). Interestingly, this def
inition also encapsulates the concept of gene essentiality, highlighting the interrelatedness
of ubiquitous expression and essentiality. Recent refinements of the conventional house
keeping gene concept have followed from two whole genome expression studies th a t have
dem onstrated th a t (i) housekeeping genes are not necessarily the m ost highly expressed
genes in all tissues and (ii) the expression of housekeeping genes can be variable across
tissues (W arrington et al., 2000; Hsiao et al., 2001).
It should be noted th a t part of the correlation between the level and b re ad th of a gene’s
expression is artefactual and stem s from the use of an arb itra ry cutoff to derive a measure
Causes of variation in the rate of protein sequence
evolution
Introduction
have been applied to meeisure breadth of expression in the context of both microarray
(Zhang and Li, 2004) and EST-based studies (Duret and Mouchiroud, 2000) leading to an
intrinsic dependence of measured expression breadth on expression level (Liao and Zhang,
2006). For m icroarray d a ta this dependency results from the use of signal intensity cutoffs
whereas for expressed sequence-based measures it is a function of the sampling depth of EST
libraries. This raises the possibility th a t previous observations of an association between
the evolutionary ra te of a protein and its tissue-specificity may have arisen due to the
confounding influence of expression level (Duret and Mouchiroud, 2000). This may be
particularly pertin en t given the fact th a t, in yeast, expression level is the strongest predictor
of the rate of protein evolution (Drum m ond et al., 2006).
This problem can be addressed using a recently proposed alternative measure of the
tissue-distribution of a gene’s expression. This “tissue-specificity index” (Yanai et al., 2005)
does not rely on the use of expression cut-offs to distinguish between presence or absence
of expression. Interestingly, this measure of tissue-s])ecificity is found not to correlate
w ith gene expression level, thus apparently overturning the long-standing assum ption th a t
housekeeping genes are expressed at high levels and in agreement with more recent results
(W arrington et al.. 2000: Hsiao et al., 2001). The lack of dependence of this measure on
gene expression level allows the effect of tissue specificitj' on protein evolution to be assessed
independently of the confounding influence of exi)ression level. In fact, a statistically sig
nificant association was found between tissue-specificity index and both the ra te of protein
evolution (m easured by
dj\j) and the strength of selective constraint (measured by
d ^ / d s )
(Liao et al., 2006). Therefore, previous claims th a t the evolutionary rate of a protein is
correlated w ith its tissue-specificity remain robust. This has been separately confirmed
using a partial correlation analysis approach: expression breadth and ra te of m am m alian
protein evolution remain significantly correlated once expression level is controlled for (M ar
tin Lercher, personal com m unication). However, the m agnitude of this association appears
to be small. A t most 3% of the ordinal variation in protein rate in explained by ordinal
variation in tissue-specificity (Liao et al., 2006).
1.2.16
T issu e-sp ecific ity and p rotein secretion
Causes o f variation in the rate o f protein sequence
evolution
Introduction
prim ary determ inant of this effect or w hether other possible properties distinguishing these
groups of genes could account for the difference in evolutionary rates. In other words,
does a classification of genes w ith respect to tissue specificity introduce a hidden bias w ith
respect to some other potential determ inant of th e ra te of protein evolution? For example,
tissue-specific genes are likely to ftmction more frequently in cell-cell com m unication and
signal transduction roles com pared to the m ore common m etabolic activity of housekeeping
genes.
Therefore, the unequivocal dem onstration th a t tissue-specificity alone is responsible
for accelerating the ra te of m am m alian protein evolution (e.g., through a reduction in
pleiotropy) would require the com parison of proteins th a t differ only with respect to their
bread th of expression bu t share all other relevant properties (e.g., have a common biochem
ical function).
One approach to disentangle the effects on protein evolution of tissue-specificity and
functional differences is to consider evolutionary rates w ithin gene families. According to
this approach, if two paralogous genes th a t differ in their rate of evolution also differ in their
expression breadth then the ra te difference can be solely a ttrib u te d to the difference in their
bread th of expression. The com nion-ancestry (and presumed comm on biochemical function)
of members of a gene family provides a control for the im pact of functional differences on
rate. An early study of this n atu re found th a t among 15 studied gene families, 14 showed
a p a tte rn of evolutionary ra te consistent w ith the effect of expression bread th (Hastings,
1996). In these 14 families the slowest evolving member was found to be expressed in the
broadest range of tissues.
More recent work has exposed one potential correlate of tissue-specificity th a t may