DOI: 10.1534/genetics.109.110155
Budding Yeast Dbf4 Sequences Required for Cdc7 Kinase Activation
and Identification of a Functional Relationship Between the Dbf4
and Rev1 BRCT Domains
Victoria Harkins,
1,2Carrie Gabrielse,
1Louise Haste
3and Michael Weinreich
4 Laboratory of Chromosome Replication, Van Andel Research Institute, Grand Rapids, Michigan 49503Manuscript received September 22, 2009 Accepted for publication October 1, 2009
ABSTRACT
Cdc7-Dbf4 is a two-subunit kinase required for initiating DNA replication. The Dbf4 regulatory subunit is required for Cdc7 kinase activity. Previous studies have shown that the C termini of Dbf4 orthologs encode a single (putative) C2H2zinc (Zn) finger, referred to as ‘‘motif C.’’ By mutational analysis we show that the Zn finger is not required for the essential function of Dbf4. However, deletion and point mutants altering conserved Zn-finger residues exhibit a substantially slowed S-phase, DNA damage sensitivity, and a hypo-mutagenic phenotype following UV irradiation. Using two-hybrid and biochemical assays, we show that the Dbf4 Zn finger interacts with Cdc7 and stimulates its kinase activity. However, a separable Dbf4 region also mediates an interaction with Cdc7 such that only the loss of both Cdc7-interacting regions results in lethality. In contrast, an N-terminal BRCT-like domain is not required for induced mutagenesis nor does it interact with Cdc7. By making chimeric Dbf4 proteins that contain known BRCT domains in
Saccharomyces cerevisiae, we show that the BRCT domain from Rev1, a translesion DNA polymerase, can uniquely substitute for the Dbf4 BRCT domain. Thus, we have mapped regions on budding yeast Dbf4 required for binding and activating Cdc7 kinase. Our data also suggest that the Dbf4 and Rev1 BRCT domains interact with a common protein or structure, although the precise function of both domains and their binding partners remains elusive.
A
two-subunit serine/threonine kinase, Cdc7-Dbf4, is required for initiating DNA replication in all eukaryotes (for reviews, see Sclafani 2000; Bell and Dutta2002). The Dbf4 regulatory protein is required for Cdc7 kinase activation and forms a stable two-subunit complex with Cdc7. Cdc7 protein is expressed at constant levels during the cell cycle, but Dbf4 protein levels are cyclical and peak near the G1/S phasetransition ( Jackson et al. 1993; Cheng et al. 1999; Oshiro et al. 1999; Weinreich and Stillman 1999; Ferreiraet al.2000; Sullivanet al.2008). Since Cdc7 kinase activity parallels Dbf4 protein abundance, it is thought that Dbf4 binding to Cdc7 is the primary mechanism for activating its kinase activity. Numerous data strongly suggest that Cdc7-Dbf4 is recruited to replication origins to promote DNA synthesis by activating the MCM helicase (Hardy and Pautz1996;
Lei et al. 1997; Sato et al. 1997; Weinreich and Stillman 1999; Sheu and Stillman 2006; Francis et al.2009). The MCM helicase is assembled at all origins during G1phase in an inactive form and then is activated
by a Cdc7-Dbf4-dependent step, which triggers origin unwinding and the initiation of DNA synthesis (Geraghty et al. 2000). The precise molecular nature of this step is unknown although Cdc7-Dbf4 kinase is required for origin recruitment of two additional initiation proteins, Cdc45 and the GINS complex, that bind to the MCM helicase (Owenset al.1997; Zouand Stillman2000; Kanemakiet al.2003; Masaiet al.2006; Yabuuchiet al.2006). Cdc45, MCM, and GINS, termed the ‘‘CMG complex,’’ are required for the initiation of DNA synthesis but also travel with the replication fork and are thought to compose the replicative helicase (Gambuset al.2006; Moyeret al.2006; Paceket al.2006). MCM and Cdc45 proteins are in vitro Cdc7-Dbf4 substrates (Weinreich and Stillman 1999; Kihara et al. 2000; Francis et al. 2009), suggesting that phos-phorylation of one or more CMG proteins promotes helicase activation.
The function of the Dbf4 regulatory subunit was discovered following its isolation as a high-copy sup-pressor of thecdc7-1temperature-sensitive (ts) allele in budding yeast (Kitada et al. 1992). Johnston and Thomas(1982) had previously isolateddbf4
temperature-Supporting information is available online athttp://www.genetics.org/ cgi/content/full/genetics.109.110155/DC1.
1These authors contributed equally to this work.
2Present address:Zoology Department, Michigan State University, East Lansing, MI 48823.
3Present address: Microbiology Ely, Thermo Fisher Scientific, Ely, Cambridgeshire CB7 4ET, United Kingdom.
4Corresponding author:Van Andel Research Institute, 333 Bostwick Ave. NE, Grand Rapids, MI 49503. E-mail: [email protected]
sensitive alleles in a screen for new DNA replication mutants. ‘‘Dbf4’’ denotesdumbbellformer4, because, upon elevation to the nonpermissive temperature,dbf4 mutants arrest as large-budded cells or ‘‘dumbbells’’ with incompletely replicated DNA, a common cell-cycle arrest phenotype for DNA replication mutants. Sub-sequently,DBF4orthologs were isolated in Schizosacchar-omyces pombe (dfp11
) (Brown and Kelly 1998), Aspergillus nidulans(nimO) ( Jameset al.1999),Drosophila melanogastor(chiffon) (Landisand Tower1999),Xenopus laevis(XDBF4) (Furukohriet al.2003), mice (MmDbf4) (Lepke et al. 1999), and humans ( Jiang et al. 1999; Kumagaiet al.1999), where it is referred to asHsDBF4or ‘‘ASK’’ foractivator ofS-phase kinase. All Dbf4 proteins bind to the Cdc7 kinase subunit and are required for its kinase activity. Multiple sequence alignment of Dbf4 proteins across species revealed low sequence conserva-tion except in three regions, referred to as motifs N, M, or C, denoting their position within the protein (Figure 1) (Masaiand Arai2000).
Genetic and comparative sequence-based evidence suggests that Dbf4 orthologs encode a single BRCT-like domain that overlaps motif N (Masaiand Arai 2000; Gabrielseet al. 2006). BRCT domains are present in many proteins that respond to DNA damage and were first discovered as a tandemly repeated sequence in the C terminus of the breast cancer susceptibility gene BRCA1 (Koonin et al. 1996), although they are also present in single copy (Borket al.1997). Tandem BRCT domains have been shown to bind phosphorylated proteins (reviewed by Glover et al. 2004). In the absence of structural information on any Cdc7-Dbf4 protein, it is not known if this Dbf4 region resembles a BRCT fold. Since deletion of this region in budding and fission yeasts results in sensitivity to a wide variety of DNA-damaging agents, it is thought that this region of Dbf4 plays a role in the response to DNA damage or replication fork arrest (Ogino et al.2001; Funget al. 2002; Gabrielse et al. 2006). Indeed, Dfp1 has been shown to interact with Swi1 (budding yeast Tof1), which is a component of the replication fork, suggesting that Dbf4 might be targeted to replication forks (Matsumotoet al. 2005). InSaccharomyces cerevisiae, however, mutants that precisely delete only the BRCT-like domain or more extensive N-terminal deletion mutants that restore a deleted nuclear localization sequence (NLS) exhibit little DNA damage sensitivity and are only moderately sensitive to replication fork stalling by the drug hydroxyurea (HU) (Gabrielseet al.2006). Interestingly, loss of the BRCT-like sequence is synthetically lethal withrad53-1 (encod-ing the Chk2 checkpoint kinase) and confers a defect in activating late vs. early replication origins (Gabrielse et al.2006). Together, these and other findings suggest that the BRCT-like domain does not play a prominent role in DNA damage repair under normal circumstances but may instead regulate kinase targeting to early vs. late origins. In addition, Dbf4 may have a redundant role at
stalled replication forks or may participate in a Rad53-mediated checkpoint under conditions yet to be defined. Interestingly, nearly 30 years agocdc7 mutants were reported to be defective in induced mutagenesis (also known as error-prone repair, one pathway of post-replicative repair, or PRR) (Njagiand Kilbey1982a,b). It is now known thatCDC7functions in theRAD6epistasis pathway for PRR (Pessoa-Brandaoand Sclafani2004). Error-prone repair occurs when the replicative DNA polymerase encounters a bulky lesion that cannot be accommodated within the polymerase active site, the lesion is by-passed and instead replicated across by lower-fidelity translesion (TLS) DNA polymerases such as Rev1 or Rev3-7 (Polz) (reviewed by Waterset al.2009). It is not known how Cdc7-Dbf4 promotes error-prone repair or whether Dbf4 targets the kinase to sites of replication fork stalling or directly to DNA lesions.
Dbf4 binds to Cdc7 to activate its kinase activity. A detailed mutational analysis ofS. pombe Dfp1 revealed that Dfp1 binds to the Hsk1 (Cdc7) subunit using motifs M and C (Oginoet al.2001). This analysis also revealed that there is a spacer region between motifs M and C (150 residues) that is not required for the mitotic function of Dfp1. Hsk1 kinase activity was stimulated by wild-type Dfp1 but was compromised by using Dfp1 mutants lacking motif M or motif C (Oginoet al.2001; Funget al.2002), and mutations within motif M caused methyl methanesulfonate (MMS) and HU sensitivity (Takeda et al. 1999). Evidence from two-hybrid assays also indicated thatS. cerevisiaeDbf4 could interact with Cdc7 using residues spanning motifs M or C (Dowell et al.1994; Hardyand Pautz1996). Human ASK also uses motifs M and C to interact with Cdc7 (Satoet al. 2003). The short, 40-residue region known as motif M does not encode a known protein domain; however, motif C encodes a C2H2-type Zn finger (Masaiand Arai
2000). Although initially defined as DNA-binding mod-ules, there are.20 types of Zn fingers that are involved in protein–protein interactions, DNA binding, RNA binding, ubiquitin binding, and other functions (re-viewed by Brayerand Segal2008).
domains in budding yeast, we find that the Rev1 BRCT domain functionally substitutes for the Dbf4 BRCT-like domain. Paradoxically, although deletion of the Dbf4 N-terminal BRCT-like domain does not result in an error-prone repair defect, we find that the BRCT domain from a known translesion DNA polymerase suppresses the associated defects in Dbf4 activity. Since the BRCT domain from mouse Rev1 promotes targeting to repli-cation foci (Guoet al.2006), this raises the possibility that yeast Rev1 and Dbf4 are targeted to the replication fork via their respective BRCT domains and may interact with the same protein.
MATERIALS AND METHODS
Construction of yeast strains, plasmids, baculoviruses, and growth media:All yeast strains (with the exception of PJ69-4A and KKY02C) are derivatives of W303 (R. Rothstein, Columbia
University) or were backcrossed at least four times to W303 from the parental strain and are listed in Table 1. Genetic manipulation and transformation of yeast was done using standard techniques. YPD denotes rich medium containing 1% yeast extract, 2% peptone, and 2% glucose. We used FOA, a synthetic complete medium containing 1 mg/ml 5-fluoroorotic acid. Drugs were added directly to media before pouring at the indicated concentrations.
All plasmids used in this study are listed in the supporting information, Table S1. DBF4deletions and point mutations were constructed by QuikChange (Stratagene, La Jolla, CA) on pMW489. pMW489 is pRS415 containing DBF4 on a genomic 2.5-kbMluI–XbaI fragment. For each mutation, the entireDBF4sequence on the plasmid was verified by sequenc-ing. BRCT-Dbf4 chimeras were constructed by amplifying single or tandem BRCT repeats from genomic DNA (Bork et al.1997) and were cloned into pCG29 (dbf4-ND221) on an
NcoI fragment. pVMH14 (CHS5) contains residues 161–273, pCG46 (DNL4, 682–942), pCG48 (DPB11, 1–228), pCG165 (DPB11, 306–592), pCG95 (FCP1, 495–595), pCG94 (NOP7, 350–454), pCG51 (POL4, 1–125), pCG45 (RAD9, 1027–1297), TABLE 1
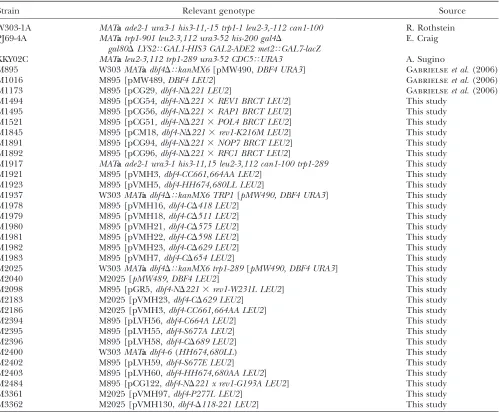
Yeast strains
Strain Relevant genotype Source
W303-1A MATaade2-1 ura3-1 his3-11,-15 trp1-1 leu2-3,-112 can1-100 R. Rothstein PJ69-4A MATatrp1-901 leu2-3,112 ura3-52 his-200 gal4D
gal80DLYS2TGAL1-HIS3 GAL2-ADE2 met2TGAL7-lacZ
E. Craig
KKY02C MATaleu2-3,112 trp1-289 ura3-52 CDC5TURA3 A. Sugino
M895 W303MATadbf4DTkanMX6[pMW490,DBF4 URA3] Gabrielseet al.(2006)
M1016 M895 [pMW489,DBF4 LEU2] Gabrielseet al.(2006)
M1173 M895 [pCG29,dbf4-ND221 LEU2] Gabrielseet al.(2006)
M1494 M895 [pCG54,dbf4-ND2213REV1 BRCT LEU2] This study
M1495 M895 [pCG56,dbf4-ND2213RAP1 BRCT LEU2] This study
M1521 M895 [pCG51,dbf4-ND2213POL4 BRCT LEU2] This study
M1845 M895 [pCM18,dbf4-ND2213rev1-K216M LEU2] This study
M1891 M895 [pCG94,dbf4-ND2213NOP7 BRCT LEU2] This study
M1892 M895 [pCG96,dbf4-ND2213RFC1 BRCT LEU2] This study
M1917 MATaade2-1 ura3-1 his3-11,15 leu2-3,112 can1-100 trp1-289 This study
M1921 M895 [pVMH3,dbf4-CC661,664AA LEU2] This study
M1923 M895 [pVMH5,dbf4-HH674,680LL LEU2] This study
M1937 W303MATadbf4DTkanMX6 TRP1[pMW490, DBF4 URA3] This study
M1978 M895 [pVMH16,dbf4-CD418 LEU2] This study
M1979 M895 [pVMH18,dbf4-CD511 LEU2] This study
M1980 M895 [pVMH21,dbf4-CD575 LEU2] This study
M1981 M895 [pVMH22,dbf4-CD598 LEU2] This study
M1982 M895 [pVMH23,dbf4-CD629 LEU2] This study
M1983 M895 [pVMH7,dbf4-CD654 LEU2] This study
M2025 W303MATadbf4DTkanMX6 trp1-289[pMW490, DBF4 URA3] This study
M2040 M2025 [pMW489, DBF4 LEU2] This study
M2098 M895 [pGR5,dbf4-ND2213rev1-W231L LEU2] This study
M2183 M2025 [pVMH23,dbf4-CD629 LEU2] This study
M2186 M2025 [pVMH3,dbf4-CC661,664AA LEU2] This study
M2394 M895 [pLVH56,dbf4-C664A LEU2] This study
M2395 M895 [pLVH55,dbf4-S677A LEU2] This study
M2396 M895 [pLVH58,dbf4-CD689 LEU2] This study
M2400 W303MATadbf4-6(HH674,680LL) This study
M2402 M895 [pLVH59,dbf4-S677E LEU2] This study
M2403 M895 [pLVH60,dbf4-HH674,680AA LEU2] This study
M2484 M895 [pCG122,dbf4-ND221 x rev1-G193A LEU2] This study
M3361 M2025 [pVMH97,dbf4-P277L LEU2] This study
pCG56 (RAP1, 121–209), pCG54 (REV1, 160–251), and pCG96 (RFC1, 148–243).
Baculovirus transfer plasmids encoding DBF4 deletions (on anNcoI–NotI fragment) were constructed in pAcSG2, and baculoviruses were generated using the Baculo Gold kit (BD Biosciences), plaque purified, and then amplified to high titer. The wild-type DBF4 andHA-CDC7 viruses have been described previously (Weinreichand Stillman1999). The dbf4-6 mutant (HH674,680LL) was integrated at the
dbf4DTkanMX6locus of M895 [dbf4D/pMW490 (DBF4 URA3)] by cotransformation of aHindIII–XbaI fragment containing full-lengthdbf4-HH674,680LL together with pRS415. Leu1
trans-formants were replica plated to FOA. Multiple FOA-resistant colonies were recovered to YPD plates and then tested on YPD plates containing 0.2 mg/ml geneticin to score loss of the
dbf4DTkanMX6 marker. The resulting GenS candidates were confirmed as correct recombinants following PCR amplification of theDBF4locus, tested for temperature sensitivity, and back-crossed to W303.
Cell-cycle analysis and preparation of whole-cell extracts: FACS analysis of DNA content was as described (Weinreich and Stillman1999). Whole-cell extracts were prepared using a trichloroacetic acid extraction method from 10 ml of cells (Foianiet al.1994). Five percent of the whole-cell extract was separated on a 10% or 15% SDS–PAGE gel, blotted, and probed with rabbit polyclonal antisera against GST-Cdc7 (1:4000), GST-Dbf4 (1:1000) (Weinreich and Stillman 1999), or yeast anti-Gal4AD (1:1000, Upstate Cell Signaling) followed by anti-rabbit HRP (1:10,000) in 13 phosphate-buffered saline containing 0.1% Tween and 1% dry milk. To detect Dbf4 proteins in Figure 5A (lanes 1–7), we used a rabbit polyclonal antisera raised against an N-terminal Dbf4 peptide spanning residues 142–153. Ponceau S staining of the blot con-firmed equal protein loading in the samples being compared. UV reversion assay: The procedure was derived from Lawrence and Christensen (1976) and other sources. Briefly, triplicate cultures containingtrp1-289were grown in YPD media to saturation and adjusted to the same number of cells per milliliter in water. Cells were diluted, irradiated with increasing UV fluences up to 80 J/m2, and plated onto SCM complete plates to score survival or onto SCM Trp plates to score TRP1colonies. Plates were kept in the dark for 3 days at 30°(4 days at 25°for thedbf4-1ts mutant) and then counted. Reversion frequencies were calculated relative to surviving cells at each UV fluence, averaging two independent experi-ments (i.e., six measurements).
Immunoprecipitation and kinase assay of Cdc7-Dbf4 pro-tein from Sf9 cells:Sf9 cells were co-infected at an MOI of 10 with baculoviruses expressing HA-Cdc7 and Dbf4 derivatives. After 48 hr, soluble whole-cell extracts were prepared and immunoprecipitated using a 12CA5 monoclonal antibody against the HA epitope. After extensive washing, the immu-noprecipitation (IP) was split in half. Half of the IP was incubated for 30 min at 30°in kinase buffer (50 mmTris–HCl, pH 7.5, 10 mm MgCl2, 1 mm DTT, 0.1 mm ATP, 10 mCi [g-32P]ATP). These samples were then separated on parallel 10% SDS–PAGE gels and either blotted and probed with Cdc7 and Dbf4 polyclonal antisera, as described above, or subjected to autoradiography.
RESULTS
Motif C is not essential for Dbf4 activity but is required for growth under stress conditions: To de-termine the Dbf4 C-terminal residues that were essential for activity, we tested a series of truncation and point
mutants spanning the C-terminal 392 amino acids of Dbf4 for their ability to rescue the viability of adbf4DTkanMX6 yeast strain by plasmid shuffling (Figure 1). Interestingly, all of the deletion mutants up to residue 418 behaved similarly in that they complemented viability but grew more poorly at 25° than wild type and were strictly temperature sensitive at 37°(Figure 1, B and C). Dele-tions that removed C-terminal residues up to 357 or 312 did not complement viability, indicating that they re-moved essential Dbf4 residues (Figure 1B). An internal deletion removing residues 312–418 also did not com-plement viability (not shown), indicating that these residues were indeed essential for Dbf4 activity and not just when the remainder of the C terminus (including motif C) was also deleted. In addition, all of the viable C-terminal deletion mutants were substantially more sensitive to the DNA-damaging agents HU, bleomycin, and MMS that cause replication fork arrest, double-strand breaks, and alkylation damage, respectively (Figure 1C).
The Zn-finger domain becomes essential when combined with additional perturbations in Dbf4 structure: To determine if the conserved ‘‘motif N’’ and motif M regions were required for Dbf4 activity in the absence of a functional Zn-finger domain, we combined several C-terminal truncations as well as a CC661,664AA mutant affecting the conserved cysteines in the Zn finger with mutations affecting motifs N or M. We found that thedbf4-1point mutation P277L within motif M and both N-terminal and internal deletions removing the BRCT-like region were lethal in combina-tion with all C-terminal mutacombina-tions (Figure 2). Unex-pectedly, although an N-terminal deletion removing the first 65 amino acids of Dbf4 was tolerated, a further deletion to amino acid 109 was also lethal in combina-tion with the C-terminal mutants. The dbf4-ND109 mutant retains the BRCT-like sequence (including motif N) and motif M, has essentially wild-type growth characteristics, kinase activity, and S-phase progression and activates replication origins as does the wild type (Gabrielseet al.2006). As far as we have determined, this mutant behaves exactly as does the wild type with the exception that it removes a nonessential region that interacts with Polo kinase to inhibit mitotic exit (Miller et al. 2009). Therefore, it is possible that, because of their slowed S-phase (see below), the C-terminal mu-tants activate fewer replication origins and depend upon G2/M checkpoint mechanisms to delay entry into
or exit from mitosis. In any event, further moderate perturbations of Dbf4 function cause lethality in the absence of the Zn-finger domain, probably due to a significant defect in kinase activation (see below).
The Dbf4 Zn finger most closely resembles a classical C2H2-type domain and physically interacts
are absolutely conserved as are several additional residues such as G659, E662, F683, and D694 (Figure 3A, blue type), as shown previously (Masai and Arai 2000). A number of additional residues are also highly conserved as well as an S/T residue between the two histidines. DNA-binding Zn-finger proteins typically contain several tandem fingers with the consensus (F/ Y)-X-C-X(2-5)-C-X3-(F/Y)-X5-C-X2-H-X(3-5)-H, whereCis
a hydrophobic residue (Wolfe et al. 2000). However, Dbf4 contains a single Zn finger with the consensus G-Y-C-E-X-C-X3-(F/Y)-X2-C-E-X-H-X5-H-X2-F. This
consen-sus resembles the C2H2-type DNA-binding Zn finger
with several important differences: (1) Dbf4 orthologs have a 9-amino-acid spacing between the last C and first H (instead of 12); (2) they have an aromatic residue immediately preceding the first C (instead of two residues before); and (3) Zn fingers that bind DNA typically have conserved basic and acidic residues that bind the phosphate backbone and bases, but these are not in the correct positions for the Dbf4-type Zn finger (Wolfeet al.2000), suggesting that it might not mediate an interaction with DNA. On the basis of crystallo-graphic information there are at least eight different structural classes of Zn fingers that function as protein– protein interaction domains and at least three of these bind the small-protein modifier ubiquitin (Krishna et al.2003). Inspection of these classes reveals that the Dbf4 Zn finger most closely resembles a classical TFIIIA-type Zn-finger domain; however, on the basis of the reasons cited above it might bind a protein instead of DNA.
Since Dbf4 contains a C2H2-type Zn finger that
defines motif C, we tested the importance of conserved residues within the Zn finger for Dbf4 activity. We made two point mutants that altered either both cysteine or both histidine residues within the Zn finger and tested their ability to complement the strain deleted forDBF4. The point mutants complemented the dbf4D but had phenotypes that were essentially as severe as the C-terminal deletion mutants (Figure 3B). The CC661, 664AA and HH674,680LL mutants were temperature sensitive and extremely sensitive to bleomycin and MMS, although they showed only a mild HU-sensitive pheno-type. An HH674,680AA mutant and a single C664A mutant had the same phenotypes, indicating that the conserved Zn-binding residues are critical for Dbf4 activity. A small 15-residue C-terminal deletion (D689-704) beyond the C2H2 cluster gave the same pattern of sensitivities,
indicating that the entire conserved region of 40 residues is important for Dbf4 activity. Finally, it was possible that the highly conserved S/T residue (S667 in budding yeast) between the two histidine residues could function as a phosphorylation site to impact Dbf4 activity. However, mutation of S677 to Ala or Glu (a phospho-mimic) had no effect on Dbf4 growth or sensitivity to DNA-damaging agents (Figure 3B).
We have described new dbf4 temperature-sensitive alleles, so it is informative to compare these to the previously describeddbf4ts alleles:dbf4-1,dbf4-2,dbf4-3, dbf4-4, anddbf4-5(Kiharaet al.2000). Thedbf4-1,dbf4-2, anddbf4-3alleles have mutations within motif M: P277L, P308L, and P308S, respectively. However, dbf4-4 con-Figure1.—The Dbf4 Zn finger is required for growth under stress condi-tions. (A) Schematic of Dbf4 showing the BRCT-like domain and motifs N, M, and C together with a de-scription of the C-terminal mutants. (B) Single-copy plasmids (LEU21)
contain-ing the indicated Dbf4 C -terminal deletions were transformed into M895 [dbf4D/pMW490 (DBF4 URA3)] and tested for their ability to complementdbf4D
tains a truncating mutation at residue 596 anddbf4-5 contains a E415K mutation in addition to a W112 nonsense mutation (TGG. TAA) that is predicted to allow formation of an N-terminally truncated protein beginning at the M120 codon but containing E415K. In summary, our newdbf4ts alleles result from the loss of a functional Zn finger either by point mutation or by a deletion analogous to that found in dbf4-4. Further-more, residues 312–418 are essential for Dbf4 function, suggesting that the E415K mutation within thedbf4-5ts allele affects that (unknown) function.
We next determined whether the Zn-finger domain mutants behaved similarly on the chromosome. Since thedbf4-4mutant encodes a representative C-terminal truncation and gives a ts phenotype, we integrated the dbf4-HH674,680LL point mutant at the endogenous DBF4locus. This allele (that we now refer to asdbf4-6) gave very similar phenotypes to the allele when present on a plasmid (Figure 3C), including a tight ts pheno-type. The temperature and DNA damage sensitivities of dbf4-6 were complemented by wild-type DBF4 as ex-pected but also by high-copy CDC7 (Figure 3C). This raises the possibility that mutations in the Dbf4 Zn finger affected the interaction with Cdc7 because in-creasedCDC7copy number rescued thedbf4-6 pheno-types. Below we show that the Zn finger interacts with Cdc7.
Dbf4 C-terminal mutations cause a significant delay in S-phase progression and are defective in UV-induced mutagenesis: We tested whether mutations within the Dbf4 C terminus affected S-phase progression by flow cytometric analysis of DNA content. Dbf4 is required for the initiation of DNA replication, and mutations
affect-ing the essential function of eitherCDC7orDBF4affect entry into and progression through S-phase ( Johnston and Thomas1982; Woodand Hartwell1982). Cdc7-Dbf4 also affects S-phase progression since it is needed throughout S-phase to activate DNA synthesis from individual replication origins (Bousset and Diffley 1998; Donaldson et al.1998). We arrested cells in G1 phase by using mating pheromone and released cells into fresh medium at the permissive temperature of 25°. Wild-type cells initiated DNA replication between 20 and 30 min following the release and had completed S-phase by 50 min (Figure 4B). Thedbf4-CC661,664AA anddbf4-CD629mutants delayed entry into S-phase to between 30 and 50 min following the G1release and did
not complete S-phase until 60 or 75 min following the release, respectively. These data indicate that the Dbf4 Zn finger is required for normal DNA replication, almost certainly by affecting the ability of Cdc7-Dbf4 to initiate DNA replication.
In contrast to the MMS and bleomycin sensitivity,dbf4 mutants affecting the Zn finger were not particularly sensitive to UV irradiation up to 80 J/m2 (Figure 4A, inset). Deletions that removed the N-terminal 65 or 221 residues of Dbf4 also did not result in UV sensitivity (not shown). We then asked whether the BRCT-like or Zn-finger domains might be required for UV-induced mutagenesis since cdc7 hypomorphic mutants are de-fective in this process (Njagiand Kilbey1982a,b). For this purpose, we measured reversion of the ochre trp1-289 allele to Trp11 prototrophy over a range of UV doses. UV-induced cyclobutane prymidine dimers and 6-4 photoproducts are repaired by the DNA transle-sion polymerases Rev1 and Polhin an error-prone and Figure 2.—The Dbf4 C terminus is essential if Dbf4 activity is further impaired. A CC661,664AA point mutation or C-terminal deletions were combined with additional mutations in
DBF4 as indicated, and the resulting mutants were tested for their ability to comple-ment the viability defect of
error-free manner, respectively (reviewed by Prakash et al.2005). Error-prone repair can result in substitution of a sense codon for the nonsense codon oftrp1-289, which in this system results in an2000-fold increase in Trp11 reversion events at 80 J/m2 (Figure 4A). In-terestingly, UV-induced Trp11reversion was abolished by deletion of the Dbf4 C terminus and was substantially diminished by the CC661,664AA mutant. In contrast, an internal deletion (D118-221) that removed the BRCT-like domain or thedbf4-1(P227L) allele within motif M had Trp11 reversion frequencies similar to the wild type. These data indicate that the Dbf4 Zn finger is required for UV-induced mutagenesis but the BRCT-like domain is dispensable for this activity.
Two separable Dbf4 regions direct binding to and activation of the Cdc7 kinase:To determine the regions of Dbf4 required for binding Cdc7 and activating its kinase activity, we expressed HA-Cdc7 together with wild-type or mutant Dbf4 proteins in Sf9 cells. We immunoprecipitated the Cdc7 subunit using an antibody against the HA tag and then examined co-IP of the Dbf4 subunit and also measured Cdc7-Dbf4 kinase activity in the IP (Figure 5A). We previously showed that N-terminal Dbf4 deletions through residue 292 (that partially deletes motif M) bound Cdc7 efficiently and showed little change in Cdc7-Dbf4 auto-phosphorylation or Mcm2 phosphor-ylation (Gabrielse et al. 2006). Cdc7 kinase activity is measured on HA-Cdc7-Dbf4 bound to beads, and we have
noted that Mcm2 phosphorylation (but not Cdc7-Dbf4 auto-phosphorylation) is diminished compared to that seen with the free kinase. In our experimental system, there was no Cdc7 kinase activity in the absence of Dbf4 expression (compare lanes 2 and 3 in Figure 5A). The C-terminal deletions to residue 629 or 418 did not affect Dbf4 binding to Cdc7 but dramatically lowered its kinase activity (lanes 4 and 5). This suggests that the Zn-finger domain plays a substantial role in Cdc7 kinase activation but is not absolutely required for Dbf4-Cdc7 binding. A further deletion mutant to residue 312 (that retains an intact motif M) eliminated the ability of Dbf4 to bind Cdc7 and stimulate its kinase activity (lane 6). There is a critical region between residues 312 and 418 that pro-moted Dbf4 binding to Cdc7 since Dbf4 containing a 312–418 internal deletion also bound very poorly to Cdc7 compared to the wild type and stimulated very little Cdc7 kinase activity (lane 7). The CC661,664AA mutation in the Dbf4 Zn-finger domain did not affect the ability of Dbf4 to bind Cdc7 in the context of full-length Dbf4 but strongly decreased Cdc7-Dbf4 kinase activation (lane 8). The CC661,664AA mutation, however, did affect the ability of Dbf4 to bind Cdc7 when the N-terminal 292 amino acids were deleted, including most of motif M (lane 10). Finally, deletion of the N-terminal 357 amino acids abolished the ability of Dbf4 to bind Cdc7 and stimulate its kinase activity (lane 11). Together, these data indicate that there are two regions of Dbf4 that bind Cdc7 Figure3.—Individual mu-tations in the Dbf4 Zn finger cause sensitivity to stress. (A) Clustal align-ment of the Dbf4 Zn finger among nine orthologs with the budding yeast number-ing above. Sc (S. cerevisiae), Sp (Schizosaccharomyces pombe), Hs (Homo sapiens), Mm (Mus musculus), Xl (Xenopus laevis), Dm ( Dro-sophila melanogastor), An ( As-pergillus nidulans), Cg (Cricetulus griseus), and Ec (Equus caballus). Highly conserved residues are in red and identical residues are in blue. (B) The indi-cated mutants were spotted onto plates as in Figure 1. Wild-type Dbf4 and mutant proteins were expressed at similar levels (Figure S1B) (C) The dbf4-6
temperature-sensitive strain (M2400) was transformed with low- or high-copy vec-tors containing theDBF4or
and stimulate its kinase activity. The Zn-finger domain binds Cdc7 and is required for full stimulation of Cdc7 kinase activity, and a region spanning residues 312–418 also contributes to Cdc7 binding. This second Cdc7-binding region also includes residues from motif M and mediates the primary physical interaction since the Zn finger alone is not sufficient for Cdc7 binding. These results are summarized in Figure 5B.
To confirm whether the Dbf4 Zn finger and/or residues 312–418 interacted independently with Cdc7, we performed a two-hybrid analysis. Dbf4 residues 630– 704 (that included the Zn finger) interacted with Cdc7 by a two-hybrid assay (Figure 5C). This interaction absolutely required the conserved cysteine and histidine residues within the C2H2 Zn finger. Residues 252–418
also bound Cdc7, but residues 312–418 alone could not interact with Cdc7 (Figure 5D). We made several two-hybrid constructs spanning only motif M, but un-fortunately, these were not expressed. A Dbf4 BRCT two-hybrid protein was expressed, but this did not interact with Cdc7 (data not shown). In summary, the two-hybrid data are consistent with our mutational analysis and
together strongly suggest that Dbf4 uses its Zn finger and an extended 160-residue sequence, including motif M, to interact with Cdc7.
The Rev1 BRCT domain can substitute for the Dbf4 BRCT-like domain:In contrast to regions required for binding and activating Cdc7 kinase, a significant Dbf4 N-terminal region (residues 1–265) is largely dispens-able for its replication activity (Gabrielseet al.2006). The Dbf4 N terminus contains a destruction box, two classic NLSs, and residues that bind Cdc5 (Hardyand Pautz1996; Milleret al.2009) and Rad53 (Kihara et al. 2000; Duncker et al. 2002). In addition, Dbf4 contains a BRCT-like domain from residues117–218. Deletion of this motif results in slow growth, DNA damage sensitivity, and slowed progression through S-phase (Gabrielse et al. 2006). However, these phenotypes are likely due to a concomitant decrease in Dbf4 nuclear localization, since a dbf4-ND221 mu-tant that contains a heterologous NLS largely reverses these phenotypes with the exception that the dbf4-NLS-ND221 mutant retains a moderate sensitivity to HU.
Figure4.—Mutations in the Dbf4 Zn finger af-fect UV mutagenesis and cause slow S-phase pro-gression. (A) Wild-type DBF4 (M2040) or the indicated mutants dbf4-CC661,664AA (M2186),
dbf4-CD629 (M2183), dbf4-P277L (M3361), and
dbf4-D118-221 (M3362) were UV irradiated and tested for survival (inset) or Trp1 reversion events. (B) Wild type (M1016), dbf4-CD629
To further investigate whether Dbf4 encoded abona fideBRCT-like repeat, we performed domain-swapping experiments using known BRCT domains inS. cerevisiae to see if any could substitute for the Dbf4 BRCT-like domain. There are 11 proteins in yeast that contain single or tandem BRCT repeats (Table 2) (Borket al. 1997). Two of these proteins, Dbp11 and Esc4, contain two and three pairs of tandem BRCT repeats, respec-tively. We found that placing any tandem BRCT repeat on the N terminus of Dbf4 was lethal (Table 2 and Figure 6B). However, chimeric Dbf4 proteins contain-ing only a scontain-ingle BRCT domain suppressed the viability defect of thedbf4Dwith the exception of the Fcp1- and Chs5-Dbf4 chimeras (Table 2 and Figure 6B). A Western blot of all the Dbf4 chimeric proteins that rescued
viability showed that they were expressed very similarly to Dbf4-ND221 protein (Figure 6C). Interestingly, the Rev1 BRCT domain swap was unique in that it rescued all of the DNA damage sensitivity of the dbf4-ND221 mutant (Figure 6D), including its HU sensitivity. Rev1 is a member of the translesion DNA polymerase family, and its BRCT domain is required for this activity (Lawrence 2004). The mouse Rev1 BRCT domain can interact with PCNA by co-immunoprecipitation and contributes to its targeting to replication foci (Guoet al.2006), but the precise function of the yeast Rev1 BRCT domain is still unknown.
within the Rev1 BRCT domain at positions 202–218, we mutated one of the conserved lysines to methionine (K216M), disrupting the match to the NLS. The Rev1(K216M)-Dbf4 still suppressed the growth and DNA damage sensitivity of thedbf4-ND221mutant, suggesting that the suppression that we observed was not due to the putative Rev1 NLS (Figure 7, A and B). Furthermore, a dbf4-NLS-ND221 mutant retains HU sensitivity, but the Rev1-Dbf4 chimera has wild-type sensitivity to HU (Figure 6D). In contrast, suppression of thedbf4-ND221 pheno-types required the structural integrity of the Rev1 BRCT domain (Figure 7, A and B). Mutations of conserved BRCT family residues (see Borket al.1997) in the Rev1-Dbf4 chimera either entirely abolished Rev1-Dbf4 activity (G193L, G193R, G193V, W231A) or eliminated the DNA damage resistance (G193A, P229A, D230A, and W231L). The G193R substitution defines the originalrev1-1mutant that is defective for UV-induced mutagenesis (Lawrence 2004). The equivalent protein levels of representative Rev1-Dbf4 point mutants are shown in Figure 7C. Two Rev1 BRCT mutations that eliminated Dbf4 activity entirely (G193R and W231A) were not expressed (Figure S1C), indicating that they are destabilizing mutations. In conclusion, our data indicate that the Rev1 BRCT domain functionally substitutes for the Dbf4 BRCT-like domain, suggesting that Dbf4 and Rev1 interact with a shared protein. The identity of this protein remains unknown for both Dbf4 and Rev1, but an informed hypothesis suggests that it is a protein present at replication forks.
DISCUSSION
In this study, we mapped two regions on budding yeast that Dbf4 required for binding and activating Cdc7
kinase. One of these regions, motif C, likely encodes a functional Zn-finger domain on the basis of our point mutational analysis, and the other region includes motif M plus significant downstream sequences. We also presented further evidence that Dbf4 contains an N-terminal BRCT-like domain that could target the kinase to a replication fork protein. Finally, we showed that the Zn finger but not the BRCT-like domain is required for induced mutagenesis in yeast.
Deletion analysis of theS. pombeDfp1 (ScDbf4) subunit showed that two regions contributed to Hsk1 (ScCdc7) binding and kinase activation (Oginoet al. 2001; Fung et al.2002). In Dfp1, regions encompassing motif M or motif C bind independently to Hsk1 and contribute to kinase activation. In fact, a mutant that deleted the intervening residues between motif M and motif C could complement Dfp1 activity, strongly suggesting that the S. pombeDfp1 required only these two regions to bind and activate Hsk1in vivo. Deletion of motif C had little effect on Cdc7 kinase activity in one study (Oginoet al.2001) but had more pronounced effects in the second (Fung et al.2002) although this did not seem to affect S-phase progression. On the basis of further mutational data it was proposed that a redundancy existed between motifs N and C since motif N or motif C could be deleted but deletion of both motifs was not tolerated (Oginoet al. 2001).
Our mutational analysis of the S. cerevisiae Dbf4 is consistent with these earlier studies but reveals some important differences regarding the budding yeast subunit. We find that C-terminal mutants removing the Dbf4 Zn finger exhibit a significant defect in Cdc7 kinase activation, which is consistent with the significant defect that these mutants have entering and progressing TABLE 2
BRCT-Dbf4 chimeric proteins
Protein name
No. of BRCT repeats
No. of BRCT repeats in chimeraa
Rescue ofdbf4D
lethalityb
Suppression of DNA damage phenotype
Dbf4 1 1 1 1
Chs5 1 1 NA
Dnl4 2 2 NA
Dpb11 4 2 (aa1-228) NA
Dpb11 4 2 (aa306-592) NA
Esc4 6 0 NA NA
Fcp1 1 1 NA
Nop7 1 1 1
Pol4 1 1 1
Rad9 2 2 NA
Rap1 1 1 1
Rev1 1 1 1 1
Rfc1 1 1 1
a
BRCT-Dbf4 chimeras were constructed as indicated in Figure 6A and as described in materials and methods.
bChimeras were tested for the ability to suppress the
through S-phase. A small 60-amino-acid region span-ning the Dfp1 motif M could interact with Hsk1 by co-IP and was auto-phosphorylated (Ogino et al. 2001). Although we were unable to detect binding of only motif M to Cdc7 for technical reasons, we found that the in vitro Dbf4-Cdc7 interaction required motif M plus 100 amino acids downstream of motif M. Our finding that a C-terminal deletion mutant to residue 312 (that retained motif M) exhibited a total loss of Cdc7 binding and kinase activation (although the mutant Dbf4 protein was abundantly expressed) supports this
conclusion. Furthermore, we previously showed that an N-terminal Dbf4 mutant that deletes most of motif M (ND292) retains the ability to bind and activate Cdc7 kinase in vitro, but a further deletion to residue 357 abolishes Cdc7 binding and kinase activation (Figure 5). Thus, both the budding and fission yeast Dbf4 subunits bind to Cdc7 kinase using a bipartite interac-tion. However, Dbf4 differs somewhat from Dfp1 in that motif M plus substantial downstream sequences com-pose the first binding site. The Zn finger comprises the second binding site as in Dfp1, but in budding yeast Figure 6.—The Rev1 BRCT domain suppresses loss of the Dbf4 BRCT-like sequence. (A) Heterologous BRCT domains were cloned on anNcoI fragment into the dbf4-ND221 gene. (B) M895 [dbf4D/pMW490 (DBF4 URA3)] was transformed with wild type (WT) DBF4, dbf4-ND221, or various chimeric dbf4-BRCT-ND221 plasmids (LEU2) and tested for the ability to rescue viability of dbf4D. (C) Western blot of total cell extracts showing wild-type Dbf4, Dbf4-ND221, and the viable BRCT-containing chimeric proteins. (D) Tenfold serial dilutions of wild type (M1016), dbf4-ND221 (M1173), and the various chimeric strains were spotted onto YPD (1/ ) and the indicated drugs and were incubated for 2 days at 30°.
the Zn finger contributes substantially to kinase activation.
Point mutations in the conserved C2H2 Zn-binding
residues of Dbf4 cause a substantial defect in cell growth and S-phase progression and also give rise to DNA damage and temperature sensitivities. The cell growth and DNA damage phenotypes likely result from a significant defect in activating Cdc7 kinase, which is required for the initiation of DNA replication. We pre-viously showed that increased MMS sensitivity is a common phenotype of replication initiation mutants (Gabrielse et al. 2006). Therefore, initiation mutants are MMS sensitive probably because too few replication origins are activated in the presence of MMS. Similarly, the bleomycin sensitivity ofdbf4N-terminal mutants was also accompanied by decreased initiation activity, a phenotype also shared by several other initiation mu-tants. Importantly, both the initiation defect and the bleomycin sensitivity of thedbf4-ND221 N-terminal mu-tant were rescued by adding back a functional NLS (Gabrielseet al.2006). Therefore, it seems possible that the MMS and bleomycin sensitivities of thedbf4Zn-finger mutants result from the initiation defect and that Cdc7-Dbf4 kinase is not directly involved in repair of alkylation damage or double-strand breaks. However, it is also pos-sible that Cdc7-Dbf4 operates in a direct pathway for repair of alkylation damage as suggested in fission yeast (Fung et al.2002; Sommarivaet al. 2005). For example, when Cdc7-Dbf4 promotes the initiation of DNA replication, this might be coupled with some Cdc7-Dbf4-dependent modification of the replication fork that enables cells to respond to particular types of DNA damage.
On the basis of multiple sequence alignments of Dbf4 orthologs and mutational data we previously suggested that Dbf4 encodes a BRCT-like domain that is dispensable for its essential mitotic activities (Gabrielseet al.2006). To further test this hypothesis, we performed domain-swapping experiments with known BRCT domains in yeast to see if we could rescue the dbf4-ND221mutant phenotypes using abona fideBRCT domain. None of the tandem BRCTrepeats in yeast even allowed viability when fused to the N terminus of Dbf4-ND221 protein;i.e., they inactivated the essential activity of Dbf4. This inviability might arise because the tandem BRCT domains target Cdc7 kinase away from its essential replication substrates to those phosphorylated substrates recognized by the respective BRCT domains. In contrast, we found that the single Rev1 BRCT domain suppressed all of the mutant phenotypes of dbf4-ND221 and that this suppression depended on conserved BRCT residues. This suggests that Dbf4 does encode a BRCT domain and that the Rev1 and Dbf4 domains interact with a common protein or protein structure. Indeed, a recent crystal structure of the Dbf4 BRCT motif confirms that it encodes a bona fide BRCT domain (A. Guarne, personal communication).
The C-terminal Dbf4 mutants are not sensitive to UV irradiation; however, they are almost completely
defective in UV-induced mutagenesis. This phenotype is similar to therev1-1mutant, which affects the Rev1 translesion DNA polymerase (Lawrence 2004). In other words, rev1-1 is not particularly sensitive to UV irradiation, but it is largely defective in UV-induced mutagenesis. Rev1 plays a minor role for the bulk of UV repair, which is instead accomplished by nucleotide excision repair or by the error-free TLS polymerase, Rad30(Polh) (Lawrence2004; Prakashet al.2005). In contrast to thedbf4C-terminal mutants, we found that the BRCT domain wasnotrequired for induced muta-genesis. Dbf4 mutants have not been previously ana-lyzed for their ability to support induced mutagenesis, butcdc7hypomorphs are largely defective in this process (Njagiand Kilbey1982a,b). Since Dbf4 is required for the mitotic activity of Cdc7 kinase, it was reasonable that dbf4hypomorphs would also affect induced mutagene-sis, but this had not been demonstrated.
Since Rev1 is responsible for the bulk of UV-induced mutagenesis in yeast, our data suggest that Dbf4 promotes a Rev1-dependent step in this process. The Dbf4 Zn finger is required for induced mutagenesis either because it stimulates Cdc7 kinase activity or because it also targets the Cdc7-Dbf4 to a protein required for induced mutagenesis. In addition, BRCT swapping data suggest that both Rev1 and Dbf4 might bind a common replication protein either during normal fork progression, when fork stalling occurs, or during G2when it is thought that nonrepairable lesions
are repaired by TLS polymerases (Lehmannand Fuchs 2006). This protein could be PCNA since the mouse Rev1 BRCT domain has been shown to mediate an interaction with PCNA by co-immunoprecipitation from cell extracts (Guoet al.2006). However, the target of the yeast Rev1 BRCT domain is unknown, and we have also not been able to detect a two-hybrid in-teraction between the budding yeast Rev1 or Dbf4 BRCT domains with PCNA or a PCNA–ubiquitin fusion protein (data not shown). Thus the binding partner for both BRCT domains remains uncertain. It will be im-portant to determine the exact binding partner for the Dbf4 BRCT domain and also to determine how Cdc7-Dbf4 kinase promotes induced mutagenesis in yeast.
We thank E. Craig and A. Sugino for yeast strains and the flow cytometry lab at the Van Andel Research Institute for technical support. We gratefully acknowledge the Van Andel Research Institute and a grant from the American Cancer Society (RSG0506301GMC), which supported this work.
LITERATURE CITED
Bell, S. P., and A. Dutta, 2002 DNA replication in eukaryotic cells.
Annu. Rev. Biochem.71:333–374.
Bork, P., K. Hofmann, P. Bucher, A. F. Neuwald, S. F. Altschul
et al., 1997 A superfamily of conserved domains in DNA damage-responsive cell cycle checkpoint proteins. FASEB J.11: 68–76.
Bousset, K., and J. F. Diffley, 1998 The Cdc7 protein kinase is
Brayer, K. J., and D. J. Segal, 2008 Keep your fingers off my DNA:
protein-protein interactions mediated by C2H2 zinc finger do-mains. Cell Biochem. Biophys.50:111–131.
Brown, G. W., and T. J. Kelly, 1998 Purification of Hsk1, a
mini-chromosome maintenance protein kinase from fission yeast. J. Biol. Chem.273:22083–22090.
Cheng, L., T. Collyerand C. F. Hardy, 1999 Cell cycle regulation
of DNA replication initiator factor Dbf4p. Mol. Cell. Biol.19: 4270–4278.
Donaldson, A. D., W. L. Fangmanand B. J. Brewer, 1998 Cdc7 is
required throughout the yeast S phase to activate replication ori-gins. Genes Dev.12:491–501.
Dowell, S. J., P. Romanowskiand J. F. Diffley, 1994 Interaction of
Dbf4, the Cdc7 protein kinase regulatory subunit, with yeast rep-lication origins in vivo. Science265:1243–1246.
Duncker, B. P., K. Shimada, M. Tsai-Pflugfelder, P. Paseroand
S. M. Gasser, 2002 An N-terminal domain of Dbf4p mediates
interaction with both origin recognition complex (ORC) and Rad53p and can deregulate late origin firing. Proc. Natl. Acad. Sci. USA99:16087–16092.
Ferreira, M. F., C. Santocanale, L. S. Druryand J. F. Diffley,
2000 Dbf4p, an essential S phase-promoting factor, is targeted for degradation by the anaphase-promoting complex. Mol. Cell. Biol.20:242–248.
Foiani, M., F. Marini, D. Gamba, G. Lucchini and P. Plevani,
1994 The B subunit of the DNA polymerase alpha-primase com-plex inSaccharomyces cerevisiaeexecutes an essential function at the initial stage of DNA replication. Mol. Cell. Biol.14:923–933. Francis, L. I., J. C. Randell, T. J. Takara, L. Uchimaand S. P. Bell,
2009 Incorporation into the prereplicative complex activates the Mcm2–7 helicase for Cdc7-Dbf4 phosphorylation. Genes Dev.23:643–654.
Fung, A. D., J. Ou, S. Buelerand G. W. Brown, 2002 A conserved
domain ofSchizosaccharomyces pombe dfp1(1)is uniquely required for chromosome stability following alkylation damage during S phase. Mol. Cell. Biol.22:4477–4490.
Furukohri, A., N. Sato, H. Masai, K. Arai, A. Sugino et al.,
2003 Identification and characterization of aXenopushomolog of Dbf4, a regulatory subunit of the Cdc7 protein kinase required for the initiation of DNA replication. J. Biochem.134:447–457. Gabrielse, C., C. T. Miller, K. H. McConnell, A. Deward, C. A.
Fox et al., 2006 A Dbf4p BRCA1 C-terminal-like domain
re-quired for the response to replication fork arrest in budding yeast. Genetics173:541–555.
Gambus, A., R. C. Jones, A. Sanchez-Diaz, M. Kanemaki, F. van
Deursenet al., 2006 GINS maintains association of Cdc45 with
MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat. Cell Biol.8:358–366.
Geraghty, D. S., M. Ding, N. H. Heintz and D. S. Pederson,
2000 Premature structural changes at replication origins in a yeast minichromosome maintenance (MCM) mutant. J. Biol. Chem.275:18011–18021.
Glover, J. N., R. S. Williamsand M. S. Lee, 2004 Interactions
be-tween BRCT repeats and phosphoproteins: tangled up in two. Trends Biochem. Sci.29:579–585.
Guo, C., E. Sonoda, T. S. Tang, J. L. Parker, A. B. Bielen et al.,
2006 REV1 protein interacts with PCNA: significance of the REV1 BRCT domain in vitro and in vivo. Mol. Cell 23: 265–271.
Hardy, C. F., and A. Pautz, 1996 A novel role for Cdc5p in DNA
replication. Mol. Cell. Biol.16:6775–6782.
Jackson, A. L., P. M. Pahl, K. Harrison, J. Rosamondand R. A.
Sclafani, 1993 Cell cycle regulation of the yeast Cdc7 protein
kinase by association with the Dbf4 protein. Mol. Cell. Biol.13: 2899–2908.
James, S. W., K. A. Bullock, S. E. Gygax, B. A. Kraynack, R. A.
Maturaet al., 1999 nimO, anAspergillusgene related to
bud-ding yeast Dbf4, is required for DNA synthesis and mitotic check-point control. J. Cell Sci.112(Pt. 9): 1313–1324.
Jiang, W., D. McDonald, T. J. Hopeand T. Hunter, 1999
Mam-malian Cdc7-Dbf4 protein kinase complex is essential for initia-tion of DNA replicainitia-tion. EMBO J.18:5703–5713.
Johnston, L. H., and A. P. Thomas, 1982 A further two mutants
de-fective in initiation of the S phase in the yeastSaccharomyces cer-evisiae.Mol. Gen. Genet.186:445–448.
Kanemaki, M., A. Sanchez-Diaz, A. Gambus and K. Labib,
2003 Functional proteomic identification of DNA replication proteins by induced proteolysis in vivo. Nature423:720–724. Kihara, M., W. Nakai, S. Asano, A. Suzuki, K. Kitada et al.,
2000 Characterization of the yeast Cdc7p/Dbf4p complex pu-rified from insect cells. Its protein kinase activity is regulated by Rad53p. J. Biol. Chem.275:35051–35062.
Kitada, K., L. H. Johnston, T. Sugino and A. Sugino,
1992 Temperature-sensitivecdc7mutations ofSaccharomyces cer-evisiaeare suppressed by theDBF4gene, which is required for the G1/S cell cycle transition. Genetics131:21–29.
Koonin, E. V., S. F. Altschuland P. Bork, 1996 BRCA1protein
products. . .Functional motifs. Nat. Genet.13:266–268. Krishna, S. S., I. Majumdarand N. V. Grishin, 2003 Structural
clas-sification of zinc fingers: survey and summary. Nucleic Acids Res. 31:532–550.
Kumagai, H., N. Sato, M. Yamada, D. Mahony, W. Seghezziet al.,
1999 A novel growth- and cell cycle-regulated protein,ASK, ac-tivates human Cdc7-related kinase and is essential for G1/S tran-sition in mammalian cells. Mol. Cell. Biol.19:5083–5095. Landis, G., and J. Tower, 1999 TheDrosophila chiffongene is
re-quired for chorion gene amplification, and is related to the yeast Dbf4 regulator of DNA replication and cell cycle. Development 126:4281–4293.
Lawrence, C. W., 2004 Cellular functions of DNA polymerase zeta
and Rev1 protein. Adv. Protein Chem.69:167–203.
Lawrence, C. W., and R. Christensen, 1976 UV mutagenesis in
radiation-sensitive strains of yeast. Genetics82:207–232. Lehmann, A. R., and R. P. Fuchs, 2006 Gaps and forks in DNA
rep-lication: rediscovering old models. DNA Repair (Amst.)5:1495– 1498.
Lei, M., Y. Kawasaki, M. R. Young, M. Kihara, A. Sugino et al.,
1997 Mcm2 is a target of regulation by Cdc7-Dbf4 during the initiation of DNA synthesis. Genes Dev.11:3365–3374. Lepke, M., V. Putter, C. Staib, M. Kneissl, C. Berger et al.,
1999 Identification, characterization and chromosomal locali-zation of the cognate human and murine DBF4 genes. Mol. Gen. Genet.262:220–229.
Masai, H., and K. Arai, 2000 Dbf4 motifs: conserved motifs in
ac-tivation subunits for Cdc7 kinases essential for S-phase. Biochem. Biophys. Res. Commun.275:228–232.
Masai, H., C. Taniyama, K. Ogino, E. Matsui, N. Kakusho et al.,
2006 Phosphorylation of MCM4 by Cdc7 kinase facilitates its interaction with Cdc45 on the chromatin. J. Biol. Chem. 281: 39249–39261.
Matsumoto, S., K. Ogino, E. Noguchi, P. Russelland H. Masai,
2005 Hsk1-Dfp1/Him1, the Cdc7-Dbf4 kinase in Schizosaccharo-myces pombe, associates with Swi1, a component of the replication fork protection complex. J. Biol. Chem.280:42536–42542. Miller, C. T., C. Gabrielse, Y. C. Chen and M. Weinreich,
2009 Cdc7p-Dbf4p regulates mitotic exit by inhibiting Polo ki-nase. PLoS Genet.5:e1000498.
Moyer, S. E., P. W. Lewisand M. R. Botchan, 2006 Isolation of the
Cdc45/Mcm2–7/GINS (CMG) complex, a candidate for the eu-karyotic DNA replication fork helicase. Proc. Natl. Acad. Sci. USA 103:10236–10241.
Njagi, G. D., and B. J. Kilbey, 1982a cdc7–1a temperature sensitive
cell-cycle mutant which interferes with induced mutagenesis in
Saccharomyces cerevisiae.Mol. Gen. Genet.186:478–481. Njagi, G. D., and B. J. Kilbey, 1982b Mutagenesis incdc7strains of
yeast: the fate of premutational lesions induced by ultraviolet light. Mutat. Res.105:313–318.
Ogino, K., T. Takeda, E. Matsui, H. Iiyama, C. Taniyama et al.,
2001 Bipartite binding of a kinase activator activates Cdc7-related kinase essential for S phase. J. Biol. Chem.276:31376–31387. Oshiro, G., J. C. Owens, Y. Shellman, R. A. Sclafaniand J. J. Li,
1999 Cell cycle control of Cdc7p kinase activity through regu-lation of Dbf4p stability. Mol. Cell. Biol.19:4888–4896. Owens, J. C., C. S. Detweilerand J. J. Li, 1997 CDC45is required in
conjunction withCDC7/DBF4to trigger the initiation of DNA replication. Proc. Natl. Acad. Sci. USA94:12521–12526. Pacek, M., A. V. Tutter, Y. Kubota, H. Takisawaand J. C. Walter,
Pessoa-Brandao, L., and R. A. Sclafani, 2004 CDC7/DBF4
func-tions in the translesion synthesis branch of theRAD6epistasis group inSaccharomyces cerevisiae.Genetics167:1597–1610. Prakash, S., R. E. Johnsonand L. Prakash, 2005 Eukaryotic
trans-lesion synthesis DNA polymerases: specificity of structure and function. Annu. Rev. Biochem.74:317–353.
Sato, N., K. Araiand H. Masai, 1997 Human andXenopuscDNAs
encoding budding yeast Cdc7-related kinases: in vitro phosphor-ylation of MCM subunits by a putative human homologue of Cdc7. EMBO J.16:4340–4351.
Sato, N., M. Sato, M. Nakayama, R. Saitoh, K. Arai et al.,
2003 Cell cycle regulation of chromatin binding and nuclear localization of human Cdc7-ASK kinase complex. Genes Cells 8:451–463.
Sclafani, R. A., 2000 Cdc7p-Dbf4p becomes famous in the cell
cy-cle. J. Cell Sci.113(Pt. 12): 2111–2117.
Sheu, Y. J., and B. Stillman, 2006 Cdc7-Dbf4 phosphorylates MCM
proteins via a docking site-mediated mechanism to promote S phase progression. Mol. Cell24:101–113.
Sommariva, E., T. K. Pellny, N. Karahan, S. Kumar, J. A. Huberman
et al., 2005 Schizosaccharomyces pombeSwi1, Swi3, and Hsk1 are components of a novel S-phase response pathway to alkylation damage. Mol. Cell. Biol.25:2770–2784.
Sullivan, M., L. Holtand D. O. Morgan, 2008 Cyclin-specific
con-trol of ribosomal DNA segregation. Mol. Cell. Biol.28:5328–5336. Takeda, T., K. Ogino, E. Matsui, M. K. Cho, H. Kumagai et al.,
1999 A fission yeast gene,him1(1)/dfp1(1), encoding a regula-tory subunit for Hsk1 kinase, plays essential roles in S-phase ini-tiation as well as in S-phase checkpoint control and recovery from DNA damage. Mol. Cell. Biol.19:5535–5547.
Thompson, J. D., T. J. Gibsonand D. G. Higgins, 2002 Multiple
se-quence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinformatics Chapter 2: Unit 2.3.
Waters, L. S., B. K. Minesinger, M. E. Wiltrout, S. D’Souza, R. V.
Woodruffet al., 2009 Eukaryotic translesion polymerases and
their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev.73:134–154.
Weinreich, M., and B. Stillman, 1999 Cdc7p-Dbf4p kinase
binds to chromatin during S phase and is regulated by both the APC and the RAD53 checkpoint pathway. EMBO J. 18: 5334–5346.
Wolfe, S. A., L. Nekludovaand C. O. Pabo, 2000 DNA recognition
by Cys2His2 zinc finger proteins. Annu. Rev. Biophys. Biomol. Struct.29:183–212.
Wood, J. S., and L. H. Hartwell, 1982 A dependent pathway of
gene functions leading to chromosome segregation in Saccharo-myces cerevisiae.J. Cell Biol.94:718–726.
Yabuuchi, H., Y. Yamada, T. Uchida, T. Sunathvanichkul, T.
Nakagawaet al., 2006 Ordered assembly of Sld3, GINS and
Cdc45 is distinctly regulated by DDK and CDK for activation of replication origins. EMBO J.25:4663–4674.
Zou, L., and B. Stillman, 2000 Assembly of a complex containing
Cdc45p, replication protein A, and Mcm2p at replication origins controlled by S-phase cyclin-dependent kinases and Cdc7p-Dbf4p kinase. Mol. Cell. Biol.20:3086–3096.
Supporting Information
http://www.genetics.org/cgi/content/full/genetics.109.110155/DC1
Budding Yeast Dbf4 Sequences Required for Cdc7 Kinase
Activation and Identification of a Functional Relationship Between
the Dbf4 and Rev1 BRCT Domains
Victoria Harkins, Carrie Gabrielse, Louise Haste and Michael Weinreich
Copyright © 2009 by the Genetics Society of America
V. Harkins et al.
2 SI
A
WT
C
!
654
C
!
629
C
!
598
C
!
575
C
!
511
*
*
*
*
*
*
"
-Dbf4
B
WT
HH674,680LL
HH674,680AA
CC661,664AA
C664A
S677E
S677A
!
689-704
Dbf4
"
-Dbf4
Harkins et al. Fig S1
C
Rev1
BRCT
Rev1-G193R
Rev1-W231A
WT Dbf4
Dbf4-N
!
221 +Rev1 BRCT
"
-Dbf4
FIGURE S1.—Expression levels of Dbf4 proteins. (A) Whole cell extracts of the indicated mutants examined in Figure 1C were blotted for Dbf4 protein using a polyclonal antibody against
GST-Dbf4. Asterisks mark the Dbf4 proteins. (B) The mutants analyzed in Figure 3B were similarly probed for Dbf4 protein. (C) M895 strains (dbf4Δ/pMW490 (DBF4 URA3) expressing Dbf4,
Rev1-G193R-Dbf4, or Rev1-W231A-Dbf4 BRCT domain chimeras on plasmids were streak purified selecting for the REV1-DBF4 chimeric plasmid. Whole cell extracts were then probed for Dbf4 protein
V. Harkins et al. 3 SI
TABLE S1
Plasmids
Name Description
pMW489 pRS415‐DBF4 pMW490 pRS416‐DBF4 pMW526 pRS415 dbf4NΔ65
pCG10 pRS415 dbf4NΔ109
pCG15 pRS415 dbf4Δ136221
pCG29 pRS415 dbf4NΔ221
pCG45 pCG29 x RAD9 BRCT pCG46 pCG29 x DNL4 BRCT pCG48 pCG29 x DPB11 BRCT #1/2 pCG51 pCG29 x POL4 BRCT pCG54 pCG29 x REV1 BRCT pCG56 pCG29 x RAP1 BRCT pCG70 pCG29 x rev1P229A BRCT pCG71 pCG29 x rev1D230A BRCT pCG72 pCG29 x rev1W231A BRCT pCG73 pCG29 x rev1G193R BRCT pCG76 pRS415 dbf4NLSNΔ221
pCG92 pAcSG2 dbf4NΔ292
pCG94 pCG29 x NOP7 BRCT pCG95 pCG29 x FCP1 BRCT pCG96 pCG29 x RFC1 BRCT
pCG119 pCG29 x rev1GG192,193AA BRCT pCG122 pCG29 x rev1G193A BRCT pCG126 pRS415 dbf4NLSaa221417 pCG165 pCG29 x DPB11 BRCT #3/4 pCG175 pGBKT7‐CDC7
pCG183 pAcSG2 dbf4NΔ357
pCG193 pAcPK30 dbf4Δ312418
pCG209 pGAD dbf4aa311418
pCG211 pAcSG2 dbf4NΔ292, CC661,684AA
V. Harkins et al.
4 SI
pLVH51 pAcPK30 dbf4CC661,664AA pLVH52 pAcPK30 dbf4CΔ629
pLVH53 pRS425‐CDC7 pLVH55 pRS415 dbf4S677A pLVH56 pRS415 dbf4C664A pLVH58 pRS415 dbf4CΔ689
pLVH59 pRS415 dbf4S677E pLVH60 pRS415 dbf4HH674,680AA pLVH62 pLVH47 HH674,680LL pLVH63 pLVH47 CC661,664AA pLVH64 pLVH47 C661A
pVMH3 pRS415 dbf4CC661,664AA pVMH5 pRS415 dbf4HH674,680LL pVMH7 pRS415 dbf4CΔ654
pVMH14 pCG29 x CHS5 BRCT pVMH16 pRS415 dbf4CΔ418
pVMH18 pRS415 dbf4CΔ511
pVMH21 pRS415 dbf4CΔ575
pVMH22 pRS415 dbf4CΔ598
pVMH23 pRS415 dbf4CΔ629
pVMH59 pRS415 dbf4CΔ312
pVMH61 pRS415 dbf4CΔ357
pVMH71 pCG76 x CΔ629
pVMH72 pCG76 x CΔ654
pVMH79 pCG76 x CC661,664AA pVMH81 pAcPK30 x dbf4CΔ418
pVMH84 pAcPK30 x dbf4CΔ312
pVMH85 pCG10 x CΔ654
pVMH86 pCG10 x CΔ629
pVMH87 pCG10 x CC661,664AA pVMH91 pMW526 x CΔ654
pVMH93 pMW526 x CΔ629
pVMH95 pMW526 x CC661,664AA pVMH97 pRS415 dbf4P277L pVMH101 pMW526 x CΔ418
pVMH107 pVMH97 x CΔ654
pVMH109 pVMH97 x CΔ629
pVMH110 pVMH97 x CΔ418
pVMH112 pVMH97 x CC661,664AA pVMH121 pCG15 x CC661,664AA pVMH123 pCG15 x CΔ654
V. Harkins et al. 5 SI
pVMH127 pCG15 x CΔ629
pVMH130 pRS415 dbf4Δ118221
pVMH131 pVMH130 x CC661,664AA pVMH132 pVMH130 x CΔ654
pVMH133 pVMH130 x CΔ418
pVMH134 pVMH130 x CΔ629
pVMH136 pRS415 dbf4Δ122221