Development of a Standardized Sanger-Based Method for
Partial Sequencing and Genotyping of Dengue Viruses
Gilberto A. Santiago,
aGlenda L. González,
aFabiola Cruz-López,
aJorge L. Muñoz-Jordan
aaCenters for Disease Control and Prevention, National Centers for Emerging and Zoonotic Infectious Diseases, Division of Vector Borne Diseases, Dengue Branch, San Juan, Puerto Rico
ABSTRACT
The global expansion of dengue viruses (DENV-1 to DENV-4) has
con-tributed to the divergence, transmission, and establishment of genetic lineages of
epidemiological concern; however, tracking the phylogenetic relationships of these
virus is not always possible due to the inability of standardized sequencing
proce-dures in resource-limited public health laboratories. Consequently, public genomic
data banks contain inadequate representation of geographical regions and historical
periods. In order to improve detection of the DENV-1 to DENV-4 lineages, we report
the development of a serotype-specific Sanger-based method standardized to
se-quence DENV-1 to DENV-4 directly from clinical samples using universal primers that
detect most DENV genotypes. The resulting envelope protein coding sequences are
analyzed for genotyping with phylogenetic methods. We evaluated the performance
of this method by detecting, amplifying, and sequencing 54 contemporary DENV
isolates, including 29 clinical samples, representing a variety of genotypes of
epide-miological importance and global presence. All specimens were sequenced
success-fully and phylogenetic reconstructions resulted in the expected genotype
classifica-tion. To further improve genomic surveillance in regions where dengue is endemic,
this method was transferred to 16 public health laboratories in 13 Latin American
countries, to date. Our objective is to provide an accessible method that facilitates
the integration of genomics with dengue surveillance.
KEYWORDS
E gene, Sanger, dengue, dengue virus, genotyping, sequencing
D
engue virus, a member of the
Flaviviridae
RNA virus family, consists of four distinct
serotypes (DENV-1, DENV-2, DENV-3, and DENV-4), all of which have been
trans-mitted in more than 100 countries. Approximately 390 million infections have
esti-mated to occur annually (1, 2). Over the last 4 decades, the range of endemic
transmission has increased significantly, as dengue is frequently introduced into
re-gions previously unexposed to the virus, often causing epidemics (3–5), and
conse-quently placing approximately 40% of the world population at risk (1, 6). This global
spread, in addition to the characteristic high rate of mutation by flavivirus polymerases,
has contributed to important evolutionary events that affect disease severity and
epidemiology, such as the divergence of subserotypic genotypes and taxonomic
lineages. Many genotypes of epidemiological relevance have been associated with
transmission in a particular geographic region (7–9), disease severity (10–12), or even
increased epidemic potential (13–16). Recent outbreak investigations have reported
molecular evidence suggesting that the introduction of genetically distinct virus strains
may lead to the emergence of new lineages directly related to epidemic transmission
(5, 17). In addition, phylogenetic and molecular epidemiological studies have identified
the formation, expansion, decline, and extinction of genotypes and subgenotypic
lineages during periods of endemic and epidemic dengue transmission (18–20);
nev-CitationSantiago GA, González GL, Cruz-López
F, Muñoz-Jordan JL. 2019. Development of a standardized Sanger-based method for partial sequencing and genotyping of dengue viruses. J Clin Microbiol 57:e01957-18.https://doi.org/ 10.1128/JCM.01957-18.
EditorYi-Wei Tang, Memorial Sloan Kettering
Cancer Center
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Address correspondence to Jorge L. Muñoz-Jordan, [email protected].
G.A.S. and G.L.G. contributed equally to this work.
Received10 December 2018
Returned for modification26 December
2018
Accepted22 January 2019
Accepted manuscript posted online13
February 2019
Published
crossm
28 March 2019
on May 17, 2020 by guest
http://jcm.asm.org/
ertheless, the drivers of these dynamics and their impact on the epidemiology of
disease are not well understood.
Even after decades of studying dengue molecular epidemiology and the modern
advances in genomics, the capacity remains inaccessible for many public health
laboratories running dengue surveillance in regions of endemicity. Although there is a
wide array of published DENV sequencing methods, significant limitations have
im-paired implementation, such as high variability between methods, cost, lack of
stan-dardization with clinical specimens, inadequate method evaluation, and high protocol
complexity, which often includes amplification of the virus by tissue culture. Only a
limited number of laboratories run some sort of genomic surveillance and have the
capacity to track the local traffic of circulating dengue strains using a diversity of partial
sequencing methods (21–25). However, this function is not frequently performed or
applied to complement surveillance. This is reflected in the public repository of dengue
genome sequences, GenBank, which currently holds an unbalanced collection of DENV
genomic data that limits phylogenetic studies. Although the GenBank collection
in-cludes abundant DENV envelope glycoprotein coding sequence (E) data, as historically
targeted for DENV genotyping, this collection is handicapped by the abundance of
redundant sequences and the misrepresentation of certain time periods and endemic
geographical locations (Virus Pathogen Resource,
https://www.viprbrc.org/brc/home
.spg?decorator
⫽
flavi_dengue
).
To increase surveillance and research laboratory capacity and support genomic
surveillance of DENV, we developed the CDC DENV E gene sequencing assay, a
standardized, serotype-specific, Sanger-based method for genotyping analyses by
se-quencing the envelope glycoprotein coding sequence (E) directly from clinical serum
specimens. We developed this assay for broad implementation in surveillance
labora-tories, including an open-platform protocol that incorporates simple, cost-effective, and
traditional molecular biology techniques to facilitate adaptability. Our assay includes
DNA primers specifically tailored and validated to detect a wide variety of genotypes of
epidemiological relevance and contemporary circulation globally. The E protein coding
sequence was selected because it provides a convenient target considering the high
abundance of E sequences published in GenBank; in addition to reports that validate E
for genotyping analyses, this fragment contains sufficient phylogenetic signal to
dis-tinguish DENV from other flaviviruses and between serotypes, genotypes, and
individ-ual strains (26–28). Furthermore, DENV phylogenetic tree reconstructions based on
E coding sequence render tree topologies similar to those based on complete
genome sequences facilitating genotyping at a significantly reduced cost and effort
(8, 26, 29, 30).
This method aims to facilitate DENV genomic surveillance, improve genotyping, and
foster molecular epidemiology by providing laboratories with a convenient,
cost-effective, and highly efficient tool for sequencing DENV circulating globally directly
from clinical specimens.
MATERIALS AND METHODS
Sequencing target selection and primer design. The DENV E protein coding sequence was selected as the target for amplification and sequencing considering that the length of this sequence, approximately 1,485 bp, contains sufficient conserved sequence regions to differentiate DENV from other flaviviruses and sufficient variability to further classify between serotypes, genotypes, and individual strains (8, 31); it is thus a historically preferred genome fragment for genotyping. The E coding sequence has been the target for genotyping DENV, so more E coding sequences are available than for any other gene in GenBank. These E coding sequences allow for wider sequence sampling for assay design and phylogenetic analysis. All amplification and sequencing primers were designed from multiple sequence alignments of each serotype considering approximately 300 sequences obtained from GenBank and comparing complementarity with 70% and 90% consensus sequences. Each alignment included repre-sentation of all clinically relevant genotypes, global geographical reprerepre-sentation, and sequences isolated between 1990 to 2017. Each alignment was revised to exclude extensive geotemporal redundancy and sylvatic lineages. Up to three noncontiguous mismatches were considered acceptable if sequence changes did not affect melting point temperatures (Tm) significantly; however, we recognize thatin silicomatches may not predict efficient primer binding accurately. The final primer sets for RT-PCR are listed in Table 1, and the sequencing primers are listed in Table 2.
on May 17, 2020 by guest
http://jcm.asm.org/
Viral strains and clinical samples.A panel of 25 presequenced DENV international isolates was obtained from the Centers for Disease Control and Prevention (CDC) Dengue Branch archived collection (Table 3). These 25 strains represent the four serotypes and genotypes of contemporary circulation globally. Sylvatic strains were not included due to the sporadic clinical relevance and the high sequence divergence from the urban-endemic strains, which results in an average of four mismatches per reverse transcription-PCR (RT-PCR) or sequencing primer and reduces the probability of amplification or se-quencing. Each isolate was amplified by tissue culture on Aedes albopictus-derived C6/36 cells, as previously described (32). In addition, a panel of 29 clinical serum specimens RT-PCR positive for all DENV serotypes was obtained from the reference specimen archive of the CDC Dengue Branch collected as part of the island-wide passive surveillance system in collaboration with the Puerto Rico Department of Health and from a dengue outbreak in American Samoa investigated by the CDC Dengue Branch in 2017 (Table 4). The CDC Institutional Review Board (IRB) authorized the use of deidentified samples for this study,
TABLE 1RT-PCR oligonucleotides for DENV E gene amplification
Serotype and primer Sequence
DENV-1 PCR-F AGC ACA TGC YAT AGG AAC ATC C DENV-1 PCR-R GCT GAT CGA ATT CCA CAC ACR CC DENV-2 PCR-F TCG CTC CTT CAA TGA CAA TGC DENV-2 PCR-R CAG CTC ACA ACG CAA CCA CTA DENV-3 PCR-F TAG CCC TAT TTC TTG CCC ATT AC DENV-3 PCR-R AGT TCA TTG GCT ATT TGC TTC CA DENV-4 PCR-F AAC AGG RAT CCA GCG AAC TGT DENV-4 PCR-R CTC GCT GGG GAC TCT GGT TG
TABLE 2DENV E gene sequencing primers
Serotype and primer Sequence
DENV-1
D1SEQ1 GCY ATA GGA ACA TCC ATC ACY CAG D1SEQ2 ACG TGT GCY AAG TTY AAG TGT GT D1SEQ3 AAG ACA GCT CAT GCA AAG AAR CAG D1SEQ4 TTG GTG AGA GYT ACA TCG TGR TAG D1SEQ5 GCT GAT CGA ATT CCA CAC ACR C D1SEQ6 CTG GTG TAC CAA TTT TCC CAC AG D1SEQ7 TGG GTC TCA GCC ACT TCY TTC T D1SEQ8 AGC CCT GTT CTA GGT GAG CAA TC DENV-2
D2SEQ1 TYG CTC CTT CAA TGA CAA TGC G D2SEQ2 ACA TGC AAA AAG AAC ATG GAA GGA D2SEQ3 AAT CCC CAY GCV AAG AAA CAG GAT D2SEQ4 CCA TTC GGR GAC AGC TAC ATC AT D2SEQ5 CAG CTC ACA ACG CAA CCA CTA D2SEQ6 TGA TGA TGT AGC TGT CTC CGA ATG D2SEQ7 CTG TGA GTG CCG TGT GCA TG D2SEQ8 CAA RTT TTC TGG TTG CAC GAC T DENV-3
D3SEQ1 AGG CAC TTC CTT GAC CCA GAA D3SEQ2 TGG CAA GGG AAG CTT GGT AA D3SEQ3 AAC AGG AAG GAG CTT CTT GTG ACA D3SEQ4 GAG CCT GTC AAT ATT GAG GCT GA D3SEQ5 TGT CAT GGA TGG GGT GAC CA D3SEQ6 TCC CTC TAT TGG TTC CAG GCA D3SEQ7 CTT GCG ATC CAA GGA CAA CTA CT D3SEQ8 GCG TTG TCT CCA ATT CCA ATY ACT DENV-4
D4SEQ1 TGT CCT AAT GAT GCT GGT CGC D4SEQ2 GAG GAG TTG TGA CAT GTG C D4SEQ3 ATG GTG ACA TTT AAG GTT CCT CAT D4SEQ4 GCT GAG AAT ACC AAC AGT GTA ACC A D4SEQ5 CTC GCT GGG GAC TCT GGT TG D4SEQ6 GCC ATD CGY TTT GCA CCT CT D4SEQ7 TTG AGA TGT CCT GCA AAC ATG T D4SEQ8 ATG GTG ACC TGG GRG TTA TCG TC
on May 17, 2020 by guest
http://jcm.asm.org/
[image:3.585.42.380.352.735.2]protocol 6874. Serotypes and the relative RNA concentration (threshold cycle [CT]ⱕ30) were confirmed by real-time RT-PCR (33).
Viral RNA extraction.For our evaluation of the CDC DENV E gene sequencing assay, we extracted viral RNA from clinical specimens (serum) or tissue culture supernatant using a QIAamp viral RNA minikit (Qiagen, catalog no. 52904) or other automated method, including the MagNA Pure LC 2.0 instrument (Roche, catalog no. 05197686001) or the MagNA Pure 96 instrument (Roche, catalog no. 05195322001), according to the manufacturer’s recommended protocols.
E coding sequence amplification.E coding sequence was amplified by endpoint RT-PCR using a SuperScript III Platinum one-step RT-PCR system (Invitrogen, catalog no. 12574026) and one set of serotype-specific amplification primers (Table 1). These primers were designed to detect DENV geno-types of contemporary circulation globally and amplify the complete E coding sequence flanked by a small portion of the premembrane (preM) and nonstructural protein 1 (NS1) coding sequences. RT-PCRs were assembled by mixing 13.0l of nuclease-free PCR-grade water, 25.0l of 2⫻SuperScript reaction mix, 20.0 M concentrations of each DENV forward and reverse primer, 2.0 l of SuperScript III RT/platinumTaqmix, and 8.0l of template RNA elution to a final reaction volume of 50.0l. RT-PCR analysis was performed in a conventional thermal cycler (SimpliAmp; Thermo Fisher) programmed as follows: reverse transcription (RT) at 55°C for 30 min and 94°C for 2 min; followed by 40 cycles of 94°C for 15 s, 55°C for 30 s, and 68°C for 2 min; and then a final extension at 68°C for 5 min. RT-PCR products were detected and visualized by agarose gel electrophoresis. A 0.8% UltraPure agarose gel (Invitrogen, catalog no. 16500500) was prepared in 1⫻Bionic buffer (Sigma-Aldrich, catalog no. B6186-4L) stained with SYBR Safe DNA stain (Invitrogen, catalog no. S33102) with wells sufficiently thick to contain the complete RT-PCR and loading buffer (approximately 55.0l). A 1-kb ladder DNA marker was used to determine the amplicon size. The RT-PCR amplicon length will vary by serotype: approximately 1,700 bp for DENV-1, 1,500 bp for DENV-2, 1,800 bp for DENV-3, and 1,600 bp for DENV-4. This RT-PCR procedure was tested with serial dilutions of reference strains of the four DENV serotypes and dilutions with a real-time RT-PCR CTvalue of⬍30 produced a band with sufficient DNA for successful sequencing (33). Reagents for RT-PCR and electrophoresis can be substituted for other compatible reagents (with the exception of the primers) of similar performance, if available.
[image:4.585.42.377.83.364.2]RT-PCR amplicon purification.RT-PCR amplicon bands should be of the expected size with a brightness equal to or greater than the bands in the 1-kb DNA ladder marker. The amplicon bands were excised with a clean scalpel under low-intensity UV light, eliminating as much agarose as possible. The excised slice of gel was placed in a 1.7-ml nuclease-free microcentrifuge tube. DNA from the gel was extracted and purified using a QIAquick gel extraction kit (Qiagen, catalog no. 28706) with the following modifications to the manufacturer’s recommended protocol: the microcentrifuge tube containing the gel slice was filled to capacity with QG buffer and incubated at 50°C for 10 min with frequent vortexing until the gel was completely melted and dissolved in the buffer. Isopropanol was not added to the tube. Half
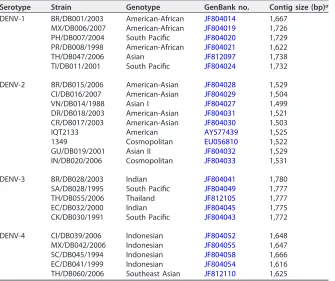
TABLE 3Panel of sequenced virus isolates
Serotype Strain Genotype GenBank no. Contig size (bp)a
DENV-1 BR/DB001/2003 American-African JF804014 1,667 MX/DB006/2007 American-African JF804019 1,726 PH/DB007/2004 South Pacific JF804020 1,729 PR/DB008/1998 American-African JF804021 1,622 TH/DB047/2006 Asian JF812097 1,738 TI/DB011/2001 South Pacific JF804024 1,732 DENV-2 BR/DB015/2006 American-Asian JF804028 1,529 CI/DB016/2007 American-Asian JF804029 1,504 VN/DB014/1988 Asian I JF804027 1,499 DR/DB018/2003 American-Asian JF804031 1,521 CR/DB017/2003 American-Asian JF804030 1,503 IQT2133 American AY577439 1,525 1349 Cosmopolitan EU056810 1,522 GU/DB019/2001 Asian II JF804032 1,529 IN/DB020/2006 Cosmopolitan JF804033 1,531 DENV-3 BR/DB028/2003 Indian JF804041 1,780 SA/DB028/1995 South Pacific JF804049 1,777 TH/DB055/2006 Thailand JF812105 1,777 EC/DB032/2000 Indian JF804045 1,775 CK/DB030/1991 South Pacific JF804043 1,772 DENV-4 CI/DB039/2006 Indonesian JF804052 1,648 MX/DB042/2006 Indonesian JF804055 1,647 SC/DB045/1994 Indonesian JF804058 1,666 EC/DB041/1999 Indonesian JF804054 1,616 TH/DB060/2006 Southeast Asian JF812110 1,625
aContig size was determined using SeqMan Pro (DNASTAR Lasergene 14).
on May 17, 2020 by guest
http://jcm.asm.org/
of the contents of the tube was transferred to the Qiagen spin columns by pipetting, and the columns were centrifuged for 1 min at 14,000 rpm. The flowthrough was discarded, and the previous step was repeated until the entire volume of the samples passed through the column. The columns were then washed with 800 l of QG buffer and centrifuged for 1 min at 14,000 rpm. The manufacturer’s recommended protocol continues at this point. Alternatively, the amplicon can be purified using enzymatic methods such as ExoSAP-IT PCR product cleanup reagent (Thermo-Fisher, catalog no. 78201.1.ML). The purified products were quantified using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific).
Sanger sequencing.Sequencing reactions were assembled using a BigDye Terminator v3.1 cycle sequencing kit (Thermo-Fisher, catalog no. 4337455). Optimal purified product input was determined experimentally to range between 40 and 60 ng/reaction. Eight sequencing reactions were performed per sample using serotype-specific primer sets: four primers that generate tiled, overlapping sequences from 5=to 3=and four primers that generate overlapping sequence from 3=to 5=for a minimum 2⫻coverage across the E coding sequence (Table 2). Sequencing reactions were assembled according to the manufacturer’s recommended protocol: 8.0l of BigDye Terminator 3.1 Ready Reaction mix, a 3.3M concentration of the corresponding single sequencing primer, 60.0 ng of purified DNA product and nuclease-free PCR-grade water to complete a final reaction volume of 20.0l. Sequencing reactions were placed on a conventional thermocycler programmed with the following protocol: denaturation at 96°C for 1 min, followed by 25 cycles of 96°C for 10 s, 50°C for 5 s, and 60°C for 4 min. The ramp rate was adjusted to 1°C/s at each step. Variations of the protocol for the BigDye sequencing reactions should be evaluated and adjusted for optimal results considering the sequencing instrument available and labo-ratory experience. Variations of the protocol may include dilution of the BigDye Terminator 3.1 Ready Reaction mix with 5⫻sequencing buffer and DNA input. BigDye reactions were purified using a BigDye XTerminator purification kit (Thermo-Fisher, catalog no. 4376484) to remove unincorporated dyes following the manufacturer’s recommended protocol. Alternatively, sequencing reactions can be purified by spin column methods like DyeEx 2.0 spin kit (Qiagen, catalog no. 63204). Sequencing was performed on an ABI 3130 XL genetic analyzer (Thermo-Fisher, catalog no. 4359571).
[image:5.585.41.373.84.403.2]Trace sequence analysis and assembly. Sequence trace files were analyzed individually using BioEdit software (www.mbio.ncsu.edu/BioEdit/bioedit.html) or Chromas 2.6.5 software (Technelysium). A minimum of 400 bp of high-quality sequence (determined by size of fluorescence signal and clarity of the peak) are expected per trace file. Contig assembly was performed with the eight trace files per sample using the SeqMan Pro program found in the DNASTAR Lasergene 14 software package. The software reports the number of contigs assembled and the number of trace files used to assemble the contigs and
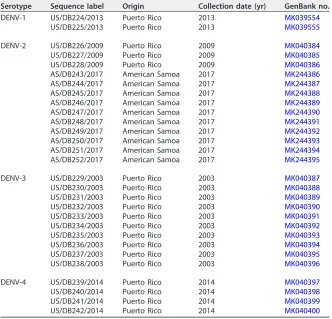
TABLE 4Panel of clinical specimens sequenced
Serotype Sequence label Origin Collection date (yr) GenBank no.
DENV-1 US/DB224/2013 Puerto Rico 2013 MK039554
US/DB225/2013 Puerto Rico 2013 MK039555
DENV-2 US/DB226/2009 Puerto Rico 2009 MK040384
US/DB227/2009 Puerto Rico 2009 MK040385
US/DB228/2009 Puerto Rico 2009 MK040386
AS/DB243/2017 American Samoa 2017 MK244386
AS/DB244/2017 American Samoa 2017 MK244387
AS/DB245/2017 American Samoa 2017 MK244388
AS/DB246/2017 American Samoa 2017 MK244389
AS/DB247/2017 American Samoa 2017 MK244390
AS/DB248/2017 American Samoa 2017 MK244391
AS/DB249/2017 American Samoa 2017 MK244392
AS/DB250/2017 American Samoa 2017 MK244393
AS/DB251/2017 American Samoa 2017 MK244394
AS/DB252/2017 American Samoa 2017 MK244395
DENV-3 US/DB229/2003 Puerto Rico 2003 MK040387
US/DB230/2003 Puerto Rico 2003 MK040388
US/DB231/2003 Puerto Rico 2003 MK040389
US/DB232/2003 Puerto Rico 2003 MK040390
US/DB233/2003 Puerto Rico 2003 MK040391
US/DB234/2003 Puerto Rico 2003 MK040392
US/DB235/2003 Puerto Rico 2003 MK040393
US/DB236/2003 Puerto Rico 2003 MK040394
US/DB237/2003 Puerto Rico 2003 MK040395
US/DB238/2003 Puerto Rico 2003 MK040396
DENV-4 US/DB239/2014 Puerto Rico 2014 MK040397
US/DB240/2014 Puerto Rico 2014 MK040398
US/DB241/2014 Puerto Rico 2014 MK040399
US/DB242/2014 Puerto Rico 2014 MK040400
on May 17, 2020 by guest
http://jcm.asm.org/
displays the trace file alignment to assemble the contig consensus sequence. Initially, we confirm that we obtained a minimum of 2⫻ coverage across the length of the E coding sequence, followed by a base-by-base curation of the sequence data to guarantee sequence quality. If an ambiguous base is called in the contig consensus sequence, the trace files are analyzed at the specific sequence position to determine the correct base. Once the contig is assembled and the sequence is curated, the consensus sequence is analyzed by BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) to confirm sequence identity and retrieve similar sequences. The top 5 to 10 sequences from the BLAST results were selected and aligned with our sample consensus sequence using Clustal W or MUSCLE in MEGA 7 software (www .megasoftware.net) to determine consensus sequence orientation and trim sequence data flanking the E coding sequence. This step also serves to identify additional sequences of high similarity that can be used in downstream phylogenetic analyses and provides a general idea of the genotype, as well as the geographic-temporal origin of the query sequence. The resulting DNA sequence was translated to amino acid sequence and compared to similar annotated amino acid sequences retrieved from GenBank. We also confirmed the absence of internal stop codons that might have been introduced by sequencing errors. There are numerous software packages that perform these functions, and we defer software selection to the user considering availability and computational capacity.
Phylogenetic analysis.The sequences obtained with this method were grouped by serotype and aligned with other genotype reference sequences obtained from GenBank (see Table S1 in the supplemental material) using the MUSCLE module in MEGA 7. The multiple sequence alignments were trimmed to the length of the shortest sequence (1,480 to 1,485 bp). Phylogenetic relationships were inferred using the maximum likelihood method based on the Tamura-Nei model and topolo-gies tested with 1,000 bootstrap replicates using IQ-TREE software (http://www.iqtree.org/). Geno-types are marked by brackets and labeled accordingly (7).
Data availability.Sequence data were deposited in GenBank under the accession numbers listed in Table 4.
RESULTS
DENV E coding sequence amplification was successful in the 54 clinical serum and
tissue culture samples evaluated (Tables 3 and 4). Amplicons containing E coding
sequence flanked by preM and NS1 coding sequence fragments were sequenced with
the CDC DENV E gene sequencing assay. The assay produced the eight expected
sequence trace files with a minimum of 400 bp of high-quality data for 29 of 29 clinical
samples and 24 of the 25 virus isolates, which assembled single contigs of more than
1,500 bp with a minimum of 2
⫻
coverage across the E coding sequence segment (Table
3). The overlapping sequencing reactions and the bidirectional sequencing strategy
increased the probability of covering the coding sequence with high-quality sequence
data and facilitated base-to-base calling. For DENV-4 isolate Ecuador 1999, GenBank
accession number
JF804054
, the assay produced seven of the eight sequence trace files
with the expected minimal size of 400 bp in length; however, a single contig of 1,438 bp
was assembled with 2
⫻
coverage across 80% of the E coding sequence fragment. A
multiple sequence alignment with 10 similar sequences showed that the missing
sequence data were at the 3
=
end of the E coding sequence. To address this issue, we
resequenced this isolate and succeeded obtaining all eight sequence trace files
assem-bling a contig of 1,616 bp. A highly conserved region of the E coding sequence across
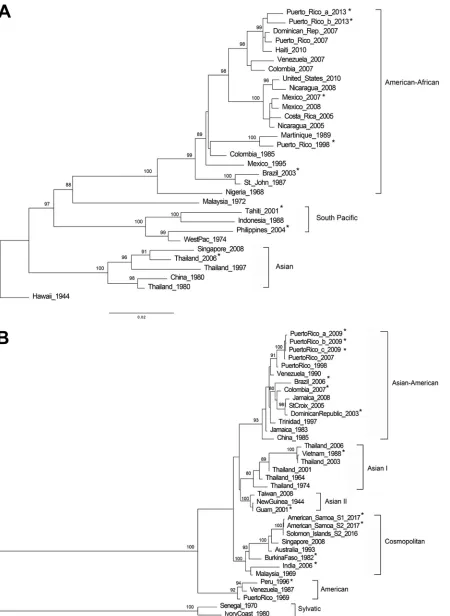
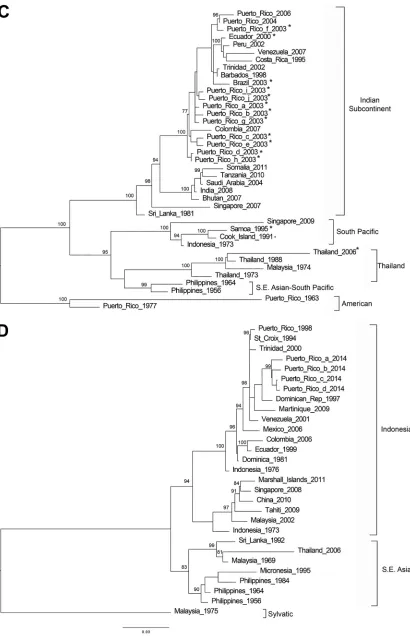
similar sequences was evaluated. Maximum-likelihood phylogenetic trees were inferred
for each DENV serotype using the E coding sequences obtained from this method in
addition to other E coding sequences considered as references for each genotype (Fig.
1). Each of the 54 E coding sequences obtained grouped with the expected genotypes
and relatedness to genotype reference sequences were supported by bootstrap scores
of higher than 75%. In addition, our genotyping results concurred with the results
obtained from a recently developed web-based online Genome Detective Dengue Virus
Typing Tool for dengue virus genotyping (
https://www.genomedetective.com/app/
typingtool/dengue/introduction
) which aligns any DENV-1 to DENV-4 sequence with
preselected genotype reference sequences and outputs a phylogenetic tree.
DISCUSSION
The transmission of DENV continues to expand globally, increasing the number of
regions exposed and people at risk of infection. Real-time monitoring of transmission
and epidemics is necessary to forecast further virus spread, which will impact the
design of preventive interventions and treatment measures. Several studies have
shown how, occasionally, foreign virus introductions or even endemic transmission can
on May 17, 2020 by guest
http://jcm.asm.org/
FIG 1Genotyping DENV using E gene sequence. Phylogenetic reconstruction was used to determine DENV genotypes by the maximum-likelihood method based on the Tamura-Nei model of nucleotide substitution. (A) DENV-1; (B) DENV-2; (C) DENV-3; (D) DENV-4. Asterisk-labeled taxa indicate sequences obtained by this method. Due to the sequence similarity and space limitations, only two of the ten DENV-2 American Samoa 2017 sequences were included in the maximum-likelihood tree.
on May 17, 2020 by guest
http://jcm.asm.org/
[image:7.585.45.495.70.686.2]FIG 1(Continued)
on May 17, 2020 by guest
http://jcm.asm.org/
[image:8.585.45.455.70.708.2]generate dengue outbreaks regardless of previous exposure to the virus (5, 17, 34). This
supports the importance of genomic surveillance to understand the epidemiology of
dengue in a region. The CDC DENV E gene sequencing assay was developed to provide
a simple and convenient standardized tool for DENV partial sequencing, for
genotyp-ing, and to harmonize genomic surveillance of dengue viruses. Such a tool will be
readily adaptable to public health laboratories and provide the opportunity to monitor
genetic changes associated with circulating genotypes or other epidemiological
fac-tors. The selection of the E coding sequence as sequencing target provides sufficient
data to perform phylogenetic inferences for genotyping and lineage classification with
tree topologies that closely resemble those obtained from complete genome
se-quences (7, 26, 35).
This assay was developed to sequence directly from diagnostic serum specimens
using a simple approach to amplify the target region and obtain reliable sequence data
taking advantage of the Sanger sequencing method. Despite the advantages that
next-generation sequencing technologies offer, the Sanger method is more accessible
to laboratories in regions where dengue is endemic and provides a nimble strategy to
process smaller numbers of samples with small genomes with high accuracy. Our
evaluation of this assay demonstrated the capacity to sequence a wide variety of
international DENV genotypes within each serotype and with wide sequence variation.
Only on one occasion was a DENV-4 isolate not sequenced entirely, with a contig
formed by seven sequence trace files, and we speculate that performance of the eighth
sequencing reaction could have been affected by insufficient loading of amplicon
product into the reaction. However, this was corrected by resequencing the isolate with
the proper concentration of DNA template. A simple genomic analysis demonstrated
that the sequences obtained by this method readily apply to phylogenetic studies
relevant to molecular and genomic surveillance such as genotype detection. Although
whole-genome sequences are more suitable for evolutionary studies and provide
higher phylogenetic resolution for lineage classification, the E coding region has been
a more popular target historically; hence, there is a larger availability and
representa-tion in public databases. Consequently, the accuracy of all phylogenetic analyses based
on this or any other sequencing method is restricted by the sequence sampling and
availability in public domain. Because all national laboratories in the Americas are
currently running the same RT-PCR procedures for DENV-1 to DENV-4 detection (33),
the RNA extracts already obtained can be readily sequenced using this procedure.
Therefore, we have deployed this procedure in collaboration with the Pan American
Health Organization, the National Health Institute of Surveillance and Reference in
Mexico, and the Institute Fiocruz in Brazil. These efforts have made it possible to
incorporate genomic surveillance components in 16 public health laboratories in 13
countries in the region.
The success rate of this assay when used directly on diagnostic serum specimens
is limited by the sample quality, i.e., maintenance of the cold chain, the time of
specimen collection (i.e., acute stage 0 to 5 days after onset of symptoms), and the
viral RNA concentration (i.e.,
C
T⬍
30 determined by real-time RT-PCR, although a
low
C
Tvalue may not reflect the integrity of the RNA genome molecule). Further
in
silico
analyses of the primer sequences showed several mismatches with modern
sequences of Asian origin. To address this, we offer a list of the primers with IUPAC
nucleic acid codes in positions where mismatches were detected (Table S2);
how-ever, due to the unavailability of Asian dengue virus isolates in our laboratory, we
were unable to validate these primer sequences with PCR testing. For further
evolutionary and molecular epidemiological studies, we recommend methods that
provide complete genome sequence.
In conclusion, surveillance and research laboratories can use this method to
collect dengue genomic information to strengthen the understanding of DENV
genotypes, diversity, and transmission dynamics and to improve disease prevention
strategies.
on May 17, 2020 by guest
http://jcm.asm.org/
SUPPLEMENTAL MATERIAL
Supplemental material for this article may be found at
https://doi.org/10.1128/JCM
.01957-18
.
SUPPLEMENTAL FILE 1
, XLSX file, 0.01 MB.
ACKNOWLEDGMENT
G.A.S. is a research scientist at the Centers for Disease Control and Prevention,
Dengue Branch, San Juan, Puerto Rico. His research is focused on the development
of molecular diagnostic tests and the study of the molecular epidemiology of
arboviruses.
REFERENCES
1. Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GR, Simmons CP, Scott TW, Farrar JJ, Hay SI. 2013. The global distribution and burden of dengue. Nature 496:504 –507.https://doi.org/ 10.1038/nature12060.
2. Gubler DJ. 2006. Dengue/dengue haemorrhagic fever: history and cur-rent status. Novartis Found Symp 277:3–16. discussion 16-22, 71-3, 251-3.
3. Leduc-Galindo D, Rincón-Herrera U, Ramos-Jiménez J, Garcia-Luna S, Arellanos-Soto D, Mendoza-Tavera N, Tavitas-Aguilar I, Garcia-Garcia E, Galindo-Galindo E, Villarreal-Perez J, Fernandez-Salas I, Santiago GA, Muñoz-Jordan J, Rivas-Estilla AM. 2015. Characterization of the dengue outbreak in Nuevo Leon state, Mexico, 2010. Infection 43:201–206.
https://doi.org/10.1007/s15010-014-0700-7.
4. Thomas DL, Santiago GA, Abeyta R, Hinojosa S, Torres-Velasquez B, Adam JK, Evert N, Caraballo E, Hunsperger E, Munoz-Jordan JL, Smith B, Banicki A, Tomashek KM, Gaul L, Sharp TM. 2016. Reemergence of dengue in southern Texas, 2013. Emerg Infect Dis 22:1002–1007.https:// doi.org/10.3201/eid2206.152000.
5. Munoz-Jordan JL, Santiago GA, Margolis H, Stark L. 2013. Genetic relat-edness of dengue viruses in Key West, Florida, USA, 2009-2010. Emerg Infect Dis 19:652– 654.https://doi.org/10.3201/eid1904.121295. 6. Guzman MG, Halstead SB, Artsob H, Buchy P, Farrar J, Gubler DJ, Hunsperger
E, Kroeger A, Margolis HS, Martinez E, Nathan MB, Pelegrino JL, Simmons C, Yoksan S, Peeling RW. 2010. Dengue: a continuing global threat. Nat Rev Microbiol 8:S7–S16.https://doi.org/10.1038/nrmicro2460.
7. Rico-Hesse R. 2003. Microevolution and virulence of dengue viruses. Adv Virus Res 59:315–341.https://doi.org/10.1016/S0065-3527(03)59009-1. 8. Holmes EC, Twiddy SS. 2003. The origin, emergence and evolutionary
genetics of dengue virus. Infect Genet Evol 3:19 –28.https://doi.org/10 .1016/S1567-1348(03)00004-2.
9. Rico-Hesse R. 1990. Molecular evolution and distribution of dengue viruses type 1 and 2 in nature. Virology 174:479 – 493.https://doi.org/ 10.1016/0042-6822(90)90102-W.
10. Leitmeyer KC, Vaughn DW, Watts DM, Salas R, Villalobos I, de C, Ramos C, Rico-Hesse R. 1999. Dengue virus structural differences that correlate with pathogenesis. J Virol 73:4738 – 4747.
11. Cologna R, Armstrong PM, Rico-Hesse R. 2005. Selection for virulent dengue viruses occurs in humans and mosquitoes. J Virol 79:853– 859.
https://doi.org/10.1128/JVI.79.2.853-859.2005.
12. OhAinle M, Balmaseda A, Macalalad AR, Tellez Y, Zody MC, Saborio S, Nunez A, Lennon NJ, Birren BW, Gordon A, Henn MR, Harris E. 2011. Dynamics of dengue disease severity determined by the interplay be-tween viral genetics and serotype-specific immunity. Sci Transl Med 3:114ra128.https://doi.org/10.1126/scitranslmed.3003084.
13. Armstrong PM, Rico-Hesse R. 2001. Differential susceptibility ofAedes aegyptito infection by the American and Southeast Asian genotypes of dengue type 2 virus. Vector Borne Zoonotic Dis 1:159 –168.https://doi .org/10.1089/153036601316977769.
14. Anderson JR, Rico-Hesse R. 2006. Aedes aegypti vectorial capacity is determined by the infecting genotype of dengue virus. Am J Trop Med Hyg 75:886 – 892.https://doi.org/10.4269/ajtmh.2006.75.886.
15. Villabona-Arenas CJ, Zanotto PM. 2013. Worldwide spread of dengue virus type 1. PLoS One 8:e62649.https://doi.org/10.1371/journal.pone .0062649.
16. Manokaran G, Finol E, Wang C, Gunaratne J, Bahl J, Ong EZ, Tan HC, Sessions OM, Ward AM, Gubler DJ, Harris E, Garcia-Blanco MA, Ooi EE.
2015. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 350:217–221.https://doi .org/10.1126/science.aab3369.
17. de Melo FL, Romano CM, de Andrade Zanotto PM. 2009. Introduction of dengue virus 4 (DENV-4) genotype I into Brazil from Asia? PLoS Negl Trop Dis 3:e390.https://doi.org/10.1371/journal.pntd.0000390. 18. McElroy KL, Santiago GA, Lennon NJ, Birren BW, Henn MR, Munoz JJL.
2011. Endurance, refuge, and reemergence of dengue virus type 2, Puerto Rico, 1986 –2007. Emerg Infect Dis 17:64 –71.https://doi.org/10 .3201/eid1701.100961.
19. Santiago GA, McElroy-Horne K, Lennon NJ, Santiago LM, Birren BW, Henn MR, Muñoz-Jordán JL. 2012. Reemergence and decline of dengue virus serotype 3 in Puerto Rico. J Infect Dis 206:893–901.https://doi.org/10 .1093/infdis/jis426.
20. Carneiro AR, Cruz ACR, Vallinoto M, Melo D. d V, Ramos RTJ, Medeiros DBA, Silva E. V P d, Vasconcelos P. F d C. 2012. Molecular characterization of dengue virus type 1 reveals lineage replacement during circulation in Brazilian territory. Mem Inst Oswaldo Cruz 107:805– 812.https://doi.org/ 10.1590/S0074-02762012000600016.
21. Dash PK, Sharma S, Soni M, Agarwal A, Parida M, Rao PV. 2013. Complete genome sequencing and evolutionary analysis of Indian isolates of dengue virus type 2. Biochem Biophys Res Commun 436:478 – 485.
https://doi.org/10.1016/j.bbrc.2013.05.130.
22. Koo C, Nasir A, Hapuarachchi HC, Lee KS, Hasan Z, Ng LC, Khan E. 2013. Evolution and heterogeneity of multiple serotypes of dengue virus in Pakistan, 2006 –2011. Virol J 10:275.https://doi.org/10.1186/1743-422X -10-275.
23. Kumar SR, Patil JA, Cecilia D, Cherian SS, Barde PV, Walimbe AM, Yadav PD, Yergolkar PN, Shah PS, Padbidri VS, Mishra AC, Mourya DT. 2010. Evolution, dispersal, and replacement of American genotype dengue type 2 viruses in India (1956 –2005): selection pressure and molecular clock analyses. J Gen Virol 91:707–720. https://doi.org/10.1099/vir.0 .017954-0.
24. Patil JA, Cherian S, Walimbe AM, Patil BR, Sathe PS, Shah PS, Cecilia D. 2011. Evolutionary dynamics of the American African genotype of den-gue type 1 virus in India (1962–2005). Infect Genet Evol 11:1443–1448.
https://doi.org/10.1016/j.meegid.2011.05.011.
25. Patil JA, Cherian S, Walimbe AM, Bhagat A, Vallentyne J, Kakade M, Shah PS, Cecilia D. 2012. Influence of evolutionary events on the Indian subcontinent on the phylogeography of dengue type 3 and 4 viruses. Infect Genet Evol 12:1759 –1769.https://doi.org/10.1016/j.meegid.2012 .07.009.
26. Koo C, Tien WP, Xu H, Ong J, Rajarethinam J, Lai YL, Ng LC, Hapuara-chchi HC. 2018. Highly selective transmission success of dengue virus type 1 lineages in a dynamic virus population: an evolutionary and fitness perspective. iScience 6:38 –51. https://doi.org/10.1016/j.isci .2018.07.008.
27. Allicock OM, Lemey P, Tatem AJ, Pybus OG, Bennett SN, Mueller BA, Suchard MA, Foster JE, Rambaut A, Carrington CV. 2012. Phylogeogra-phy and population dynamics of dengue viruses in the Americas. Mol Biol Evol 29:1533–1543.https://doi.org/10.1093/molbev/msr320. 28. Bennett SN, Holmes EC, Chirivella M, Rodriguez DM, Beltran M, Vorndam
V, Gubler DJ, McMillan WO. 2006. Molecular evolution of dengue 2 virus in Puerto Rico: positive selection in the viral envelope accompanies clade reintroduction. J Gen Virol 87:885– 893.https://doi.org/10.1099/vir .0.81309-0.
29. Klungthong C, Putnak R, Mammen MP, Li T, Zhang C. 2008. Molecular
on May 17, 2020 by guest
http://jcm.asm.org/
genotyping of dengue viruses by phylogenetic analysis of the sequences of individual genes. J Virol Methods 154:175–181. https://doi.org/10 .1016/j.jviromet.2008.07.021.
30. Zhang H, Zhang Y, Hamoudi R, Yan G, Chen X, Zhou Y. 2014. Spatio-temporal characterizations of dengue virus in mainland China: insights into the whole genome from 1978 to 2011. PLoS One 9:e87630.https:// doi.org/10.1371/journal.pone.0087630.
31. Twiddy SS, Holmes EC, Rambaut A. 2003. Inferring the rate and time-scale of dengue virus evolution. Mol Biol Evol 20:122–129.https://doi .org/10.1093/molbev/msg010.
32. Medina F, Medina JF, Colon C, Vergne E, Santiago GA, Munoz-Jordan JL. 2012. Dengue virus: isolation, propagation, quantification, and storage. Curr Protoc Microbiol Chapter 15:Unit 15D.2.
33. Santiago GA, Vergne E, Quiles Y, Cosme J, Vazquez J, Medina JF, Medina F, Colon C, Margolis H, Munoz JJL. 2013. Analytical and clinical perfor-mance of the CDC real time RT-PCR assay for detection and typing of dengue virus. PLoS Negl Trop Dis 7:e2311. https://doi.org/10.1371/ journal.pntd.0002311.
34. Villabona-Arenas CJ, Miranda-Esquivel DR, Jimenez RE. 2009. Phylogeny of dengue virus type 3 circulating in Colombia between 2001 and 2007. Trop Med Int Health 14:1241–1250.https://doi.org/10.1111/j.1365-3156 .2009.02339.x.
35. Weaver SC, Vasilakis N. 2009. Molecular evolution of dengue viruses: contributions of phylogenetics to understanding the history and epide-miology of the preeminent arboviral disease. Infect Genet Evol 9:523–540.https://doi.org/10.1016/j.meegid.2009.02.003.