Journal Name
Cite this: DOI: 10.1039/c0xx00000x
www.rsc.org/xxxxxx
Dynamic Article Links
►
ARTICLE TYPE
Synthesis and Structural Characterization of a Crystal to
Single-Crystal Transformable Coordination Polymer
Yuyang Tian,
aPhoebe K. Allan,
aCatherine L. Renouf,
aXiang He,
bLaura J. McCormick
aand Russell E.
Morris*
aReceived (in XXX, XXX) Xth XXXXXXXXX 20XX, Accepted Xth XXXXXXXXX 20XX
5
DOI: 10.1039/b000000x
A single-crystal to single-crystal transformable coordination polymer compound was hydrothermally synthesized. The structural rearrangement is induced by selecting a ligand that contains both strong and weaker coordinating groups. Both 10
hydrated and dehydrated structures were determined by single crystal X-ray analysis.
In the last two decades, coordination polymer and Metal-Organic Frameworks have developed into a hot research topic,1-3 due to
15
their potential application in areas such as gas adsorption, drug delivery, separation and catalysis.4-11 Compared with traditional
adsorbents and catalysts including zeolites and molecular sieves, MOFs present many unique properties due to their chemical nature and their intrinsic structures. The network structures of
20
MOF materials are built up by coordinate bonds between organic ligands and metal centres. It is possible, in a way, to target the synthesis of MOF materials with particular structures or properties by carefully selecting and designing the organic ligands.12, 13
25
One particularly unusual feature of MOFs is their structural flexibility. This can lead to remarkable effects such as ‘breathing’,14 pore-discriminating adsorption 15 and reversible single crystal to single crystal transformations.16 The latter class
of MOF materials responds to external stimuli such as heating or
30
guest exchange and reversible structure transformations can occur.17-19 This kind of material is classified as “third
generation”20 and has attracted increasing attention in technical
and research fields of gas adsorption,21, 22 sensing,23 and structure
investigation.24, 25
35
One strategy by which the transformations can be achieved uses ligands which form coordinating bonds of different binding strength. Once external stimulus is applied, the weaker bonds in the structure are more likely to be broken (and possibly reformed on removal or change of the stimulus), resulting in changes of
40
crystal structure. Meanwhile, the stronger coordinating bonds are more stable and strongly hold the whole framework so that the original structure can be recovered when the stimulus is stopped. One example of this strategy is the synthesis of Cu-SIP-3, a copper 5-sulfoisophthalate MOF that was reported by Xiao et
45
al.24, 26, 27. The ligand molecule, 5-sulfoisophthalate, contains one
sulfonate and two carboxylate groups. The sulfonate group is weaker than the strongly binding carboxylate groups. Coordinated water molecules leave the copper atoms upon
dehydration and the sulfonate group changes its coordinating
50
mode, resulting in a structural transformation of Cu-SIP-3. We have dubbed this type of material a ‘hemilabile MOF’ by analogy with homogenous catalysis where multidentate ligands can have different coordinating groups of varying strengths. This property of hemilability leads directly to an unusual property for
Cu-SIP-55
3; ultraselectivity towards the adsorption of coordinating gases such as NO, which is particularly interesting because of NO’s biological activity.28-30
In this paper, we report a new hemilabile MOF material that possesses the property of reversible crystal to
single-60
crystal transformation. The aim of the study was to show that targeted hemilabile MOFs can be prepared from ligands other than 5-sulfoisophthalate, and that the synthetic strategy may be general.
65
Experimental
All chemicals were commercially purchased and not further purified. Cu(NO3)2·3H2O (0.242 g, 1 mmol), 2-sulfoterephthalic
acid monosodium salt (0.268g, 1 mmol) and 4, 4’-bipyridyl (0.156 g, 1 mmol) were dissolved in the mixed solvent of water
70
(7.2 g, 0.4 mol) and ethanol (9.2 g, 0.2 mol). The solvent was stirring at room ambient condition for 15 min and transferred to a 50 ml Teflon-lined autoclave. After heating at 383K for 3 days, blue prism crystals were obtained. The products were filtered, washed with distilled water and ethanol and dried at 343K. 0.274
75
g of 1 was obtained. (Yield of 90.7 % (based on Cu).)
Thermogravimetric analysis (TGA) was run in a Netzsch TG 209 instrument under the atmosphere of air. The sample was heated in aluminium crucible at a heating rate of 10 Kmin-1. The hydrated
crystals were filled in a glass capillary and laid in a Schlenk
80
apparatus. According to the result of TGA, the dehydration was carried out at 503 K under vacuum. The colour of sample completely changed to dark green within 12 hours and gave the dehydrated sample. The capillary was quickly sealed under Ar to prevent the rehydration. The dehydrated sample was exposed to
85
open air for 24 hours to give the rehydrated sample. All the powder diffraction data of hydrated, dehydrated and rehydrated samples were collected in a STOE diffractometer under CuKα
radiation in Debye-Scherrer mode. The elemental analysis on C, H, N and S of the products was taken place on a Carlo Erba
90
collected at Station 11.3.1 of the Advanced Light Source, Lawrence Berkeley National Laboratory, using a wavelength of λ
=0.7749 Å. A Bruker AXS APEXII diffractometer was used for data collection, and the corresponding Bruker AXS APEXII software was used during data collection and reduction. Data of
5
hydrated samples are collected at 150 K and dehydrated samples at 500 K. Variation in crystal temperature was achieved via the cryostream with a ramp rate of 360 Kh-1. All the structures were
solved using direct method by the program SHELXS-97 and then refined on F2 using SHELXTL-97 software. (Sheldrick, G. M.
10
University of Gottingen, Germany, 1997.) Non-hydrogen atoms were refined anisotropically for hydrated structure. In the dehydrated structure, all carbon atoms were refined isotropically for a satisfactory refinement at high temperature. Other non-hydrogen atoms of the dehydrated structure were refined
15
anisotropically. Hydrogen atoms of the coordinated water molecules in the hydrated compound were fixed with DFIX restrains. Other hydrogen atoms were fixed based on idealized coordinates and refined with values of Uiso set to 1.2 times that of
the carrier atoms.
20
Results and Discussion
The materials were prepared from copper nitrate and two ligands; 2-sulfoterephthalate (STP), which contains one sulfonate group and two carboxylate groups, is used as the main ligand and 4, 4’-bipyridine (Bpy) is used as an auxiliary ligand. After
25
hydrothermal treatment at 383K for 3 days in a water/ethanol mixed solvent the ligands coordinate to copper ions to form a novel 3-dimensional compound, Cu3(STP)2(Bpy)·4H2O
(Compound 1).
Thermal gravimetric analysis (TGA) indicated a loss of
30
coordinated guest molecules at about 473K and powder X-ray diffraction (PXRD) showed the hydrated Compound 1
transformed to a crystalline dehydrated form, Cu3(STP)2(Bpy)
(Compound 2), after heating to 500K under vacuum. The reverse
transformation between 2 and 1 was also verified by PXRD after
35
rehydration (Figure 1).
Fig. 1 PXRD pattern of the crystal of (a) hydrated phase, (b) dehydrated phase and (c) rehydrated phase showing the changes in peak position on dehydration.
Variable temperature single-crystal X-ray analysis was used to solve the structures of hydrated 1 and dehydrated 2. The result of
40
variable temperature single-crystal X-ray analysis indicated the
reversible single-crystal to single-crystal transformation is caused by the variable coordinating modes of the weaker sulfonate ligand to the metal ions (Scheme 1) and supports the synthetic strategy. Full details of the synthesis and structural
45
characterisation are in the supporting online information. The TGA curve of 1 shows a weight loss of 7.42% ranging from 473K to 513K in air atmosphere. This weight loss corresponds to the removal of the coordinated molecules. The second step of weight loss started at about 543K is considered to be the
50
decomposition of organic ligands and the crystal starts to lose its crystallinity. (See Supporting information, Fig. S1.)
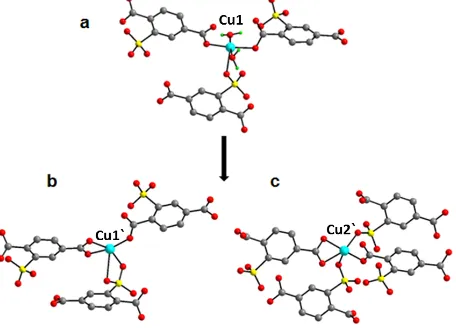
Fig.2 The coordination geometry of Cu1 in hydrated structure (a) and the changes of the geometries of Cu1` and Cu2` after dehydration (b and c). (Cu: turquoise, O: red,
S: yellow, C: grey, H: green, only water hydrogen is present for clarity.)
55
To investigate the dehydrated material the sample was heated at 500K under vacuum to investigate any structural change. The PXRD patterns of hydrated, dehydrated and rehydrated samples are shown in Fig. 1. The presence of the sharp peaks of the PXRD patterns indicates all the samples are crystalline. The
60
changes of the pattern peaks of the dehydrated sample indicate that a structural rearrangement occurs (Fig. 1b). The sample is reversibly rehydrated when the dehydrated sample is exposed to the moist air, which is proved by the identical powder pattern of rehydrated sample to that of the hydrated one (Fig. 1c).
65
In order to characterize the structures of hydrated and dehydrated phases and the structural rearrangement between them, variable temperature single crystal X-ray analysis was carried out. Data collection of the dehydrated sample was at 500 K.
The crystal structure of hydrated 1 contains two
70
crystallographically independent copper atoms, one a 5-coordinated (Cu1) and one 6-5-coordinated (Cu2). Each Cu1 atom is coordinated by three independent ligand molecules of STP and two water molecules (Fig. 2a). Three ligand oxygen atoms coordinate to Cu1, one of which comes from a sulfonate group
75
and the other two come from two individual carboxylate groups. The bond lengths of copper to carboxylate oxygen are 1.9912(16) Å and 1.9321(17) Å respectively. Compared to the Cu-O(C) bond, the bond length of copper to sulfonate oxygen is Cu1-O(S) = 2.2893(16) Å, consistent with the weaker binding of sulfonate
80
[image:2.595.307.535.172.336.2] [image:2.595.45.284.445.634.2]likely to be in the order of O(C) > O(water) > Cu1-O(S). The Cu2 atoms are coordinated by two STP molecules and two Bpy molecules. Each STP molecule chelates to the Cu2 by one sulfonate oxygen and carboxylate oxygen. The distance of
Cu2 to sulfonate oxygen is 2.2886(17) Å and bond length of
Cu2-5
[image:3.595.57.284.103.336.2]O(C) is 2.0433(16) Å.
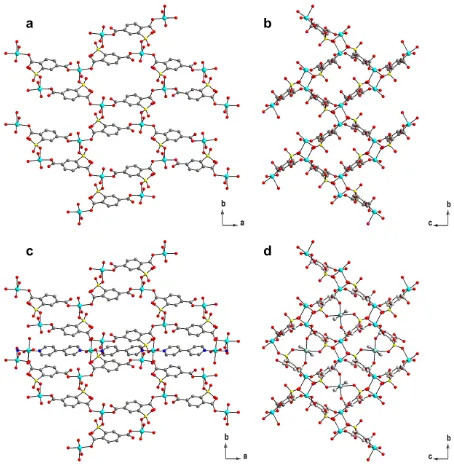
Fig. 3.Structures of compound 1. (a) Infinite layer structure (view along c axis) (b) The same layer viewed along a axis. (c) One layer with a Cu-Bpy-Cu chain that penetrates through this layer. (view along c axis) (d) One layer with four Cu-Bpy-Cu
chains penetrated through it. (view along a axis). (Cu: turquoise, O: red, S: yellow,
10
C: grey, N: blue. Hydrogen is not shown for clarity)
The overall crystal structure of hydrated compound 1 is shown in Fig. 3 and Fig. 4. Each Cu1 atom is coordinated by three surrounding STP ligands. A 2-dimensional layer structure is formed by repeating these Cu1-STP connections (Fig. 3a and 3b).
15
In addition to coordinating to the Cu1 atoms, each STP molecule also chelates to a Cu2 atom with one sulfonate and one carboxylate group. Every Cu2 atom is coordinated by two STP ligands that belong to two neighbouring layers. The Cu2 atoms link each 2-D layers to a 3-dimensional structure. The adjacent
20
Cu2 atoms in the same layer are linked by the 4, 4’-bipyridine molecule. The linear Cu-Bpy-Cu chains penetrate through the layer. Three layers and the Cu-Bpy-Cu chains are shown in Fig. 4 in different colours.
After carefully heating, two coordinated water molecules are
25
removed. Hydrated compound 1 transforms to its dehydrated phase of compound 2. An obvious change of the coordinating mode of the STP ligand is that one sulfonate group, which originally coordinated to two Cu atoms, now coordinates to three Cu atoms (Scheme 1). The changes of coordination environment
30
of Cu1 are shown in Fig. 2. In 2, four independent copper atoms are present. Two of them are originated from Cu1 of 1, denoted as Cu1` and Cu2`. The coordination geometries of two copper atoms are shown in fig. 2b and fig. 2c. Both of them are 5-coordinated. Cu1` is coordinated by three STP ligand molecules.
35
One STP molecule provides a bidentate carboxylate group. Another STP provides a bidentated sulfonate group and the third STP keeps the same monodentated carboxylate group as Cu1 of
Compound 1. Three bond lengths of Cu1` to carboxylate oxygen are 1.825(8) Å, 1.907(10) Å and 1.948(9) Å, respectively. One of
40
the Cu1`-O(S) bond is 1.960(9)Å long while the other bond is of 2.5354(116) Å, indicating the weaker binding strength of this sulfonate oxygen. Cu2` is coordinated by four STP molecules. One sulfonate group which was not coordinated to this copper in Compound 1 now monodentate to it and link the adjacent
45
parallelogram unit together. A previously monodentate carboxylate coordinates to Cu2` in a bidentate way. The bond lengths of Cu2`-O(C) are 1.856(9) Å, 1.869(12) Å and 2.121(14) Å. The bond lengths of Cu2`-O(S) are 1.975(10) Å and 2.193(10) Å repeectively, the former being considerably shorter than the
50
Cu-(O)S bond in the Compound 1. The quite similar bond lengths of Cu2`-O(C) and Cu2`-O(S) indicate the sulfonate group strongly coordinate to metal site after removal of water. The other two crystallographically indendent copper atoms, denoted as Cu3` and Cu4`, are originated from the Cu2 in Compound 1.
55
Similar to Cu2, they are still 6-coordinated by two STP molecules and two Bpy molecules in an inverse way. However, the bond lengths of copper to sulfonate oxygen increase to 2.334(10) Å of Cu3`-O(S) and 2.6635(92) Å of Cu4`-O(S). Meanwhile the
Cu-Bpy-Cu chain is twisted. The angle between two pyridine plane is
60
19.131(821)⁰. (see Supporting Information)
Fig.4. (a) three adjacent infinite layers (blue, purple and green) and (b) Cu-Bpy-Cu
[image:3.595.312.538.516.681.2]Scheme 1. Reversible changes of coordination modes of 2-sulfoterephthalate (Cu: turquoise, O: red, S: yellow, C: grey. Hydrogen on ligand is not shown for clarity)
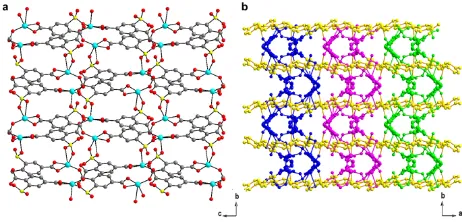
Fig. 5 (a) Double layer structure of compound 2 viewed along a axis. (Cu: turquoise, O: red, S: yellow, C: grey. Hydrogen is not shown for clarity) (b) crystal structure of
compound 2 view along c axis. Three double layers are shown in cyan, purple and
5
green. The Cu-Bpy-Cu chains are shown in yellow
A structural transformation is caused by the changes of the coordination mode of sulfonate groups. In compound 1, Cu1 and STP molecules form a infinite 2-D layer. When it is dehydrated, every Cu2` atom is coordinated by two sulfonate groups from the
10
adjacent layers. In this case a double-layer structure is formed (Fig. 5a). The copper atoms on the Cu-Bpy-Cu chains link the double layers to a 3-D structure. The Cu-Bpy-Cu chains pass through the whole structure. (Fig. 5b)
Conclusions
15To achieve a reversible single-crystal to single-crystal transformation of MOF materials, one synthetic strategy is to choose ligands that contain both weak and strong coordinating groups. Targeting this property, 2-sulfoterephthalate is selected as an eligible ligand. A copper MOF compound is hydrothermally
20
synthesized with this ligand and 4, 4’-Bipyridine. The as-synthesized compound lost the coordinating water when heated above 473K and a structural transformation occurred. The single crystal remains intact after dehydration so a variable temperature single-crystal analysis is feasible. Single crystal X-ray diffraction
25
and crystallography indicated the rearrangement of the coordination bonds, especially the coordination mode of sulfonate group to Cu. In the hydrated compound, the Cu-O bond lengths are in the order of Cu1-O(C) > Cu1-O(water) > Cu1-O(S). After dehydration, the sulfonate groups coordinated to the Cu which
30
lost the coordinating water more strongly than before. Obvious decrease of bond length of Cu-O(S) was observed. Though synthesis of metal organic framework with targeted property is still a challenge, dynamic transformation can be achieved by the strategy of selecting ligands. This kind of materials is interested
35
in selective gas adsorption, sensing and mechanism investigation. The work successfully demonstrates that the strategy of choosing ligands containing groups with varying coordination groups (e.g. those containing phosphonate groups31 or other unusual linkers in hybrid materials32 as well as sulfonate/carboxylate) is potentially
40
a general strategy for the preparation of hemilabile MOFs.
Acknowledgements
Y. T. acknowledges the support from China Scholarship Council. R.E.M. is a Royal Society Industry Fellow and thanks the British
45
Heart Foundation (NH/11/8/29253) and the EPSRC for support (EP/K005499/1 and EP/K025112/1). The Advanced Light Source is supported by the Director, Office of Science, Office of
Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02- 05CH11231.
50
Notes and references
a EaStChem School of Chemistry, University of St Andrews, Purdie Building, St Andrews KY16 9ST, UK. Fax: (+44) 1334-463-808 Prof. Russell E. Morris E-mail: [email protected]
b Department of Chemistry, College of Sciences, Shanghai University,
55
Shanghai, China, 200444
† Electronic Supplementary Information (ESI) available: Cif files of hydrated compound 1(CCDC 941460) and dehydrated compound 2(CCDC 941461); details of experimental sections; results of elemental analysis; PXRD pattern at different temperature; TGA curve and
60
crystallographic data can be found in supporting information. See DOI: 10.1039/b000000x/
1. J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213-1214.
65
2. S. Kitagawa, R. Kitaura and S.-i. Noro, Angew. Chem. Int. Ed., 2004, 43, 2334-2375.
3. S. L. James, Chem. Soc. Rev., 2003, 32, 276-288.
4. L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294-1314.
70
5. R. E. Morris and P. S. Wheatley, Angew. Chem. Int. Ed., 2008, 47, 4966-4981.
6. A. C. McKinlay, R. E. Morris, P. Horcajada, G. Férey, R. Gref, P. Couvreur and C. Serre, Angew. Chem. Int. Ed., 2010, 49, 6260-6266.
75
7. P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J.-S. Chang, Y. K. Hwang, V. Marsaud, P.-N. Bories, L. Cynober, S. Gil, G. Ferey, P. Couvreur and R. Gref, Nat Mater, 2010, 9, 172-178. 8. J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38,
80
1477-1504.
9. H. Guo, G. Zhu, I. J. Hewitt and S. Qiu, J. Am. Chem. Soc., 2009, 131, 1646-1647.
10. Y.-S. Li, F.-Y. Liang, H. Bux, A. Feldhoff, W.-S. Yang and J. Caro, Angew. Chem. Int. Ed., 2010, 49, 548-551.
85
11. J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450-1459.
12. M. O'Keeffe, Chem. Soc. Rev., 2009, 38, 1215-1217.
13. M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469-472.
90
14. C. Serre, C. Mellot-Draznieks, S. Surblé, N. Audebrand, Y. Filinchuk and G. Férey, Science, 2007, 315, 1828-1831.
15. M. I. H. Mohideen, B. Xiao, P. S. Wheatley, A. C. McKinlay, Y. Li, A. M. Z. Slawin, D. W. Aldous, N. F. Cessford, T. Düren, X. Zhao, R. Gill, K. M. Thomas, J. M. Griffin, S. E. Ashbrook
95
and R. E. Morris, Nat Chem, 2011, 3, 304-310.
16. K. Uemura, R. Matsuda and S. Kitagawa, J. Solid State Chem., 2005, 178, 2420-2429.
17. J.-P. Zhang, Y.-Y. Lin, W.-X. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2005, 127, 14162-14163.
100
18. E. Y. Lee and M. P. Suh, Angew. Chem. Int. Ed., 2004, 43, 2798-2801.
[image:4.595.48.279.52.163.2]20. S. Kitagawa and R. Matsuda, Coord. Chem. Rev., 2007, 251, 2490-2509.
21. X. Zhao, B. Xiao, A. J. Fletcher, K. M. Thomas, D. Bradshaw and M. J. Rosseinsky, Science, 2004, 306, 1012-1015.
22. K. Uemura, S. Kitagawa, M. Kondo, K. Fukui, R. Kitaura, H.-C.
5
Chang and T. Mizutani, Chem. Eur. J., 2002, 8, 3586-3600. 23. Y. Qi, F. Luo, Y. Che and J. Zheng, Cryst. Growth Des., 2007, 8,
606-611.
24. P. K. Allan, B. Xiao, S. J. Teat, J. W. Knight and R. E. Morris, J. Am. Chem. Soc., 2010, 132, 3605-3611.
10
25. X.-Y. Wang, M. Scancella and S. C. Sevov, Chem. Mater., 2007, 19, 4506-4513.
26. P. K. Allan, K. W. Chapman, P. J. Chupas, J. A. Hriljac, C. L. Renouf, T. C. A. Lucas and R. E. Morris, Chem. Sci., 2012, 3, 2559-2564.
15
27. B. Xiao, P. J. Byrne, P. S. Wheatley, D. S. Wragg, X. Zhao, A. J. Fletcher, K. M. Thomas, L. Peters, S. O. EvansJohn, J. E. Warren, W. Zhou and R. E. Morris, Nat Chem, 2009, 1, 289-294.
28. P. S. Wheatley, A. R. Butler, M. S. Crane, S. Fox, B. Xiao, A. G.
20
Rossi, I. L. Megson and R. E. Morris, J. Am. Chem. Soc., 2005, 128, 502-509.
29. R. M. J. Palmer, A. G. Ferrige and S. Moncada, Nature, 1987, 327, 524-526.
30. R. F. Furchgott and J. V. Zawadzki, Nature, 1980, 288, 373-376.
25
31. (a) G. B. Hix, D. S. Wragg, P. A. Wright and R. E. Morris, Dalton Trans., 1998, 3359-3361; (b) V. J. Carter, P. A. Wright, J. D. Gale, R. E. Morris, E. Sastre and J. PerezPariente, J. Mater. Chem., 1997, 7, 2287-2292
32. D. S. Wragg, G. B. Hix and R. E. Morris, J. Am. Chem. Soc., 1998,
30