2 8 0 7 7 1 3 5 8 3
EXPRESSION SYSTEMS FOR STRUCTURAL STUDIES
OF MEDICALLY IMPORTANT PROTEIN DOMAINS
IN PAPILLOMAVIRUSES AND COM PLEM ENT
BY
CHRISTOPHER GRAEME ULLMAN
A THESIS SUBMITTED FOR THE
DEGREE OF DOCTOR OF PHILOSOPHY
IN THE
UNIVERSITY OF LONDON
• « •>. ■ c A D .
Department of Chemistry and Biochemistry, Royal Free Hospital School of Medicine, London NW3 2PF
ProQuest Number: 10106669
All rights reserved
INFORMATION TO ALL USERS
The quality of this reproduction is dependent upon the quality of the copy submitted.
In the unlikely event that the author did not send a complete manuscript
and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uest.
ProQuest 10106669
Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author.
All rights reserved.
This work is protected against unauthorized copying under Title 17, United States Code.
Microform Edition © ProQuest LLC.
ProQuest LLC
789 East Eisenhower Parkway
P.O. Box 1346
ABSTRACT
ACKNOWLEDGEMENTS
I wish to acknowledge the following people for their assistance and support during the course of this thesis. Certainly, without their patience many of these studies would not have been possible.
I would like to thank my supervisors, Dr. S.J. Perkins and Dr. V.C. Emery for providing me with the opportunity to study for this thesis at the Royal Free Hospital School of Medicine (RFHSM). I also thank them for giving me much of their valuable time and keeping me on the right tracks in my studies. In the same breath, I would like to acknowledge Dr. R.B. Sim, Oxford, who has been invaluable in providing me with encouragement and technical advice throughout my endeavours.
In the laboratory, I would like to thank Dr. P.I. Haris (RFHSM) for his assistance with FT-IR experiments; Dr. K.F. Smith and Mr. A.S. Nealis (RFHSM) for the help with computers and secondary structure analyses; Dr. A. Steinkasserer and Dr. A.J. Day (Oxford) for assistance with the yeast a
-factor expression system; Dr. 0. Bauer (National Institute for Medical Research) for his assistance with NMR measurements; Dr. P. Bavin (RFHSM) for HPV clones; Dr. J. Cason and Mrs. B. Kell (St. Thomas' Hospital School of Medicine) for providing HPV-16 E5 peptides; Dr. D.A. Galloway (Fred Hutchinson Cancer Research Center, Seattle) for providing the HPV-16 E7 yeast strain. I would also like to thank all the fellow inmates, past and present, of the Molecular Laboratory, Virology Dept. RFHSM, who have sweated blood and tears with me over the past few years (can I have my tips, buffers and pipettes back now please?).
Finally, where would I be without the support of my family and friends who have kept me sane throughout the course of this thesis? Thank you all.
TABLE OF CONTENTS
Page CHAPTER 1
RECOMBINANT PROTEINS AND EXPRESSION S YS TE M S ... 1
1.1 INTRODUCTION... 1
1.2 VIRAL EXPRESSION SYSTEMS FOR USE WITHIN ANIMAL CELLS ... 3
1.3 THE USE OF TRANSGENIC ANIMALS FOR HETEROLOGOUS GENE EXPRESSION . . . 4
1.4 THE BACULOVIRUS SYSTEM FOR EXPRESSION WITHIN INSECT CELLS ...5
1.5 EXPRESSION OF RECOMBINANT PROTEINS IN PLANT CELLS ... 5
1.6 FILAMENTOUS FUNGI EXPRESSION SYSTEMS...6
1.7 YEAST EXPRESSION SYSTEMS ... 6
1.7.1 Yeast expression plasm ids... 7
1.7.2 Yeast p rom oters... 6
1.7.2.1 Glucose inducible promoters ...8
1.7.2.2 Glucose repressible prom oters...8
1.7.2.3 Phosphate regulated pro m o te rs...9
1.7.3 Post-translational glycosylation of p ro te in s...11
1.7.4 The yeast a-factor expression system ...13
1.8 ESCHERICHIA COL/EXPRESSION SYSTEMS...18
1.8.1 E. coli plasmids and p ro m o te rs... 19
1.8.1.1 The coliphage X promoter (PJ and expression of recombinant proteins 19 1.8.1.2 The bacteriophage T7 promoter ...19
1.8.1.3 The fac promoter and the pKK expression system ...21
1.8.2 E. coli secretion systems ...21
1.8.3 E. coli fusion expression systems and the pGEX fusion system ...22
1.8.3.1 pGEX expression p la s m id s ...23
1.8.3.2 Immunological studies using purified GST fusion proteins ...25
1.8.3.3 Protein-protein interaction studies using GST fusion p ro te in s... 25
1.8.3.4 Structure/function studies on proteins expressed by the pGEX fusion protein system ...27
1.9 CONCLUSIONS ... 27
CHAPTER 2 PROTEIN DOMAINS... 29
2.1 DEFINITION OF A D O M AIN ... 29
2.2 TRANSCRIPTION FACTOR AND ZINC-BINDING DO M AINS... 30
2.2.1 The leucine zipper m o tif... 30
2.2.2 The helix-turn-helix (HTH) motif and the homeodomain ( H D )...30
2.2.3 The helix-loop-helix (HLH) domains ...31
2.2.4 The TATA-box binding protein domain ...32
2.2.5 Regulatory zinc-binding dom ains...33
motifs) ... 35
2.2.5 3 The GAL4 DNA-binding d o m a in ...36
2.2 5.4 The GATA-1 zinc binding d o m a in ... 37
2.2.5 5 The retroviral-like zinc finger domains ... 39
2.2.5 6 The RING finger d o m a in s ...40
2.2.5.7 The zinc ribbon nucleic acid binding domain ... 40
2.2.5.8 S u m m a ry ... 41
2.3 PAPILLOMAVIRUSES ... 42
2.3.1 The papillomavirus E6 and E7 proteins...43
2.3.1.1 The papillomavirus E7 protein...43
2.3.1.2 The papillomavirus E6 Protein ...46
2.3.2 The papillomavirus E5 p ro te in ...47
2.4 COMPLEMENT DOMAINS... 49
2.4.1 The complement cascade and domain stru cture ... 49
2.4.2 The short consensus repeat (S C R )...52
2.4.3 The epidermal growth factor domain ...54
2.4.4 The C3a, 04a and C5a d om ains... 54
2.4.5 The serine protease domain ... 55
2.4.6 Factor I - a complement control protein ...56
2.4.6.1 The factor I light c h a in ...58
2.4.6 2 The factor I heavy chain ...59
2.5 CONCLUSIONS ... 63
CHAPTER 3 TECHNIQUES FOR ANALYSIS OF PROTEIN STRUCTU RE... 64
3.1 STRUCTURE PREDICTION ... 64
3.1.1 The Chou-Fasman algorithm ...65
3.1.2 The Robson (Gamier, Osguthorpe and Robson or GOR) method ...66
3.1.3 Use of averaging methods with multiple sequence alignm ents...69
3.1.4 The PHD (Profile network from HeiDelberg) algorithm ... 70
3.1.5 Secondary structure predictions for membrane p ro te in s... 71
3.2 EXPERIMENTAL TECHNIQUES TO DETERMINE PROTEIN STRUCTURE ...71
3.2.1 X-ray crystallography ... 72
3.2.2 Nuclear magnetic resonance spectroscopy ...73
3.2.3 Circular dichroism spectroscopy ...73
3.2.4 Raman spectroscopy... 74
3.2.5 Infrared spectroscopy... 74
3.3 SUMMARY...79
CHAPTER 4 EXPRESSION AND SECRETION OF HPV-16 E7 PROTEIN BY S. CEREVISIAE AND DETERMINATION OF HPV EG and E7 SECONDARY S TR U C TU R E ... 80
4.1 INTRODUCTION...80
4.2 METHO DS...81
4.2.1 Sequence and secondary structure analyses for zinc finger p ro te in s ...81
4.2.2 Preparation of HPV-16 E7 p ro te in ...84
4.2.2.1 Expression of HPV-16 E7 in Saccharomyces cerevisiae ...84
4.2.3 Synthetic peptides of HPV-16 E7 pRb binding region and of a HPV-18 E6 zinc
finger d o m a in ... 89
4.2.4 FT-IR spectroscopy data analyses ...90
4.3 RESULTS AND DISCUSSION... 91
4.3.1 Alignment and averaged structure prediction for zinc finger CySjHiSj sequences in transcription factors ... 92
4.3.2 Alignment and averaged structure prediction for Cys^ zinc finger sequences in nuclear receptors... 100
4.3.3 Alignment and averaged structure prediction for zinc finger Cys^ sequences in papillomavirus E7 proteins... 112
4.3.4 Alignment and averaged structure prediction for zinc finger Cys^ sequences in papillomavirus E6 proteins... 126
4.3.5 FT-IR spectroscopy of HPV-16 E7 protein ...136
4.4 CONCLUSIONS ...144
CHAPTER 5 EXPRESSION OF THE FACTOR I HEAVY CHAIN LDLR2 AND FIM DOMAINS CLONED WITHIN THE SACCHAROMYCES CER£V/S/AEa-FACTOR EXPRESSION S Y S T E M 149 5.1 INTRODUCTION...149
5.2 METHODS ...151
5.2.1 Method for the large-scale preparation of factor I cDNA plasmid ... 151
5.2.1.1 Preparation of the bacterial cu ltu re ...152
5.2.1.2 Isolation and purification of plasmid DNA from the bacterial culture ... 152
5.2.2 Amplification of the factor I heavy chain domains by P C R ... 154
5.2.2.1 Primer preparation... 154
5.2.2 2 PCR amplification of the factor I heavy chain module coding sequences from the cDNA c lo n e ...154
5.2.3 Preparation of the PCR amplified factor I LD Lrl, LDLr2, LDLrl/2 and FIM coding sequences for cloning ... 159
5.2.4 Preparation and transformation of competent Escherichia coli (E. coli) bacterial cells with the plasmids pKNa and pMA91 ... 159
5.2.4.1 Preparation of JM105 competent c e lls ...161
5.2 4.2 Transformation of JM105 competent cells with pKNa and pMA91 . . 161
5.2.5 Preparation of the pKNa vector for Cloning Step 1 (Figure 5.2) ... 162
5.2.6 Ligation of the PCR amplified factor I heavy chain genes to p K N a ...163
5.2.7 Minipreparation (Miniprep) procedure for purifying plasmid DNA from transformed E. coli colonies ...163
5.2.7.1 PCR analysis of recombinant bacteria ...163
5.2.7.2 Small-scale (Miniprep) purification of plasmid DNA from recombinant bacteria ... 164
5.2.8 Analysis of miniprep purified recombinant plasmid DNA by restriction enzyme d ig estio n ... 164
5.2.9 Sequence analysis of pKNaLDLr2 and pKNaFIM recombinant plasmids ...165
5.2.9.1 Dénaturation of double-stranded DNA templates ... 165
5.2.9 2 Annealing of single stranded DNA templates to the sequencing primer ... 167
5.2.9.3 Labelling of the complementary DNA s tra n d ... 167
5.2.9.4 Termination of the labelling reaction...168
5.2.9.5 Analysis of DNA sequence by polyacrylamide gel electrophoresis . . . 168
5.2.10 Purification of the a-factor leader sequence/factor I heavy chain domain gene fusions from the recombinant plasmids pKNaFIM and pKNaLDLr2 . . . 170
5.2.11.1 Bgl\\ digestion of pMA91 ... 170
5.2.11.2 Dephosphorylation of linearized p M A 91... 172
5.2.12 Ligation of aFIM and aLDLr2 into Bgl\\ cut and dephosphorylated pMA91 . . . 172
5.2.13 Preparation of competent yeast cells ... 173
5.2.14 Transformation of competent yeast cells with pMALDLr2 and pMAFIM recombinant vectors...175
5.2.15 Small-scale protein expression from recombinant yeast c e lls ... 175
5.2.16 Recovery of proteins from the culture supernatants... 176
5.2.17 Glycerol stocks of recombinant y e a s t...176
5.2.16 Method for the large-scale production of the LDLr2 and FIM d o m a in s 176 5.2.19 Purification procedures tested for the harvested proteins ... 177
5.2.19.1 Reverse phase HPLC using a CIS column ... 178
5.2.19.2 Anion exchange on a Mono Q column using a FPLC s y s te m 178 5.2.19.3 Mono S cation exchange chromatography... 178
5.2.20 Analysis of binding of FIM to C l 8 reverse phase b e a d s...179
5.2.21 Effect of time and glucose concentration on FIM expression... 179
5.2.22 PCR analysis of the glycerol stocks of FIM y e a s t... 180
5.2.23 Analysis of intracellular proteins ... 181
5.3 RESULTS ...182
5.3.1 Results of PCR Amplification of the Factor I Heavy Chain G e n e s ... 182
5.3.2 Cloning of PCR Amplified Genes into the pKNa v e c to r... 183
5.3.3 DNA sequence analysis of pKNaLDLr2 and p K N a F IM ...189
5.3.4 Cloning of the aLDLr2 and aFIM DNA fusions with the yeast vector pMA91 . . . 191
5.3.5 Expression of recombinant proteins in y e a s t...194
5.3.5.1 Small-scale production of recombinant p ro te in s ... 195
5.3.5.2 Large-scale protein production of recombinant protein domains . . . . 195
5.3.5.3 Effect of time and glucose concentration on protein secretion ...198
5.3.5.4 Intracellular analysis of protein expression... 200
5.3.6 PCR analysis of the FIM glycerol s to c k s ... 200
5.4 DISCUSSION ... 204
CHAPTER 6 EXPRESSION OF FACTOR I HEAVY CHAIN DOMAINS WITHIN THE pKK233-2 AND pGEX-3X E. COU EXPRESSION SYSTEMS ...210
6.1 INTRODUCTION... 210
6.2 M ETHO DS... 211
6.2.1 Phosphorylation of PCR primers for use in cloning the factor I heavy chain genes within Esherichia coli expression systems... 211
6.2.2 Preparation of the pKK expression v e c to r... 212
6.2.3 Preparation of the pGEX expression vector ... 213
6.2.4 Ligation of factor I heavy chain genes into pKK233-2 ... 213
6.2.5 Ligation of factor I heavy chain genes into pGEX-3X ...213
6.2.6 Small-scale expression of factor I domains within the pKK233-2 and the pGEX-3X expression system s...214
6.2.7 Large-scale protein expression of the GST:factor I domain fusion proteins . . . . 215
6.2.8 Purification of the GST:factor I fusion proteins ... 216
6.2.9 Functional analysis of the factor I heavy chain dom ains... 216
6.2.10 Preparation of rabbit polyclonal antisera to the LDLr2, LD Lrl/2 and FIM GST fusion proteins ... 218
6.2.11 ELISA tests on GST and factor I using the antisera raised in rabbits to the recombinant factor I heavy chain d o m a in s ... 218
S-transferase fusion protein using the Factor Xa e n z y m e ... 221
6.2.14 Analysis of Factor Xa linked Sepharose-4B beads and cleavage of fusion proteins... 222
6.2.15 Cleavage of LDLrl/2:GST and LDLr2;GST fusion proteins by the proteases clostripain, elastase, Ancrod and soluble Factor Xa ...223
6.2.15.1 Cleavage by clostripain...223
6.2.15.2 Cleavage by elastase ...224
6.2.15.3 Cleavage by Ancrod ...224
6.2.15.4 Cleavage by soluble Factor X a ... 224
6.2.16 Purification of the LDLrl/2 and FIM domains by C l 8 reverse phase chromatography... 224
6.3 RESULTS... 226
6.3.1 PCR of phosphorylated factor I heavy chain domains ... 226
6.3.2 Preparation of the pKK233-2 plasmid and cloning of the factor I heavy chain domains ... 227
6.3.3 Preparation of the pGEX-3X plasmid and cloning of the factor I heavy chain dom ains...230
6.3.4 Small-scale protein expression of the recombinant pKK233-2 plasmids pKKLDLrl, pKKLDLr2 and pKKFIM ...230
6.3.5 Small scale protein expression form recombinant pGEXLDLr2, pGEXLDLr1/2 and the pGEXFIM bacteria ...232
6.3.6 Analysis of the factor I heavy chain gene sequence within the pGEX-3X vector . 235 6.3.7 Purification of the FIM:GST, LDLr2:GST and LDLrl/2:GST fusion proteins . . . . 235
6.3.8 Functional analysis of the factor I heavy chain using the LDLr2, LDLrl/2 and FIM glutathione S-transferase fusion proteins ... 237
6.3.9 Analysis of polyclonal antibodies raised to LDLr2:GST, LDLrl/2:GST and FIM:GST fusion pro te in s...239
6.3.10 Western blot analysis of the anti-factor I domain antisera ... 243
6.3.11 Activity of immobilized Factor Xa on Sepharose-4B beads on the GST fusion proteins... 245
6.3.12 Activity of soluble proteases against the fusion proteins... 246
6.3.12.1 Activity of clostripain on the LDLr2:GST and LDLrl/2:GST fusion p ro te in s ... 246
6.3.12.2 Activity of elastase on the LDLrl/2:GST fusion protein ...249
6.3.12.3 Activity of Ancrod on the LDLrl/2:GST fusion protein ...249
6.3.12.4 Activity of soluble Factor Xa on the LDLrl/2:GST fusion protein .. . 249
6.3.13 Purification of the isolated factor I heavy chain domains from the cleaved GST protein by C l 8 reverse phase chromatography... 250
6.4 CONCLUSIONS ... 254
CHAPTER 7 STRUCTURAL ANALYSIS OF A FULL-LENGTH HPV-16 E5 P E P T ID E ... 258
7.1 INTRODUCTION... 258
7.2 M ETHODS...259
7.2.1 Sequence alignments and prediction of hydrophobic and helical regions of H P V E 5 ... 259
7.2.2. Peptide synthesis and FT-IR spectroscopy ... 259
7.3 RESULTS ...261
7.3.1 Consensus sequence analysis of HPV-16 E5 protein ... 261
7.3.2 Prediction of transmembrane helices in the HPV E5 p ro te in ... 267
7.3.3 FT-IR Spectroscopy of HPV-16 E5 peptides... 271
CHAPTER 8
LIST OF ABBREVIATIONS
2D-NMR two dimensional nuclear magnetic resonance
Ad adenovirus
ADH alcohol dehydrogenase
APS ammonium persulphate
BPV bovine papillomavirus
BSA bovine serum albumin
CaMV cauliflower mosaic virus
CCP complement control protein
CD circular dichroism
GRPV cottontail rabbit papillomavirus
DBD DNA-binding domain
DEAE diethylaminoethyl
DNA deoxyribonucleic acid
DPV deer papillomavirus
DTT dithiothreitol
E6-AP E6-associated protein
EDTA ethylenediaminetetraacetic acid
EEPV European elk papillomavirus
EGF epidermal growth factor
EGFR epidermal grov/th factor receptor
ENO enolase
ER endoplasmic reticulum
EV epidermodysplasia verruciformis
FIM factor 1 module
FPLC fast performance liquid chromatography
FT-IR Fourier transform infra red
GADPH glyceraldehyde-3-phosphate dehydrogenase
GAL galactose
GST glutathione S-transferase
HD homeodomain
HEPES N-2-hydroxethylpiperazine-N'-2-ethanesulfonic acid
HIV human immunodeficiency virus
hnRNA heteronulear RNA
HPLC high performance liquid chromatography
HPV human papillomavirus
HSV herpes simplex virus
HTH helix-turn-helix
LATp latency promoter
LB Luria Bertani
LDLr low density lipoprotein receptor
LSB low salt buffer
LSK low salt Klenow
MHC major histocompatibility complex
MMPV Micromys minutus papillomavirus
MNPV Mastomys natalensis papillomavirus
mRNA messenger RNA
MSK medium salt Klenow
NCP nucleocapsid protein
CD optical density
ORF open reading frame
p-NA p-nitroanilide
PBS Dulbecco's A ' phosphate buffered saline
PCPV pygmy chimpanzee papillomavirus type 1
PCR polymerase chain reaction
PGK phosphoglycerate kinase
POU Pit-1, Oct-1, Oct-2 and uncBQ
pRb 105 kDa retinoblastoma gene product
RhPV Rhesus monkey papillomavirus type 1
RNA ribonucleic acid
RFHSM Royal Free Hospital School of Medicine
SBTI soya bean trypsin inhibitor
SCR short consensus repeat
SDS sodium dodecyl sulphate
SDS-PAGE sodium dodecyl sulphate polyacrylamide gel electrophoresis
SDW sterile deionized and distilled water
SH2, SH3 src-homology 2, (and 3)
SV simian virus
TBP TATA-box binding protein
TEMED NNN'N'-tetramethylethylenediamine
TFA trifluoroacetic acid
TFE trifluoroethanol
t-PA tissue-type pasminogen activator
TPI those phosphate isomerase
UAS upstream activation site
UV ultraviolet
vWF von Willebrand factor
LIST OF FIGURES
Page Figure 1.1: Common N-linked and 0-linked modifications of proteins in eukaryotic and
yeast cells...12 Figure 1.2: Structure of the Saccharomyces cerevisiae a-factor protein... 14 Figure 2.1: Structures of three DNA-binding domains containing z in c ... 34 Figure 2.2: Three recently identified classes of zinc-binding domain showing regions
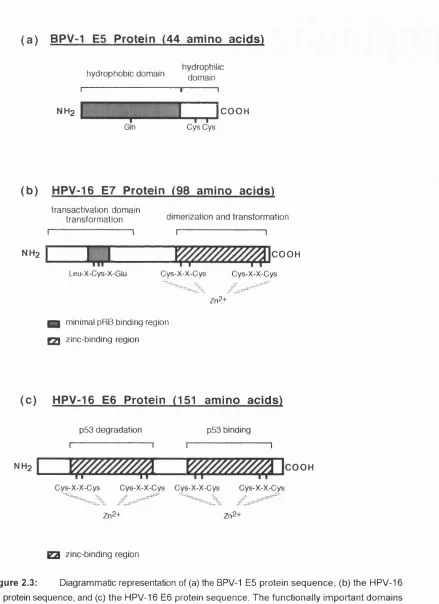
of p-sheet and a-helix...38 Figure 2.3: Diagrammatic representation of (a) the BPV-1 E5 protein sequence, (b) the
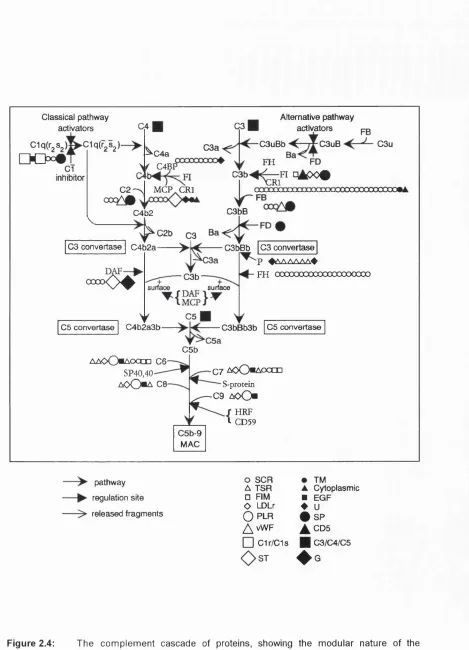
HPV-16 E7 protein sequence, and (c) the HPV-16 E6 protein sequence...44 Figure 2.4: The complement cascade of proteins, showing the modular nature of the
complement components ...50 Figure 2.5: Ribbon structures of the short consensus repeat (SCR) (a), and the
epidermal growth factor (EGF) (b)... 53 Figure 2.6: Possible domain structures for the factor I molecule as determined by solution
scattering experim ents...60 Figure 2.7: Conservation of amino acid residues in module superfamilies...61
Figure 3.1: Directional informational values (Y-axis) plotted against position relative to
j (X-axis) for the residues (a) Ala, (b) Gly, (c) Glu and (d) Lys... 67 Figure 3.2: Schematic representation of a Michelson interferometer ... 76 Figure 4.1 : An alignment of 160 transcription factor zinc finger sequences from 25
transcription factor proteins...93 Figure 4.2: Averaged predictions of the CySjHiSj zinc finger structure by the Robson 1987
(a), Robson 1978 (b), and the Chou-Fasman (c) algorithms generated from the multiple sequence alignment shown in Figure 4.1... 96 Figure 4.3: Assessment of correctly predicted secondary structure from nine different
structures of CySgHiSg zinc fingers ...97 Figure 4.4: An alignment of 72 nuclear hormone receptor DNA-binding domain
sequences... 102 Figure 4.5: Averaged predictions of the nuclear hormone receptor DNA-binding domain
structure generated from the Robson 1987, Robson 1978 and Chou-Fasman prediction algorithms from the multiple alignment shown in Figure 4.4... 104 Figure 4.6: Assessment of correctly predicted secondary structure from five known
structures of the Cys„ zinc-binding d o m a in s... 106 Figure 4.7a: Alignment of 25 E7 protein sequences from different human and primate
Figure 4.7b: Alignment of 24 E7 proteins that have greater than 30% sequence similarity to the HPV-16 E7 sequence from the PHD structure prediction
algorithm ... 114 Figure 4.8a: Robson 1987 (GOR III) prediction of human papillomavirus E7 protein structure
generated from the multiple sequence alignment shown in Figure 4.7a... 119 Figure 4.8b: Chou-Fasman prediction of human papillomavirus E7 protein structure
generated from the multiple sequence alignment shown in Figure 4.7a. . . . 120 Figure 4.8c: PHD prediction of the structure of the human papillomavirus E7 protein
generated from a multiple sequence alignment shown in Figure 4 .7 b ...121 Figure 4.9: A schematic model of the C-terminal region of HPV-16 E7 to illustrate the
proposed secondary structure elements of the zinc-binding region of E7
protein as predicted by the Robson 1987 secondary structure algorithm. . . . 125 Figure 4.10a: Alignment of 37 papillomavirus E6 proteins...127 Figure 4.1 Ob: Alignment of 22 papillomavirus E6 proteins that have greater than 30%
sequence similarity to the HPV-16 E6 sequence as produced by the PHD
secondary structure algorithm ... 129 Figure 4.11a: Robson 1987 (GOR III) prediction of papillomavirus E6 protein structure
generated from the alignment shown in Figure 4.10a... 133 Figure 4.11b: Chou-Fasman prediction of papillomavirus E6 protein structure generated
from the alignment shown in Figure 4.10a...134 Figure 4.11c: PHD prediction of papillomavirus E6 structure generated from the alternative
alignment shown in Figure 4.10b ...135 Figure 4.12: 15% SDS-PAGE gels of the purification of HPV-16 E7 secreted by
S. cerevisiae...138 Figure 4.13: The FT-IR absorbance spectra (a and c) and the second derivative spectra
(b and d) of HPV-16 E7 protein in HgO (a and b) and ^HgO (c and d)... 139 Figure 4.14a: Secondary structure of five zinc-binding domains ... 147 Figure 4.14b: Predicted secondary structure elements of the papillomavirus E6 and E7
proteins ... 148 Figure 5.1 : The nucleotide sequence of factor I heavy chain cDNA and the translated
sequence... 150 Figure 5.2: Representation of the cloning steps involved to direct the expression of
protein (e.g. LDLr2) from yeast cells using the a-factor leader sequence . . . 160 Figure 5.3: Preparation of the CL-6B Sepharose column used to desalt a denatured
DNA tem plate ... 166 Figure 5.4: Diagram of the Biotrap system (Schleicher and Schuell) for the
purification of DNA fragments from agarose gel slices...171 Figure 5.5: Analysis of the products from PCR of the factor I heavy chain coding
Figure 5.6: Plasmid map of pKNa and sequence of the prepro-a-factor le a d e r... 185 Figure 5.7: Identification of recombinant pKNa plasmids containing the FIM (a) and
LDLr2 (b) coding sequences of factor 1... 187
Figure 5.8: Restriction mapping of pKNa and pKNa containing the LDLr2
coding sequence that had undergone rearrangement... 188 Figure 5.9: Autoradiographs of recombinant FIM and LDLr2 DNA sequence... 190 Figure 5.10: Diagram showing sizes of the fragments expected from digestion of
pMA91 and recombinants pMAFIM and pMALDLr2 with Bgl\\ and H/ndlll
restriction endonucleases ... 192 Figure 5.11 : Preparations of pMAFIM and pMALDLr2 digested by H/ndlll and Bgl\\
restriction endonucleases... 193 Figure 5.13: 15% SDS-PAGE of the concentrated and partially purified proteins of
small-scale production of the recombinant proteins... 196 Figure 5.14: Analysis of proteins eluted from the C l 8 reverse phase beads in the
large-scale preparation of LDLr2 and FIM proteins by 15% SDS-PAGE
and silver stain... 197 Figure 5.15: Analysis of binding of the FIM domain to reverse phase beads in culture
by SDS-PAGE and silver s ta in ... 199 Figure 5.16: PCR of yeast colonies streaked from glycerol stocks onto YNB agar... 203 Figure 5.17: Conservation of amino acid residues in the LDLr domain found within the
complement proteins ...206 Figure 6.1: Plasmid maps for pKK233-2 (a) and pGEX-3X (b)... 228 Figure 6.2: Recombinant pKK233-2 isolates digested with Bamh\ and analysed by
electrophoresis through a 1.6% agarose gel...229 Figure 6.3: Recombinant pGEX-3X colonies digested with BamHI and analysed by
agarose gel electrophoresis ...231 Figure 6.4: 15% SDS-PAGE gels of the uninduced (Un) and 3 hours post-induction (Ind)
samples of the pKK233-2 recombinants...233 Figure 6.5: Time course studies on the expression of the FIM, LD Lrl/2 and LDLr2 GST
fusion proteins analysed by SDS-PAGE... 234 Figure 6.6: Illustration of the mutation found within isolate 7 of pGEXFIM...236 Figure 6.7: SDS-PAGE analysis of unreduced FIM:GST, LDLr2:GST and LDLrl/2:GST
purified from E. co//lysates using glutathione Sepharose-4B m a trix... 238 Figure 6.8: Autoradiographs of the inhibition studies performed using the FIM:GST fusion
protein...240 Figure 6.9: Graph to show the stimulation of cleavage of C3u by factor I with its
cofactor, factor H, in the presence of the fusion proteins FIM:GST,
Figure 6.10: Graphs of the results obtained from ELISAs using both GST (a) and factor I purified from blood plasma (b) as ta rg e ts ... 242
Figure 6.11 : Western blots of the antisera raised to the factor I heavy chain domains in
fusion with GST...244
Figure 6.12: Graph of Factor Xa protease activity against a test peptide, S-2222... 247
Figure 6.13: Time course study of activity of proteases on factor I GST fusion proteins. .. 248
Figure 6.14: Separation of the FIM (a) and the LDLr1/2 (b) domains from GST using CIS
reverse phase chromatography following cleavage by Factor Xa... 252
Figure 6.15: FIM and LD Lrl/2 domains purified by reverse phase chromatography... 253
Figure 7.1 : Multiple sequence alignment of twelve human papillomavirus E5 protein
sequences and the pygmy chimpanzee papillomavirus E5 protein sequence. 262
Figure 7.2: Alignment of 13 E5 protein of papillomaviruses with the sequences of the
gap junction (GJ) protein connexin ... 264
Figure 7.3: Kyte and Doolittle plot over a 9 residue window for the consensus HPV E5
sequence ... 265
Figure 7.4: Prediction of membrane helical regions of HPV E5 using PERSCAN v7.0. . 268
Figure 7.5: Helical wheels (100° angle between residues) of the 1®‘ (a), the 2"*^ (b) the
and 3''* (c) predicted helices of HPV-16 E5 protein... 269
Figure 7.6: FT-IR spectra of whole HPV-16 E5 synthetic peptide (a & b) and partial
LIST OF TABLES
Page
Table 1.1 : Examples of heterologous proteins expressed in Saccharomyces
cerevisiae and the promoters u s e d ...10
Table 1.2: Recombinant proteins secreted from yeast a-factor secretion system s...17
Table 1.3: Examples of recombinant proteins expressed in various E. coli expression
system s... 20
Table 1.4: Examples of the uses of GST fusion proteins... 24
Table 2.1 : Summary of the physiological concentrations and domain structures
of the complement components...51
Table 4.1: Stock 'amino acid mixture' containing 13 amino acids, a purine and a pyrimidine
nucleotide used to supplement yeast nitrogen base broth ... 85
Table 4.2: Sodium dodecyl sulphate-polyacrylamide gel electrophoresis system for the
resolution of proteins by their molecular w e ig h t... 86
Table 4.3: Comparison of the quantification of HPV-16 E7 structure by the Chou-Fasman,
the PHD and the Robson 1987 secondary structure prediction algorithms and by FT-IR spectroscopy... 141
Table 5.1 : List of primers for the amplification of the factor I heavy chain domains
by P C R ... 155
Table 5.2: Mixture used for the amplification of factor I heavy chain domains from
factor I cDNA by PCR ... 157
Table 5.3: PCR program used for the amplification of the factor I heavy chain
domains from factor I cDNA ...158
Table 5.4: PCR analysis of the FIM gene within yeast cultures at different glucose
concentrations and at various time intervals... 202
Table 7.1 : Regions of the consensus sequence (Figure 7.1 ) above the Kyte and
CHAPTER 1
RECOMBINANT PROTEINS AND EXPRESSION SYSTEMS
1.1 INTRODUCTION
Proteins have many crucial biological functions. These functions include enzyme catalysis, transport and storage, movement, mechanical support, immune protection, signal transduction and growth and differentiation (Stryer, 1988). Proteins are encoded by the cellular genome which is composed of deoxyribonucleic acid (DNA). DNA is transcribed to ribonucleic acid (RNA) and finally translated in the cytoplasm of the cell to protein. The protein sequence is composed of subunits known as amino acid residues which have a common backbone but different sidechains. Amino acids have the common structure NHgC^HRCOOH where R is the sidechain. The a-carbon is optically active, but only the L-isomer is found within proteins. Amino acids are linked end to end in a chain by peptide bonds formed by the condensation of the amino (-NHj) and carboxylic groups (-COOH) of neighbouring amino acid residues. Hence, proteins are also termed polypeptides. Twenty amino acids exist which are each encoded by specific DNA sequences of three bases (codon). These codons form a genetic code' that enables the amino acid sequence of a protein to be interpreted directly from the DNA sequence. The specific sequence of the amino acids in the protein is known as its primary structure. The three-dimensional folding of the polypeptide mainchain and the formation of periodic structures such as a-helix and p-sheet is termed the secondary structure of the protein. The full tertiary structure is formed from the interactions of the amino acid sidechains that are near or far apart in the polypeptide chain as defined by its secondary structure. The folding of several polypeptide chains within a molecule constitute the quartenary structure of the protein (Stryer,
1988).
1.2 VIRAL EXPRESSION SYSTEMS FOR USE WITHIN ANIMAL CELLS
Viruses are infectious agents that, in their simplest form, are composed of a viral genome (either single or double stranded DNA or RNA) surrounded by a protein coat and in some viruses a lipid envelope. Upon infection, viruses enter cells by either binding to a specific receptor on the cell surface or by fusion with the envelope to the plasma membrane. Following entry, the viral genome is uncoated and sequesters the translational and/or transcriptional machinery of the host to direct the synthesis of more virus particles, although the genome may exist in a latent, non-lytic stage. Because of the high level of expression of the viral encoded proteins from strong promoter elements within the virus genome, virus vectors are good vehicles for the expression of recombinant proteins within cultured animal cells. In addition, the advantage of mammalian proteins receiving authentic post- translational modifications is retained. Viruses that have been used for the expression of recombinant proteins include viruses that have both double stranded DNA and double and single (positive and negative sense) stranded RNA genomes.
study as gene transfer vectors (Majors, 1992). Likewise, the herpesvirus HSV-1 (herpes simplex virus- type 1), which is a neurotrophic human virus, has also been investigated as a gene transfer expression vector because of its ability to exist extrachromosomally in a latent, non-lytic, state within neurones and direct expression from promoters (LATp1 and LATp2) within a region of the genome responsible for latency (Glorioso et a i, 1992). The poxviruses encode their own RNA polymerases and transcription factors which makes the expression of recombinant proteins possible by introducing a plasmid carrying a poxvirus promoter and the recombinant gene (flanked by regions of poxvirus DNA) into cells infected with poxvirus. The foreign gene can be introduced into the poxvirus genome by recombination between the genome and regions of poxvirus DNA carried on the plasmid. Insertional inactivation of the thymidine kinase gene or cotransfer of a (3-galactosidase gene allows selection of recombinants (Moss, 1992). Amongst the RNA viruses, the single-stranded positive sense alphaviruses are the most versatile for the expression of recombinant proteins. Heterologous sequences can be engineered within the genome to provide recombinants that are replication and packaging competent or in place of the structural or non-structural genes to provide packaging defective or replication defective viruses respectively. Recombinant proteins expressed from these systems include human transferrin receptor, mouse dihydrofolate reductase and chicken lysozyme (Bredenbeek and Rice, 1992).
1.3 THE USE OF TRANSGENIC ANIMALS FOR HETEROLOGOUS GENE EXPRESSION
Vectors described in the previous section can be used to introduce foreign DNA sequences into the embryos of animals to result in a whole animal system, known as a transgenic animal (Jânne
et ai, 1992). The retroviruses are particularly useful in this respect because of their ability to integrate
study, it was calculated that a transgenic goat expressing recombinant tissue plasminogen activator in its milk at a level of 3 mg/ml would produce, in a day's milk, the equivalent to a daily harvest of a 1000 litre cell culture bioreactor (Janne et a i, 1992).
1.4 THE BACULOVIRUS SYSTEM FOR EXPRESSION WITHIN INSECT CELLS
Viruses also naturally infect insects. This virus family is known as the Baculoviridae which contain several viruses which infect a large range of insect hosts (Kidd and Emery, 1993). The
Autographs californica multiple nuclear polyhedrosis virus (AcMNPV) which infects Autographs
califomica (the fail army-worm) has been developed into a successful expression system. Infection
involves the production of large quantities of occluded viral nucleocapsids (polyhedra) which accumulate intracellularly. Polyhedra are composed of polyhedrin proteins which can constitute up to 50% of the total cellular protein. However, these proteins are dispensable in cell culture and can be replaced by heterologous genes placed under the control of the p10 or polyhedrin promoters. These genes are introduced by recombination with bacterial plasmids containing the promoter and termination sequences. Multiple gene sequences have also been expressed. The post-translational modifications are similar to animal cells including myristylation, acétylation of the N-terminal residue, phosphorylation, amidation, O- and N-linked glycosylation of short and long mannose forms. However, because the proteins are expressed in the late stage of infection (18 hours post-infection), modifications do not always occur (Bishop, 1992; Kidd and Emery, 1993).
1.5 EXPRESSION OF RECOMBINANT PROTEINS IN PLANT CELLS
monocotyledonous and dicotyledonous plants (Topfer et a i, 1993). Recently, these vectors have been utilised to express and secrete monoclonal antibodies in plant cells. The monoclonal antibodies were functional and were identical to the native proteins except for the glycosylation of the heavy chain. This has implications for cheap and almost limitless supply of monoclonal antibodies for passive immunization (Hiatt and Ma, 1993).
1.6 FILAMENTOUS FUNGI EXPRESSION SYSTEMS
Sections 1.2-1.5 have described the use of higher eukaryotes to express recombinant proteins. However, systems which utilize filamentous fungi or single cell organisms may be advantageous because they require simple and cheap growth media and can be cultured easily in large-scale fermentations. Industrial applications using strains of the filamentous fungi Aspergillus,
Pénicillium and Triichoderma are capable of secreting over 30 grams/litre of a specific protein e.g.
glucoamylase. This technology has been useful to direct the secretion of recombinant proteins from the glucoamylase gene (glaA) promoter and the glucoamylase signal sequence to a level of 0.2-10 mg/l for mammalian proteins such as prochymosin and human interleukin-6 (Cees et a i, 1992).
1.7 YEAST EXPRESSION SYSTEMS
In general, when unicellular expression systems are compared, yeast systems produce less recombinant protein than bacterial systems, but the product is usually correctly folded and purification can be relatively simple in comparison. Post-translational modifications such as O- and N-linked glycosylation occur within yeast cells. In the following section, both the intracellular and secretory yeast expression systems are described, with particular emphasis on the yeast a-factor expression system. This was used to express the human papillomavirus type 16 (HPV-16) E7 protein and the protein domains of the factor I heavy chain (Chapters 4 and 5).
for this yeast. Recently, however, other yeast strains (e.g. Pichia pestons) have been successfully adapted to produce very high levels of protein expression not possible with S. cerevisiae (Romanos
et ai, 1992; Gellissen et ai., 1992). There are many types of systems available for the production of
protein both within the cell and for secretion into the culture medium. However, secretion systems are advantageous because of easier purification procedures especially as S. cerevisiae naturally only secretes about 0.5% of its total cell protein (Bitter et ai, 1987). Correct folding and formation of the intramolecular disulphide bonds is catalysed by the enzyme disulphide isomerase, which is located on the luminal side of the endoplasmic reticulum in yeast (Freedman, 1984).
1.7.1 Yeast expression plasmids
There are three basic types of yeast plasmid used in molecular biology. The first is the yeast integrating plasmid (Yip). This plasmid is used to study eukaryotic DNA which can be introduced into the yeast genome by homologous recombination. The second system is the yeast centromere plasmid (YCp) which carries a copy of the yeast centromere. It is a low copy number, autonomously replicating, stable, properly segregated plasmid. The plasmid system most used for protein expression is based on the yeast episomal plasmid (YEp). This plasmid is a high-copy number, autonomously replicating, relatively stable, efficiently segregated plasmid carrying the replication origin from the yeast 2-micron plasmid (Botstein and Fink, 1988; Romanos et ai., 1992).
The expression plasmids used for heterologous protein expression within yeast are E. coii
regulation of mRNA production. Downstream of the UAS, the heterologous protein sequence may or may not contain signal sequences necessary for secretion of the final polypeptide product. Finally, termination sequences end the transcription process (Shuster, 1989).
1.7.2 Yeast promoters
The promoter elements in the expression cassette are derived from yeast since heterologous promoters are not fully functional (Kiss et al., 1982). Promoters from genes involved in carbon metabolism are often used for protein expression. These promoters are both inducible and repressible and are derived from genes known to produce large amounts of protein within the host (Shuster, 1989).
1.7.2.1 Glucose inducible promoters
Glycolytic gene promoters (Table 1.1) are regulated by glucose and the addition of glucose induces the promoter tenfold for the alcohol dehydrogenase I promoter (ADH1), twentyfold for enolase-2 (EN02) and two to seventyfold for glyceraldehyde-3-phosphate dehydrogenase (GAPDH)
gene promoters (Shuster, 1989). The large discrepancy noted for GAPDH corresponds to widely different induction values found in S. cerevislae and Candida respectively (Denis and Young, 1983).
1.7.2.2 Glucose repressible promoters
The regulation of the ADH2 UAS is well understood, and leads to the use of the activation sequence for the regulatable expression of heterologous gene products from different glycolytic promoters. These hybrids are regulated as wild type UAS/promoter sequences (Shuster, 1989). An
ADH2 UASfGAPDH hybrid promoter has been engineered for glucose-repressible expression and
successfully used for the expression of human insulin, HIV viral proteins, Plasmodium
circumsporozoite proteins and interferon (Table 1.1) (Barr eta!., 1987a, b, 1988). As this hybrid promoter can be fully regulated, expression of proteins that are toxic to the host strain can be controlled so the heterologous gene is retained by the host and expressed at an appropriate time.
The system based on the galactose promoter is also repressed by high levels of glucose but can be induced by the addition of galactose. The genes involved in this system include galactokinase
(GAL1), galactose transferase (GALT) and epimerase (GAL10) (Table 1.1) (Oshima, 1982). These
three GAL genes are linked on the S. cerevislae genome, with transcription of GAL1 and GAL10
under the control of a single UAS, known as UAS-G. UAS-G is regulated both by a positive activator and negative effector protein, GAL4 and GAL80 respectively (Romanos ef a/.,1992). The GAL1/10
gene promoter sequence Is also bidirectional and has been successfully used to produce simultaneously the heavy and light chains of the catalytic antibody 1F7 with chorismate mutase activity. The antibody appeared to be correctly folded and had the same enzymic activity as the hybridoma-derived catalytic antibody (Browdish et ai., 1991).
1.7.2.3 Phosphate regulated promoters
Table 1.1: Examples of Heterologous Proteins Expressed in Saccharomvces
cerevisiae and the Promoters Used
Protein Promoter References
Drosophila alcohol dehydrogenase GAL10 Atria n etal., 1990
calf chymosin PGK Mellor etal., 1983
GAL1 Gott etal., 1984
GAL1/10 Smith etal., 1985
Epstein-Barr virus envelope protein GAL1/10 Schulz etal., 1987
Aspergillus glucoamylase ENO Innis etal., 1985
hepatitis B antigen ADH1 Valenzuela etal., 1982
human tissue-type plasminogen activator P H 05 Hinnen etal., 1989
HIV envelope antigens GAPDH Barr etal., 1987b
human interferon-a PGK Hitzeman etal., 1983
human interferon-y PGK Derynck etal., 1983
GAL1/10 Bitter & Egan, 1988
human P-450IIC GAL1/10 Yasumori etal., 1989
human insulin TPI Thim etal., 1986
plant plasma H'^-ATPase GAL1 Villalba etal., 1992
Abbreviations
ADM alcohol dehydrogenase
ENO enolase
GAPDH glyceraldehyde-3-phosphate dehydrogenase
GAL galactose inducible promoters (see text)
PGK phosphoglycerate kinase
PHD phosphatase
TPI those phosphate isomerase
containing 35 cysteines, of which at least 24 are involved in disulphide bond formation (Hinnen etal.,
1989). Although most of the recombinant protein accumulated in the Golgi of the yeast cells, its activities were comparable with human t-PA in assays measuring the conversion of plasminogen to plasmin suggesting a correct conformation. However, the level of glycosylation of t-PA produced in yeast cells was four times that of human t-PA derived from a melanoma cell line (Hinnen etal.,
1989).
1.7.3 Post-translational glvcosvlation of proteins
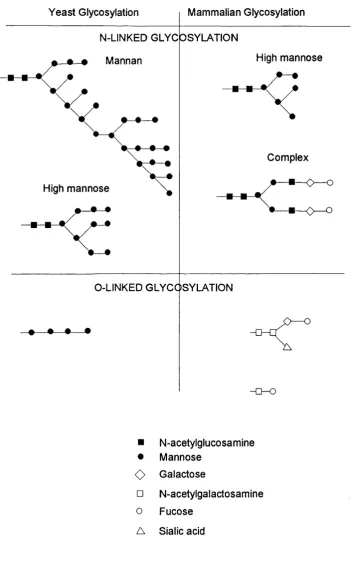
Unlike bacteria, yeast have the potential to glycosylate heterologous proteins. O-linked glycosylation on the hydroxyl groups of serine or threonine residues with short manno- oligosaccharides occurs within the endoplasmic reticulum of yeast cells. Modification of this carbohydrate, (Ser/Thr)-Man-Mano.3, is performed in the Golgi apparatus. There is no marker sequence for yeast O-linked glycosylation although proline is thought to be advantageous because It increases the accessibility of the site for glycosylation. However, this type of glycosylation in yeast differs markedly from mammalian O-linked glycosylation which may involve the addition of a variety of sugars in a branched chain form (Figure 1.1) (Delvin, 1992).
Yeast Glycosylation Mammalian Glycosylation
N-LINKED GLYC OSYLATION Mannan
High mannose
High mannose
Complex
<
O — oO-LINKED GLYCOSYLATION
• — • •
■ N-acetylglucosamine
• Mannose
<0 Galactose
□ N-acetylgalactosamine
0 Fucose
A Sialic acid
Figure 1.1 : Common N-linked and O-linked modifications of proteins in eukaryotic and yeast
1.7.4 The yeast «-factor expression system
Yeast secrete protein in a way that is very similar to that of mammalian cells. The proteins are translated into inactive precursors containing a signal peptide which is cleaved at the plasma membrane to result in an active secreted product. This conservation in the mechanism of protein secretion between the different organisms has been demonstrated by the use of heterologous signal peptides to achieve protein secretion in S. cerevisiae. Recombinant human interferons have been secreted by yeast under the control of the PGK promoter using the interferon signal sequences. In contrast, no secretion was observed when the mature IFN gene was expressed, without the signal sequence (Hitzeman et ai., 1983). However, more recent work has made use of homologous yeast signal peptides from the yeast «-factor pheromone precursor, invertase and acid phosphatase
{PH05-) precursors for the secretion of recombinant proteins.
Although the yeast a-factor system is the most favoured of the secretion systems, it is not the only system developed. In one of the first accounts of the use of a protein secretion system, the invertase gene (SUC2) encoding the first 60 amino acids of the invertase protein (including the signal sequence) was fused to (3-galactosidase. Unfortunately, p-galactosidase activity was associated with the endoplasmic reticulum rather than being secreted (Emr et ai., 1984). The yeast invertase signal sequence (11 amino acids), used in combination with the GAL1 promoter, was shown to secrete prochymosin, however a major proportion of recombinant protein again remained within the yeast cytoplasm. The secreted prochymosin was soluble and fully active compared to internally located prochymosin which had an incorrect conformation (Smith et a i, 1985). Both the PH05- and the invertase signals have been successfully used to secrete biologically active leech hirudin, a 7 kDa protein with three disulphide bonds (see section 1.7.2.3) (Hinnen et ai., 1989).
The yeast a-factor expression system has been studied in great detail. It is based upon the secreted yeast pheromone, known as the a-factor protein, encoded at the MAT locus within the yeast genome (Nasmyth, 1983). The a-factor is a 13 amino acid peptide secreted by haploid mating type
23 57 67
hydrophobic signal sequence
leader sequence s i (xl s2 <t2 s3 « 3 S4 (x4
Lys Arg GI u A la As p A laG lu A la TrpHis TrpLeuLysProGlyGlnProMetTyr
SPACER PEPTIDE MATURE a-FACTO R PHEROMONE
a-factor arrests the cell cycle in MATa cells at a specific point as a prelude to mating. The small a - factor peptide is the final active product of a large precursor protein encoded by tv\/o separate structural genes MFa1 and MFo2. The major structural gene, MFa1, is translated into a 165 amino acid prepro-a-factor containing an 83 residue leader sequence with three Asn-linked glycosylation sites. Following this are four tandem repeats of the a-factor peptide, each preceded by a short spacer sequence of Lys-Arg-(Glu/Asp-Ala)2^ (Figure 1.2). The protein encoded by the minor a-factor structural gene, MFo2, has only two a-factor peptide repeats. Pheromone precursors from other
Saccharomyces strains may contain between three and five repeats of the mature a-factor and a
homologous protein found within the yeast Kluyveromyces lactis contains four repeats (Kurjan and Herskowitz, 1982; Julius etal., 1983; Brake etal., 1984; Brake, 1989).
The processing of prepro-a-factor involves four different enzymes acting at various stages of the transport to the cell membrane. Initial core glycosylation of the pro- segment takes place early after translocation into the endoplasmic reticulum and is followed by outer chain glycosylation and the first proteolytic cleavage step in the Golgi apparatus. This cleavage removes the pre segment and is catalysed by a signal peptidase. The second cleavage of the precursor is instigated by a membrane-bound, calcium-dependent endoprotease. This protease, KEX2, cleaves on the carboxyl side of the Lys-Arg sequence found within the spacer region preceding each a-factor repeat (Julius
et al., 1984). Two other proteases process the precursor within secretory vesicles. These are a
dipeptidyl aminopeptidase (STE13) that removes the amino terminus of each repeat, and a carboxypeptidase (KEX1) that removes the arginyl and lysyl residues at the C-terminus of each of the first three repeats (Dmochowska etal., 1987).
hEGF synthesized within the recombinant yeast was secreted. Bitter et al. (1984) expressed the small peptide p-endorphin (31 amino acids) and a-interferon, IFN-a (166 amino acids), within the a-factor secretion system. In addition to the normal processing of the a-factor leader sequence from the recombinant protein domain, additional cleavages occurred to the carboxyl side of the Lys residues in p-endorphin probably during the passage of the hybrid protein through the normal secretory pathway. As some of the cleavage sites were digested preferentially, it was suggested that cleavage is conformation dependent. Most of the recombinant IFN-a remained intact on secretion. Further studies concerning the secretion of recombinant p-endorphin, calcitonin and IFN-a demonstrated that the smaller proteins (p-endorphin and calcitonin) are secreted more efficiently than the larger protein (IFN-a) which remained cell-associated. Secreted IFN-a had the correct disulphide arrangement but a proportion of unprocessed pro-a-factor/IFN-a fusion proteins were also found within the medium. Inclusion of (Glu-Ala); in this case increased the amount of fusion processed at the cell surface but did not increase the efficiency of recombinant protein secretion (Zsebo etal., 1986). Other workers using an a-factor system with a Lys-Arg-(Glu-Ala)z processing site have reported that 50% of the recombinant IFN-a was secreted (Singh et al., 1984).
Human parathyroid hormone expressed in and secreted from the a-factor expression system was also found to contain internal cleavage sites especially after a pair of dibasic amino acids. The combination of a protease-deficient yeast strain and increasing the amount of Casamino acids within the growth medium increased the level of intact product (Gabrielsen etal., 1990). Expression of the insulin precursor within the system has taken advantage of the a-factor processing enzyme by including a spacer region (containing dibasic residues) in place of the native C peptide between the A and B chains of proinsulin. The proinsulin was found to be processed at the spacer region to yield insulin (Thim etal., 1986).
Table 1.2 Recombinant Proteins Secreted from Yeast a-factor Secretion Systems
Recombinant proteins secreted from Saccharomyces cerevisiae:
Epidermal growth factor Interferon-a
Calcitonin Prochymosin Carboxypeptidase A Interleukin 2
a-amylase
Insulin-like growth factor I Atrial natriuretic peptide
Connective tissue activating peptide
Granulocyte-macrophage colony stimulating factor Proinsulin and analogues
Epstein-Barr virus glycoprotein Fibroblast grovyth factors Human parathyroid hormone
Brake et al. (1984) Bitter et al. (1984) Singh et al. (1984) Bitter et al. (1984) Smith et al. (1985) Gardell et al. (1985) Shaw et al. (1985) Miyajima et al. (1985) Filho étal. (1986) Ernst (1986)
Vlasuk et al. (1986) Mullenbach et al (1986) Miyajima et al. (1986) Ernst et al. (1987) Thim et al. (1986) Schultz etal. (1987)
Barr etal. (1988)
Gabrielsen et al. (1990)
Recombinant proteins secreted from non S. cerevislae yeast:
Kluveromyces lactis
Chymosin Rietveld et al. (1988)
PIchIa pastorls
connective tissue activating peptide-lll (CTAP-III), enzymes such as a-amylase, and viral glycoproteins such as the major envelope glycoprotein (gp350) of Epstein-Barr virus. Recombinant GM-CSF and gp350 exhibit heterogeneous glycosylation but still retained biological activity (Schultz
etal., 1987).
The yeast a-factor system has also been successfully used to express various protein domains rather than whole proteins themselves. Protein domains that have been expressed include the human epidermal growth factor in isolation and from human factor IX (Cooke at a!., 1990; Dudgeon etal., 1990; Handford etal., 1990), the fibronectin type I module (Baron etal., 1990), the human fibronectin type III module (Main etal., 1992), and the S*'’, the 16^\ and the 15*^-16*^ pair of short consensus repeats (SCRs) of factor H (Barlow etal., 1991,1992, 1993; Norman etal., 1991). These domains have been expressed in large enough quantities for their structures to be determined by ’H nuclear magnetic resonance (NMR) techniques.
1.8 ESCHERICHIA CPU EXPRESSION SYSTEMS
domains (Chapter 6).
1.8.1 E. coli plasmids and promoters
The plasmids used for expression of recombinant proteins in E. coli are often derived from the general cloning vector pBR322 (Bolivar etal., 1977). They contain an origin of replication and an antibiotic resistance gene (ampiciiiin or tetracyciin) so that cells carrying the plasmid can be selected. In general, although E coli promoters (described in section 1.1) are weak, promoters such as /acuvS
(lac), trp, tac (a trp-lac hybrid promoter), the X phage promoter and the T7 phage promoter have
been used to direct the efficient expression of recombinant genes within bacterial cells (see below).
1.8.1.1 The coliohaae X promoter (P.) and expression of recombinant proteins
The leftwards operator and promoter of phage X, (PJ are tightly regulated by the X repressor protein, ci. The P^ promoter sequence has been engineered into pBR322 derived plasmids (e.g. pHE6 (Milman, 1987)) and can be controlled by a single temperature shift from 28°C to 42°C in temperature sensitive mutants that synthesize thermolabile repressors (such as pc/857). The temperature shift disables the repressor protein and expression is directed from the promoter sequence (Bernard etal., 1979). Examples of recombinant proteins expressed in this system are given in Table 1.3.
1.8.1.2 The bacteriophage T7 promoter
The bacteriophage T7 promoter is solely recognized by the T7 RNA polymerase providing a means of controlling gene expression. Therefore, by insertion of a recombinant gene sequence into a vector carrying the T7 promoter, only the cloned gene is transcribed by RNA polymerase genes. In the pET series of vectors (and pRSET (Schoepfer, 1993)), the T7 gene 10 promoter, the T7 gene
Table 1.3: Examples of Recombinant Proteins Expressed in Various E. coli Expression Systems
System Recombinant protein References
A-Pt promoter SV40 small antigen Derom etal., 1982
human interferon-y Remault etal., 1987 human tumour necrosis factor Remault etal., 1987 human interleukin 2 Remault etal., 1987
77 promoter HIV-1 protease Cheng etal., 1990
HSV-1 ribonucleotide reductase small unit (R2)
Yang etal., 1991
human retinoic acid receptor Lankinen etal., 1991
papain Taylor etal., 1992
tac promoter (pKK vector) hepatitis B pre-S antigen Amann & Brosius, 1985 chicken those phosphate
isomerase
Straus & Gilbert, 1985
protein-tyrosine phosphatase Hashimoto etal., 1992 high molecular weight kininogen Kunapuli etal., 1992 lipoprotein secretion system human growth hormone Duffaud etal., 1987
human galactosyltransferase Aoki etal., 1990 phosphatase secretion anti-digoxin antibody variable Anthony etal., 1992
system region fragment
human epidermal growth factor Oka etal., 1985
Stapfi. protein A secretion insulin-like growth factors 1 & II Lôwenadier etal., 1987
system Uhlén & Abrahamsen, 1989
RNA polymerase is encoded by T7 gene 1 which may be provided on a bacteriophage X, vector or by insertion of the gene into the E. coli chromosome (Tabor and Richardson, 1985; Studier and Moffat, 1986). The T7 gene 1, and therefore expression, can be placed under the control of a lac
promoter and the X c/857 temperature sensitive repressor (Studier and Moffat, 1986). Both native protein sequences and protein fusions with the T7 gene 10 protein can be produced from the various pET vectors, however the expressed fusion proteins are insoluble (Rosenberg etal., 1987). This vector system has been used on many occasions to produce recombinant proteins. Some examples of these are given in Table 1.3.
1.8.1.3 The tac promoter and the pKK expression svstem
Both the lac and the trp promoter sequences have been used to direct the synthesis of recombinant proteins in E. coll (Danley etal., 1989). However, a trp-lac hybrid promoter has been engineered that has the -35 trp promoter region, the -10 lacZ promoter region and the lac operator. The hybrid promoter (named the tac or trc promoter) is both strong and can be regulated by the addition of isopropyl-p-D-thiogalactoside (IPTG) (Amann etal., 1983; de Boer etal., 1983). The tac
promoter, a lacZ ribosome-binding site (RBS) followed by an ATG translation initiation codon eight nucleotides downstream of the RBS have been cloned into a pBR322 based plasmid to generate the pKK series of expression vectors. An ATG codon located within a Nco\ restriction endonuclease site allows the cloning of heterologous genes within the pKK plasmids either by sticky-ended or by blunt- ended ligation and the expression of the product in an unfused state within E. coll (Amann and Brosius, 1985). The pKK expression system has been used to express a variety of proteins from viruses, proteins involved in signal transduction pathways, plasma proteins and biologically active enzymes (Table 1.3).
1.8.2 E. coli secretion svstems
the production of proteins harmful to the cell. Also, the periplasmic space is an oxidizing environment compared to the reducing environment of the cytoplasm (Duffaud etal., 1987).
Vectors have been engineered (pIN) which contain the bacterial lipoprotein or the OmpA (both major outer membrane proteins) signal peptide sequences. The efficient lipoprotein gene (/pp) promoter combined with the lac promoter operator allows the inducible expression of the cloned genes in the presence of a lac inducer such as IPTG. This leads to recombinant protein levels of possibly higher than 20% of the total cellular protein (Duffaud etal., 1987). Other secretion systems have used the alkaline phosphatase promoter (phoA) and signal sequence to direct the synthesis of recombinant proteins into the periplasm (Oka etal., 1985). The staphylococcal protein A promoter, signal sequence and protein have been used within E. coli to direct the secretion of fused products into the bacterial growth medium (Lôwenadier etal., 1987; Uhlén and Abrahamsen, 1989).
1.8.3 E. coli fusion expression svstems and the pGEX fusion svstem
In E coli expression systems, foreign proteins may be rapidly degraded by host proteases. The stability and the level of expression of recombinant domains can be improved by fusion with a stable and non-toxic protein that can be over-expressed within bacterial cells. The fusion protein can also aid purification of the recombinant protein from bacterial cell lysates using affinity matrices specific for the fusion domain. A number of fusion systems exist for E. co//which use proteins (e.g. P-galactosidase, chloramphenicol acetyltransferase, glutathione transferase, maltose binding protein and phosphate binding protein) or synthetic peptides (e.g. poly-Arg, -Glu or -His residues). These fusion components may be placed either N-terminal or C-terminal to the recombinant gene sequence. It is possible to cleave the fusion component from the expressed product following purification, either chemically, or enzymatically in vectors that have been engineered to contain the appropriate enzyme cleavage site (Uhlén and Moks, 1990). For example, in the p-galactosidase
1.8.3.1 pGEX expression plasmids
The pGEX expression system has been widely used and citations in the literature describe the versatility of the system (Table 1.4). The pGEX expression system is based upon the inducible expression of the 26 kDa Schistosoma japonicum glutathione S-transferase (GST) protein within bacterial cells. The gene encoding the protein is carried on a plasmid vector with expression of GST under the control of the tac promoter (section 1.8.1.3). Unique restriction endonuclease sites exist at the 3' end of the GST gene to allow the insertion of heterologous gene sequences. Three vectors were originally designed, pGEX-1, pGEX-2T and pGEX-3X. The pGEX-2T and pGEX-3X vectors contain a thrombin and Factor Xa cleavage site, respectively, at the C-terminus of the GST protein, although pGEX-3X has been engineered to contain a thrombin cleavage site, yielding the vector pGEX-3T (Frorath etal., 1992). These sites enable the recombinant domain to be cleaved from GST after purification. The GST fusion protein can easily be purified by glutathione-Sepharose affinity chromatography. The fusion protein binds to reduced glutathione linked to a Sepharose matrix and is eluted by the addition of soluble reduced glutathione. Alternatively, the fusion protein can be cleaved on the glutathione Sepharose matrix (Abath and Simpson, 1991; Olsen and Mohapatra, 1992). Cleavage of the fusion protein by the appropriate protease separates GST from the recombinant domain (Smith and Johnson, 1988).
Table 1.4: Examples of the uses of GST Fusion Proteins
Function Description Reference
Immunological studies Development of an ELISA sandwich technique using immobilized glutathione
Rabin etal., 1992
Epitope mapping of HPV-33 minor capsid protein epitopes
Volpers etal., 1993
Protection of sheep against larval tapeworms
Johnson etal., 1989
As antigens to raise antibodies against proteins to determine orientations and locations of proteins
Baines etal., 1993 Tomlinson etal., 1993 Vanscheeuwijck et al.,
1993 Studies of protein-nucleic
acid interaction
Recombinant GST:GCN4 (yeast DNA-binding protein) used in detection of HIV in blood samples
Kemp etal., 1989
GST:HIV p i 5 protein (containing zinc binding domains) stimulated dimerization of viral DMA
Weiss etal., 1992
Identification of the specific DMA consensus sequence bound by GST:N-Myc protein
Alex etal., 1992
Studies of protein-protein interactions
Screening of cell lysates for proteins that interact with GST:pRb (see text)
Kaelin etal., 1991
Interactions of SH2 domains with tyrosine-phosphorylated peptides of PDGF p-receptor (see text)
Panayotou etal., 1993
Structural studies Crystals of E. coli F^ portion of ATPase
Codd etal., 1992
NMR structure of the T lymphocyte CD2 antigen
Driscoll etal., 1991
Deletion mutagenesis to characterize both high and low affinity calcium binding site in calreticulin
The pGEX-3X plasmid has also been engineered so that it can be used for the expression of proteins from dicistronic genes. In the recombinant plasmids, the GST protein has a ribosome binding site (RBS), a start codon and a stop codon, followed by the heterologous gene sequence which also contains a RBS and a start and stop codon. Upon induction, this construct results in the production of both the GST protein and the recombinant protein as discrete products (Ito and Kurosawa, 1992).
1.8.3.2 Immunological studies using purified GST fusion proteins
Since GST fusion proteins are easily produced, are soluble and have affinity for immobilized glutathione, they are ideal to use as antigens either in immunological assays or to elicit an immune response (Table 1.4). The GST fusion proteins have been used to develop an ELISA sandwich technique which has bovine serum albumin modified with reduced glutathione linked to the plastic which binds the antigen as a GST fusion protein. This technique reduces the loss of epitopes on the antigen which occur on direct binding to the plastic (Rabin etal., 1992). The GST fusion proteins have also been used for mapping epitopes and have been employed as vaccines against larval tapeworms to give host-protective immunity in sheep where a similar p-galactosidase fusion protein failed (Johnson et a!., 1989; Fikrig et a!., 1990, 1993; Tomlinson et a!., 1993; Volpers et al., 1993). However, it has been noticed that different adjuvants give different antibody responses to GST fusion proteins in mice (Varley etal., 1992).
1.8.3.3 Protein-protein interaction studies using GST fusion proteins