Acta Cryst.(2001). E57, o143±o144 DOI: 101107/S1600536801000800 Bond, Edwards and Jones C9H16O4
o143
organic papers
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
Azelaic acid
Andrew D. Bond,* Marc R. Edwards and William Jones
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, England
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 180 K
Mean(C±C) = 0.002 AÊ
Rfactor = 0.037
wRfactor = 0.096
Data-to-parameter ratio = 17.0
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2001 International Union of Crystallography Printed in Great Britain ± all rights reserved
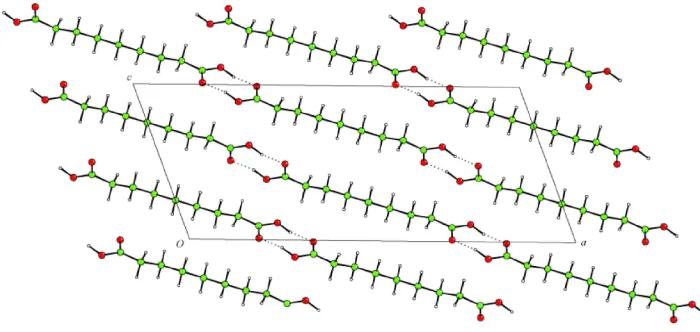
The crystal structure of heptane-1,7-dicarboxylic acid (azelaic acid), C9H16O4, has been redetermined at 180 K. The
molecular units have twofold symmetry and are linked via
the ubiquitoussyn±syncarboxylic acid dimer to form in®nite chains running approximately along the [401] vector.
Comment
Two polymorphs of azelaic acid, (I), have been reported previously: the form crystallizes in P21/c (Caspari, 1928;
Housty & Hospital, 1967) and theform crystallizes inC2/c
(Housty & Hospital, 1967). For both polymorphs, the struc-tures present in the CSD (AZELAC10 and AZELAC01; Allen & Kennard, 1993) are derived from room-temperature
data withRfactorsca10% and ambiguities in the treatment of H atoms. We have, therefore, re-examined azelaic acid and report here the structure of theform measured at 180 K to signi®cantly greater precision.
Experimental
Azelaic acid was obtained from Aldrich and recrystallized from ethanol.
Crystal data C9H16O4
Mr= 188.22 Monoclinic,C2/c a= 22.622 (2) AÊ
b= 4.7348 (2) AÊ
c= 9.6864 (7) AÊ = 110.559 (3) V= 971.5 (1) AÊ3
Z= 4
Dx= 1.287 Mg mÿ3 MoKradiation Cell parameters from 2161
re¯ections = 1.0±27.5
= 0.10 mmÿ1
T= 180 (2) K Plate, colourless 0.250.120.06 mm
Received 12 December 2000 Accepted 9 January 2001 Online 30 January 2001
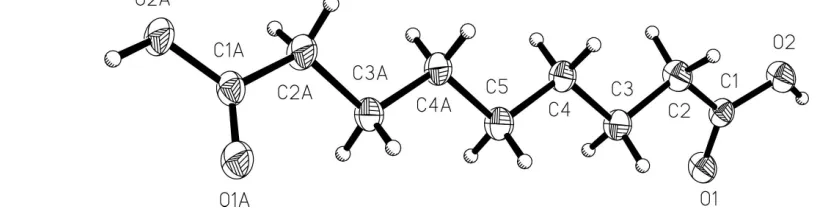
Figure 1
Data collection
Nonius KappaCCD diffractometer Thin-slice!and'scans Absorption correction: multi-scan
(SORTAV; Blessing, 1995)
Tmin= 0.907,Tmax= 0.994
3154 measured re¯ections 1085 independent re¯ections
913 re¯ections withI> 2(I)
Rint= 0.039
max= 27.4
h= 0!28
k=ÿ6!6
l=ÿ12!11
Re®nement Re®nement onF2
R[F2> 2(F2)] = 0.037
wR(F2) = 0.096
S= 1.09 1085 re¯ections 64 parameters H atoms: see below
w= 1/[2(F
o2) + (0.0328P)2 + 0.5150P]
whereP= (Fo2+ 2Fc2)/3 (/)max= 0.010
max= 0.25 e AÊÿ3
min=ÿ0.19 e AÊÿ3
Table 1
Hydrogen-bonding geometry (AÊ,).
DÐH A DÐH H A D A DÐH A
O2ÐH1 O1i 0.96 (2) 1.70 (2) 2.6576 (12) 173.5 (17)
Symmetry code: (i)1
2ÿx;12ÿy;ÿz.
The H atom of the carboxylic acid group was located in a differ-ence Fourier map and re®ned without restraint. All other H atoms were placed geometrically and allowed to ride during subsequent
re®nement with an isotropic displacement parameter ®xed at 1.2 times that for the C atom to which they are attached.
Data collection:COLLECT(Nonius, 1998); cell re®nement:HKL SCALEPACK(Otwinowski & Minor, 1997); data reduction: HKL DENZO and SCALEPACK (Otwinowski & Minor, 1997); program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to re®ne structure:SHELXL97 (Sheldrick, 1997); molecular graphics:XP(Sheldrick, 1993) andCAMERON(Watkinet al., 1996); software used to prepare material for publication:
SHELXL97.
We thank the EPSRC for ®nancial assistance with purchase of the CCD diffractometer.
References
Allen, F. H. & Kennard, O. (1993).Chem. Des. Autom. News,8, 1, 31±37. Blessing, R. H. (1995).Acta Cryst.A51, 33±38.
Caspari, W. A. (1928).J. Chem. Soc.pp. 3235±3241. Housty, J. & Hospital, M. (1967).Acta Cryst.22, 288±295.
Otwinowski, Z. & Minor, W. (1997).Methods Enzymol.276, 307±316. Nonius (1998).COLLECT. Nonius BV, Delft, The Netherlands. Sheldrick, G. M. (1993).XP. University of GoÈttingen, Germany.
Sheldrick, G. M. (1997). SHELXL97 and SHELXS97. University of GoÈttingen, Germany.
Watkin, D. J., Prout, C. K. & Pearce, L. J. (1996).CAMERON. Chemical Crystallography Laboratory, University of Oxford, England.
Figure 2
supporting information
sup-1
Acta Cryst. (2001). E57, o143–o144
supporting information
Acta Cryst. (2001). E57, o143–o144 [doi:10.1107/S1600536801000800]
Azelaic acid
Andrew D. Bond, Marc R. Edwards and William Jones
S1. Comment
Two polymorphs of azelaic acid, (I), have been reported previously: the α form crystallizes in P21/c (Caspari, 1928;
Housty & Hospital, 1967) and the β form crystallizes in C2/c (Housty & Hospital, 1967). For both polymorphs, the
structures present in the CSD (AZELAC10 and AZELAC01; Allen & Kennard, 1993) are derived from room-temperature
data with R factors ca 10% and ambiguities in the treatment of H atoms. We have, therefore, re-examined azelaic acid and
report here the structure of the β form measured at 180 K to significantly greater precision.
S2. Experimental
Azelaic acid was obtained from Aldrich and recrystallized from ethanol.
S3. Refinement
The H atom of the carboxylic acid group was located in a difference Fourier map and refined without restraint. All other
H atoms were placed geometrically and allowed to ride during subsequent refinement with an isotropic displacement
[image:3.610.77.485.422.531.2]parameter fixed at 1.2 times that for the C atom to which they are attached.
Figure 1
Figure 2
Projection onto (010) showing hydrogen-bonded chains running approximately along the [401] vector.
heptane-1,7-dicarboxylic acid
Crystal data
C9H16O4
Mr = 188.22
Monoclinic, C2/c a = 22.622 (2) Å b = 4.7348 (2) Å c = 9.6864 (7) Å β = 110.559 (3)° V = 971.5 (1) Å3
Z = 4 F(000) = 408
Dx = 1.287 Mg m−3
Melting point = 382–384 K Mo Kα radiation, λ = 0.71073 Å Cell parameters from 2161 reflections θ = 1.0–27.5°
µ = 0.10 mm−1
T = 180 K Plate, colourless 0.25 × 0.12 × 0.06 mm
Data collection
Nonius KappaCCD diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
Thin–slice ω and φ scans
Absorption correction: multi-scan (SORTAV; Blessing, 1995) Tmin = 0.907, Tmax = 0.994
3154 measured reflections 1085 independent reflections 913 reflections with I > 2σ(I) Rint = 0.039
θmax = 27.4°, θmin = 3.9°
h = 0→28 k = −6→6 l = −12→11
Refinement
Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.037
wR(F2) = 0.096
S = 1.09 1085 reflections 64 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(F
o2) + (0.0328P)2 + 0.515P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.010
Δρmax = 0.25 e Å−3
supporting information
sup-3
Acta Cryst. (2001). E57, o143–o144
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O1 0.18109 (4) 0.17201 (19) 0.01920 (10) 0.0312 (3) O2 0.24341 (4) 0.54364 (19) 0.11100 (10) 0.0314 (3)
H1 0.2682 (9) 0.465 (4) 0.057 (2) 0.079 (6)*
C1 0.19218 (5) 0.3923 (3) 0.09015 (12) 0.0244 (3) C2 0.14794 (6) 0.5227 (3) 0.15576 (14) 0.0307 (3)
H2A 0.1264 0.6833 0.0927 0.037*
H2B 0.1732 0.6004 0.2536 0.037*
C3 0.09798 (5) 0.3284 (3) 0.17462 (14) 0.0282 (3)
H3A 0.1183 0.1874 0.2523 0.034*
H3B 0.0767 0.2257 0.0813 0.034*
C4 0.04919 (5) 0.4949 (3) 0.21683 (13) 0.0270 (3)
H4A 0.0715 0.6108 0.3048 0.032*
H4B 0.0272 0.6258 0.1353 0.032*
C5 0.0000 0.3150 (4) 0.2500 0.0294 (4)
H5A −0.0213 0.1920 0.1643 0.035*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.0272 (5) 0.0337 (5) 0.0397 (5) −0.0021 (4) 0.0205 (4) −0.0071 (4) O2 0.0262 (5) 0.0340 (5) 0.0429 (5) −0.0043 (4) 0.0235 (4) −0.0055 (4) C1 0.0213 (6) 0.0289 (6) 0.0267 (6) 0.0018 (5) 0.0132 (4) 0.0030 (5) C2 0.0269 (6) 0.0322 (7) 0.0424 (7) −0.0027 (5) 0.0240 (6) −0.0064 (5) C3 0.0244 (6) 0.0299 (7) 0.0373 (7) 0.0008 (5) 0.0196 (5) −0.0003 (5) C4 0.0222 (6) 0.0300 (7) 0.0348 (7) 0.0002 (5) 0.0175 (5) −0.0012 (5) C5 0.0251 (9) 0.0317 (9) 0.0389 (9) 0.000 0.0206 (7) 0.000
Geometric parameters (Å, º)
O1—C1 1.2255 (15) C3—H3A 0.9900
O2—C1 1.3166 (14) C3—H3B 0.9900
O2—H1 0.96 (2) C4—C5 1.5224 (14)
C1—C2 1.4944 (16) C4—H4A 0.9900
C2—C3 1.5172 (16) C4—H4B 0.9900
C2—H2A 0.9900 C5—C4i 1.5224 (14)
C3—C4 1.5244 (15)
C1—O2—H1 110.9 (12) C2—C3—H3B 109.4
O1—C1—O2 123.02 (10) C4—C3—H3B 109.4
O1—C1—C2 123.67 (10) H3A—C3—H3B 108.0
O2—C1—C2 113.25 (10) C5—C4—C3 114.78 (11)
C1—C2—C3 116.05 (10) C5—C4—H4A 108.6
C1—C2—H2A 108.3 C3—C4—H4A 108.6
C3—C2—H2A 108.3 C5—C4—H4B 108.6
C1—C2—H2B 108.3 C3—C4—H4B 108.6
C3—C2—H2B 108.3 H4A—C4—H4B 107.5
H2A—C2—H2B 107.4 C4i—C5—C4 111.97 (14)
C2—C3—C4 111.04 (10) C4i—C5—H5A 109.2
C2—C3—H3A 109.4 C4—C5—H5A 109.2
C4—C3—H3A 109.4
O1—C1—C2—C3 19.17 (18) C2—C3—C4—C5 −175.40 (9)
O2—C1—C2—C3 −163.41 (10) C3—C4—C5—C4i −176.92 (11)
C1—C2—C3—C4 −170.14 (11)
Symmetry code: (i) −x, y, −z+1/2.
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
O2—H1···O1ii 0.96 (2) 1.70 (2) 2.6576 (12) 173.5 (17)