organic papers
Acta Cryst.(2005). E61, o3511–o3513 doi:10.1107/S1600536805029934 Butcheret al. C
11H15NO2
o3511
Acta Crystallographica Section E Structure Reports
Online
ISSN 1600-5368
2-Formylthymol oxime
Ray J. Butcher,a* Ratnamala S. Bendreband Anil S. Kuwarb
a
Department of Chemistry, Howard University, 525 College Street NW, Washington DC 20059, USA, andbSchool of Chemical Sciences, Noth
Maharashtra University, Jalgaon 425 001, India
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 293 K
Mean(C–C) = 0.002 A˚
Rfactor = 0.045
wRfactor = 0.137
Data-to-parameter ratio = 19.6
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography Printed in Great Britain – all rights reserved
The structure of the title compound [systematic name: 2-hydroxy-6-methyl-3-(1-methylethyl)benzaldehyde oxime] C11H15NO2, exhibits intra- as well as intermolecular hydrogen
bonding, involving participation of the phenolic OH group in intramolecular hydrogen bonding and the hydroxyl group of the oxime in intermolecular hydrogen bonding. The H atom of the phenolic hydroxyl group forms a strong O—H N intramolecular hydrogen bond with an O N distance of 2.5788 (14) A˚ , which is in the middle of the expected range for such hydrogen bonds. The H atom of the hydroxyl group (in the oxime functionality) forms a weaker hydrogen bond with the phenolic hydroxyl group of a neighboring molecule [O O = 2.8317 (14) A˚ ], forming an extended chain, as expected for phenolic aldoximes which have bulky substitu-ents on the aryl ring.
Comment
Thymol is a naturally occurring phenolic monoterpenoid. It possesses an ecological role and shows a broad spectrum of biological activities (Desai & Shah, 2003). In order to enhance the overall biological activity of thymol, derivatives such as nitroso, amino, azomethine, 4-thiazolidinones, 2-azetidinones and 4-imidazolinones have been prepared (Vashaiet al., 1995). On the other hand, derivatization of the hydroxyl group of thymol to ethers and esters has resulted in an increase in biological activities. A structure–activity correlation has also been established in this series of compounds and the overall activity has been found to depend on the nature and position of the functional groups. Thus, thymol was derivatized to 2-formyl thymol oxime, (I), to use it for the preparation of metal complexes, as a number of metal–oxime complexes are known to have biological significance (Chakravorty, 1974; Lummeet al., 1984; Jayaraju & Kondapi, 2001). We present the structure of (I) here.
Compound (I) is a member of a general class of phenolic oximes (Smith et al., 2003). These compounds have found extensive use in industry, mainly as extractants for copper
(Kordosky, 2002), but also as anticorrosives in protective coatings (Thorpeet al., 1999). Another feature of the phenolic oxime ligands is their propensity (Chaudhuriet al., 1993; Billet al., 1997) to form polynuclear complexes in which both the oxime and phenolate functions can act as bridging units.
Elemental analysis for (I) gave a satisfactory fit to the formula C11H15NO2. Table 1 contains selected bond lengths
and angles. A view of the molecule and unit-cell contents are shown in Figs. 1 and 2, respectively. The average length of the benzene ring bonds is 1.396 (12) A˚ , which is in good agree-ment with generally accepted values.
Hydrogen bonding is a major feature of the structures of phenolic oximes. This results from the high density of hydrogen-bonding donors and acceptors per molecule. Invariably, the phenolic H atom forms an intramolecular hydrogen bond to the N atom of the oxime group, giving a six-membered ring. Since the phenolic H atom is often not found in Fourier difference maps, this interaction is usually char-acterized in terms of the phenolic O to oxime N separation. This distance varies little between structures, with a maximum value of 2.65 A˚ and a minimum of 2.51 A˚. However, a general trend is that aldoximes have a greater phenolic O N distance than the ketoximes (Smithet al., 2003). In all of the free ligand structures, the molecules associate via intermolecular hydrogen bonding. These structures fall into two categories. Dimers result from the interaction of the oxime H atom with an adjacent phenolic O atom to produce a pseudomacrocyclic ligand with a 14-membered inner ring. This structure is seen only for aldoximes with no substituents or only monoatomic substituents on the aromatic ring (Smithet al., 2003).
The introduction of groups which remove planarity in the molecule appears to stop efficient packing of dimeric units in the crystal structure and, instead, a polymeric structure, [(H2sal)n], is observed. This is true for all phenolic ketoximes
and for phenolic aldoximes which have bulky substituents on the aryl ring (Smithet al., 2003).
The title compound exhibits intra- as well as intermolecular hydrogen bonding (Table 2), involving participation of the phenolic OH group in intramolecular hydrogen bonding and the hydroxyl group of the oxime in intermolecular hydrogen bonding, as indicated above. The H atom of the phenolic hydroxyl group forms a strong O1—H N1 intramolecular hydrogen bond with an O1 N1 distance of 2.5788 (14) A˚ , which is in the middle of the expected range for such hydrogen bonds (Smithet al., 2003).
The H atom of the hydroxyl group (in the oxime func-tionality) attached to atom N1 forms a weaker hydrogen bond with the phenolic hydroxyl group of a neighboring molecule [O2 O1 = 2.8317 (14) A˚ ], forming an extended chain, as expected for phenolic aldoximes which have bulky substi-tuents on the aryl ring (Smithet al., 2003).
Experimental
The title compound was prepared by the condensation of 2-formyl-thymol (obtained byortho-formylation of thymol) (3.56 g, 20 mmol) with hydroxylamine hydrochloride (1.4 g, 20 mmol) in ethanol (150 ml). Yellow crystals of (I) suitable for X-ray diffraction were obtained upon slow evaporation of the reaction mixture.
Crystal data
C11H15NO2 Mr= 193.24 Monoclinic, P21=n a= 8.8517 (7) A˚
b= 9.0145 (7) A˚
c= 13.5956 (10) A˚
= 101.698 (2)
V= 1062.31 (14) A˚3 Z= 4
Dx= 1.208 Mg m
3
MoKradiation Cell parameters from 4359
reflections
= 2.5–28.2 = 0.08 mm1 T= 293 (2) K
Irregular fragment, pale yellow 0.550.450.32 mm
organic papers
o3512
Butcheret al. C [image:2.610.46.299.67.288.2]11H15NO2 Acta Cryst.(2005). E61, o3511–o3513
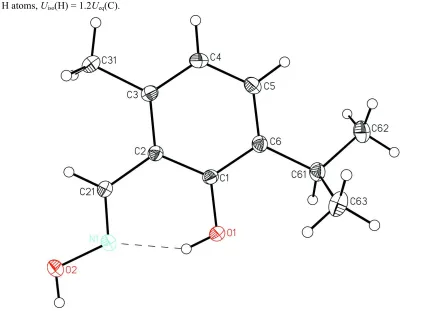
Figure 1
[image:2.610.197.552.68.296.2]A view of the title compound, showing the atom-labeling scheme. Displacement ellipsoids are drawn at the 20% probability level and H atoms are represented by circles of arbitrary size. The dashed line indicates a hydrogen bond.
Figure 2
Data collection
Bruker SMART 1K CCD area-detector diffractometer
’and!scans
Absorption correction: multi-scan (SADABS; Sheldrick, 2002)
Tmin= 0.716,Tmax= 0.928
8098 measured reflections
2592 independent reflections 1897 reflections withI> 2(I)
Rint= 0.026
max= 28.3
h=10!11
k=10!11
l=17!18
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.046
wR(F2) = 0.137 S= 1.07 2592 reflections 132 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0689P)2
+ 0.1206P]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.007
max= 0.25 e A˚
3
min=0.17 e A˚
3
Table 1
Selected geometric parameters (A˚ ,).
O1—C1 1.3710 (14)
O2—N1 1.3982 (14)
N1—C21 1.2743 (16)
C21—N1—O2 112.32 (11) O1—C1—C6 117.15 (11)
O1—C1—C2 120.36 (11) N1—C21—C2 121.68 (11)
Table 2
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
O1—H1O N1 0.82 1.85 2.5788 (14) 148
O2—H2O O1i
0.82 2.02 2.8317 (14) 169
Symmetry code: (i)xþ1 2;y
1 2;zþ
3 2.
H atoms were positioned geometrically and constrained to ride on their parent atoms. For methyl H atoms, C—H = 0.96 A˚ andUiso(H) = 1.5Ueq(C); each group was allowed to rotate freely about its C—C
bond. For other H atoms, O—H = 0.82 A˚ , aromatic C—H = 0.93 A˚ and methine C—H = 0.98 A˚ , andUiso(H) = 1.2Ueq(C,O).
Data collection:SMART(Bruker, 2001); cell refinement:SAINT
(Bruker, 2001); data reduction:SAINT; program(s) used to solve structure:SHELXS97(Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics:
SHELXTL(Sheldrick, 1998); software used to prepare material for publication:SHELXTL.
RJB acknowledges the US Department of Defense for funds to upgrade the diffractometer. RSB and ASK acknowledge financial assistance from the UGC, New Delhi, India.
References
Bill, E., Krebs, C., Winter, M., Gerdan, M., Trautwein, A. X., Flo¨rke, U., Haupt, H.-J. & Chaudhuri, P. (1997).Chem. Eur. J.3, 193–201.
Bruker (2001). SMART (Version 5.624) and SAINT (Version 6.04) for Windows NT. Bruker AXS Inc., Madison, Wisconsin, USA.
Chakravorty, A. (1974).Coord. Chem. Rev.13, 1–46.
Chaudhuri, P., Winter, M., Fleischhauer, P., Haase, W., Flo¨rke, U. & Haupt, H.-J. (1993).Inorg. Chim. Acta,212, 241–249.
Desai, J. M. & Shah, V. H. (2003).Indian J. Chem.42B, 382–385. Jayaraju, D. & Kondapi, K. (2001).Curr. Sci.81, 782–792.
Kordosky, G. A. (2002).Proceedings of the International Solvent Extraction Conference, Cape Town, South Africa, 17–21 March 2002, edited by K. C. Sole, P. M. Cole, J. S. Preston & D. A. Robinson, pp. 853–862. Johannesburg: South African Institute of Mining and Metallurgy.
Lumme, P., Elo, H. & Ja¨nne, J. (1984).Inorg. Chim. Acta,92, 241–251. Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of
Go¨ttingen, Germany.
Sheldrick, G. M. (1998).SHELXTL. Version 5.10. Bruker AXS Inc., Madison, Wisconsin, USA.
Sheldrick, G. M. (2002).SADABS. Version 2.06. University of Go¨ttingen, Germany.
Smith, A. G., Tasker, P. A. & White, D. J. (2003).Coord. Chem. Rev.241, 61–85. Thorpe, J. M., Beddoes, R. L., Collison, D., Garner, C. D., Helliwell, M., Holmes, J. M. & Tasker, P. A. (1999).Angew. Chem. Int. Ed.38, 1119–1121. Vashai, B. S., Mehta, D. S. & Shah, V. H. (1995).Indian J. Chem.34B, 802–808.
organic papers
Acta Cryst.(2005). E61, o3511–o3513 Butcheret al. C
supporting information
sup-1
Acta Cryst. (2005). E61, o3511–o3513supporting information
Acta Cryst. (2005). E61, o3511–o3513 [doi:10.1107/S1600536805029934]
2-Formylthymol oxime
Ray J. Butcher, Ratnamala S. Bendre and Anil S. Kuwar
S1. Comment
Thymol is a naturally occurring phenolic monoterpenoid. It possesses an ecological role and shows a broad spectrum of
biological activities (Desai & Shah, 2003). In order to enhance the overall biological activity of thymol, derivatives such
as nitroso, amino, azomethine, 4-thiazolidinones, 2-azetidinones and 4-imidazolinones have been prepared (Vashai et al.,
1995). On the other hand, derivatization of the hydroxyl group of thymol to ether and esters has resulted in an increase in
biological activities. A structure–activity correlation has also been established in this series of compounds and the overall
activity has been found to depend on the nature and position of the functional groups. Thus, thymol was derivatized to
2-formyl thymol oxime, (I), to utilize it for the preparation of metal complexes, as a number of metal–oxime complexes are
known to have biological significance (Chakravorty, 1974; Lumme et al., 1984; Jayaraju & Kondapi, 2001). We present
the structure of (I) here.
Compound (I) is a member of a general class of phenolic oximes (Smith et al., 2003). These compounds have found
extensive use in industry, mainly as extractants for copper (Kordosky, 2002), but also as anticorrosives in protective
coatings (Thorpe et al., 1999). Another feature of the phenolic oxime ligands is their propensity (Chaudhuri et al., 1993;
Bill et al., 1997) to form polynuclear complexes in which both the oxime and phenolate functions can act as bridging
units.
Elemental analysis for (I) gave a satisfactory fit to the formula C11H15NO2. Table 1 contains selected bond lengths and
angles. A view of the molecule and unit-cell content are shown in Figs. 1 and 2. The average length of the benzene ring
bonds is 1.396 (12) Å, which is in good agreement with generally accepted values.
Hydrogen bonding is a major feature of the structures of phenolic oximes. This results from the high density of
hydrogen-bonding donors and acceptors per molecule. Invariably, the phenolic H atom forms an intramolecular hydrogen
bond to the N atom of the oxime group, giving a six-membered ring. Since the phenolic H atom is often not found in
Fourier difference maps, this interaction is usually characterized in terms of the phenolic O to oximic N separation. This
distance varies little between structures, with a maximum value of 2.65 Å and a minimum of 2.51 Å. However, a general
trend is that aldoximes have a greater phenolic O···N distance than the ketoximes (Smith et al., 2003). In all of the free
ligand structures, the molecules associate via intermolecular hydrogen bonding. These structures fall into two categories.
Dimers result from the interaction of the oximic H atom with an adjacent phenolic O atom to produce a
pseudomacrocyclic ligand with a 14-membered inner great ring. This structure is only seen for aldoximes with no
substituents or only monatomic substituents on the aromatic ring (Smith et al., 2003).
The introduction of groups which remove planarity in the molecule appears to stop efficient packing of dimeric units in
the crystal structure and, instead, a polymeric structure, [(H2sal)n], is observed. This is true for all phenolic ketoximes and
supporting information
sup-2
Acta Cryst. (2005). E61, o3511–o3513The title compound exhibits intra- as well as intermolecular hydrogen bonding (Table 2), involving participation of the
phenolic OH moiety in intramolecular hydrogen bonding and the hydroxyl group of the oxime in intermolecular
hydrogen bonding, as indicated above. The H atom of the phenolic hydroxyl group forms a strong O1—H···N1
intramolecular hydrogen bond with an O1···N1 distance of 2.5788 (14) Å, which is in the middle of the expected range
for such hydrogen bonds (Smith et al., 2003).
The H atom of the hydroxyl group (in the oxime functionality) attached to atom N1 forms a weaker hydrogen bond with
the phenolic hydroxyl group of a neighboring molecule [O2···O1 = 2.8317 (14) Å], forming an extended chain, as
expected for phenolic aldoximes which have bulky substituents on the aryl ring (Smith et al., 2003).
S2. Experimental
The title compound was prepared by the condensation of 2-formyl thymol (obtained by ortho-formylation of thymol)
with hydroxylamine hydrochloride in ethanol. [Please give brief details of quantities or molar ratios] Yellow crystals of
(I) suitable for X-ray diffraction were obtained upon slow evaporation of the reaction mixture.
S3. Refinement
H atoms were included in their idealized geometries and constrained to ride on their attached atoms. Methyl H atoms
were constrained to an ideal geometry, with C—H distances of 0.96 Å and Uiso(H) = 1.5Ueq(C), but each group was
allowed to rotate freely about its C—C bond. All other H atoms were placed in geometrically idealized positions and
constrained to ride on their parent atoms, with O—H = 0.82 Å, aromatic C—H = 0.93 Å and methine C—H = 0.98 Å; for
[image:5.610.82.506.355.666.2]these H atoms, Uiso(H) = 1.2Ueq(C).
Figure 1
A view of the title compound, showing the atom-labeling scheme. Displacement ellipsoids are drawn at the 20%
supporting information
[image:6.610.128.482.71.389.2]sup-3
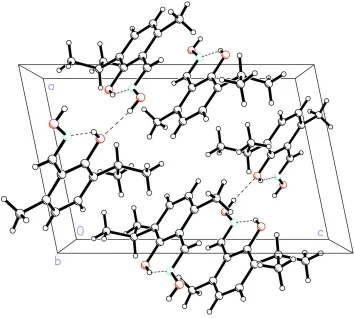
Acta Cryst. (2005). E61, o3511–o3513Figure 2
The molecular packing of the title compound, viewed along the b axis. Dotted lines indicate the hydrogen-bonding
interactions.
2-Formyl thymol oxime
Crystal data
C11H15NO2 Mr = 193.24 Monoclinic, P21/n a = 8.8517 (7) Å
b = 9.0145 (7) Å
c = 13.5956 (10) Å
β = 101.698 (2)°
V = 1062.31 (14) Å3 Z = 4
F(000) = 416
Dx = 1.208 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 4359 reflections
θ = 2.5–28.2°
µ = 0.08 mm−1 T = 293 K
Irregular, pale yellow 0.55 × 0.45 × 0.32 mm
Data collection
Bruker SMART 1K CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ and ω scans
Absorption correction: multi-scan
(SADABS; Sheldrick, 2002)
Tmin = 0.716, Tmax = 0.928
8098 measured reflections 2592 independent reflections 1897 reflections with I > 2σ(I)
Rint = 0.026
θmax = 28.3°, θmin = 2.5° h = −10→11
k = −10→11
supporting information
sup-4
Acta Cryst. (2005). E61, o3511–o3513Refinement
Refinement on F2 Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.046 wR(F2) = 0.137 S = 1.07 2592 reflections 132 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0689P)2 + 0.1206P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.007 Δρmax = 0.25 e Å−3 Δρmin = −0.17 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O1 0.10567 (11) 0.43403 (10) 0.72775 (7) 0.0449 (3)
H1O 0.1386 0.3528 0.7149 0.067*
O2 0.17161 (13) 0.04888 (11) 0.61086 (9) 0.0596 (3)
H2O 0.2440 0.0243 0.6556 0.089*
N1 0.11426 (14) 0.18593 (12) 0.63462 (9) 0.0427 (3)
C1 −0.02431 (14) 0.46760 (14) 0.65762 (9) 0.0335 (3)
C2 −0.08023 (14) 0.36816 (13) 0.57850 (9) 0.0328 (3)
C21 −0.00284 (15) 0.22718 (14) 0.56953 (10) 0.0389 (3)
H21A −0.0400 0.1660 0.5149 0.047*
C3 −0.21369 (15) 0.40662 (14) 0.50728 (9) 0.0370 (3)
C31 −0.28054 (18) 0.30561 (17) 0.42058 (11) 0.0483 (4)
H31A −0.3700 0.3513 0.3803 0.073*
H31B −0.3091 0.2127 0.4461 0.073*
H31C −0.2049 0.2886 0.3802 0.073*
C4 −0.28386 (16) 0.54065 (15) 0.51771 (11) 0.0449 (4)
H4A −0.3713 0.5676 0.4708 0.054*
C5 −0.22720 (16) 0.63643 (16) 0.59653 (11) 0.0444 (3)
H5A −0.2779 0.7257 0.6011 0.053*
C6 −0.09694 (15) 0.60262 (14) 0.66870 (10) 0.0368 (3)
C61 −0.03602 (17) 0.70251 (15) 0.75815 (11) 0.0441 (3)
H61A 0.0766 0.7046 0.7672 0.053*
C62 −0.0929 (2) 0.86189 (18) 0.74286 (15) 0.0673 (5)
H62A −0.0397 0.9223 0.7971 0.101*
H62B −0.2017 0.8650 0.7414 0.101*
supporting information
sup-5
Acta Cryst. (2005). E61, o3511–o3513C63 −0.0748 (2) 0.6381 (2) 0.85431 (12) 0.0634 (5)
H63A −0.0372 0.5381 0.8633 0.095*
H63B −0.1846 0.6387 0.8491 0.095*
H63C −0.0268 0.6972 0.9108 0.095*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.0445 (5) 0.0375 (5) 0.0444 (5) 0.0053 (4) −0.0104 (4) −0.0082 (4)
O2 0.0600 (7) 0.0405 (6) 0.0675 (7) 0.0174 (5) −0.0130 (5) −0.0116 (5)
N1 0.0456 (6) 0.0319 (6) 0.0464 (6) 0.0046 (5) −0.0009 (5) −0.0042 (5)
C1 0.0318 (6) 0.0333 (7) 0.0333 (6) −0.0012 (5) 0.0014 (5) 0.0005 (5)
C2 0.0337 (6) 0.0309 (6) 0.0327 (6) −0.0015 (5) 0.0040 (5) 0.0000 (5)
C21 0.0403 (7) 0.0351 (7) 0.0382 (7) −0.0007 (5) 0.0005 (5) −0.0048 (5)
C3 0.0362 (7) 0.0393 (7) 0.0334 (6) −0.0020 (5) 0.0022 (5) 0.0009 (5)
C31 0.0472 (8) 0.0516 (9) 0.0400 (7) −0.0009 (6) −0.0059 (6) −0.0066 (6)
C4 0.0378 (7) 0.0458 (8) 0.0457 (8) 0.0056 (6) −0.0042 (6) 0.0019 (6)
C5 0.0421 (7) 0.0364 (7) 0.0527 (8) 0.0081 (6) 0.0051 (6) −0.0012 (6)
C6 0.0370 (7) 0.0334 (7) 0.0400 (7) −0.0019 (5) 0.0077 (5) −0.0035 (5)
C61 0.0431 (7) 0.0377 (7) 0.0502 (8) −0.0022 (6) 0.0062 (6) −0.0109 (6)
C62 0.0791 (12) 0.0423 (9) 0.0757 (12) 0.0060 (8) 0.0039 (9) −0.0190 (8)
C63 0.0747 (12) 0.0678 (11) 0.0490 (9) −0.0089 (9) 0.0155 (8) −0.0172 (8)
Geometric parameters (Å, º)
O1—C1 1.3710 (14) C4—C5 1.388 (2)
O1—H1O 0.8200 C4—H4A 0.9300
O2—N1 1.3982 (14) C5—C6 1.3879 (19)
O2—H2O 0.8200 C5—H5A 0.9300
N1—C21 1.2743 (16) C6—C61 1.5213 (18)
C1—C6 1.3989 (18) C61—C62 1.523 (2)
C1—C2 1.4105 (17) C61—C63 1.531 (2)
C2—C3 1.4105 (17) C61—H61A 0.9800
C2—C21 1.4605 (18) C62—H62A 0.9600
C21—H21A 0.9300 C62—H62B 0.9600
C3—C4 1.3790 (19) C62—H62C 0.9600
C3—C31 1.5119 (18) C63—H63A 0.9600
C31—H31A 0.9600 C63—H63B 0.9600
C31—H31B 0.9600 C63—H63C 0.9600
C31—H31C 0.9600
C1—O1—H1O 109.5 C6—C5—C4 121.83 (13)
N1—O2—H2O 109.5 C6—C5—H5A 119.1
C21—N1—O2 112.32 (11) C4—C5—H5A 119.1
O1—C1—C6 117.15 (11) C5—C6—C1 116.68 (12)
O1—C1—C2 120.36 (11) C5—C6—C61 122.97 (12)
C6—C1—C2 122.49 (11) C1—C6—C61 120.33 (12)
supporting information
sup-6
Acta Cryst. (2005). E61, o3511–o3513C3—C2—C21 119.89 (11) C6—C61—C63 110.71 (12)
C1—C2—C21 121.24 (11) C62—C61—C63 110.44 (14)
N1—C21—C2 121.68 (11) C6—C61—H61A 107.4
N1—C21—H21A 119.2 C62—C61—H61A 107.4
C2—C21—H21A 119.2 C63—C61—H61A 107.4
C4—C3—C2 118.48 (12) C61—C62—H62A 109.5
C4—C3—C31 119.64 (12) C61—C62—H62B 109.5
C2—C3—C31 121.88 (12) H62A—C62—H62B 109.5
C3—C31—H31A 109.5 C61—C62—H62C 109.5
C3—C31—H31B 109.5 H62A—C62—H62C 109.5
H31A—C31—H31B 109.5 H62B—C62—H62C 109.5
C3—C31—H31C 109.5 C61—C63—H63A 109.5
H31A—C31—H31C 109.5 C61—C63—H63B 109.5
H31B—C31—H31C 109.5 H63A—C63—H63B 109.5
C3—C4—C5 121.64 (12) C61—C63—H63C 109.5
C3—C4—H4A 119.2 H63A—C63—H63C 109.5
C5—C4—H4A 119.2 H63B—C63—H63C 109.5
O1—C1—C2—C3 −179.59 (11) C31—C3—C4—C5 179.52 (14)
C6—C1—C2—C3 0.55 (19) C3—C4—C5—C6 0.2 (2)
O1—C1—C2—C21 0.55 (19) C4—C5—C6—C1 0.6 (2)
C6—C1—C2—C21 −179.31 (12) C4—C5—C6—C61 −177.47 (14)
O2—N1—C21—C2 −179.12 (12) O1—C1—C6—C5 179.15 (12)
C3—C2—C21—N1 −177.13 (13) C2—C1—C6—C5 −0.99 (19)
C1—C2—C21—N1 2.7 (2) O1—C1—C6—C61 −2.70 (19)
C1—C2—C3—C4 0.29 (19) C2—C1—C6—C61 177.16 (12)
C21—C2—C3—C4 −179.85 (12) C5—C6—C61—C62 −20.2 (2)
C1—C2—C3—C31 −179.89 (13) C1—C6—C61—C62 161.78 (14)
C21—C2—C3—C31 −0.03 (19) C5—C6—C61—C63 104.51 (16)
C2—C3—C4—C5 −0.6 (2) C1—C6—C61—C63 −73.52 (17)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
O1—H1O···N1 0.82 1.85 2.5788 (14) 148
O2—H2O···O1i 0.82 2.02 2.8317 (14) 169