organic papers
Acta Cryst.(2005). E61, o2775–o2777 doi:10.1107/S1600536805023925 Huet al. C
17H16O4
o2775
Acta Crystallographica Section EStructure Reports
Online
ISSN 1600-5368
2,2
000-(Propane-1,3-diyldioxy)dibenzaldehyde
Pu-Zhou Hu, Lu-Fang Ma, Jian-Ge Wang, Bang-Tun Zhao* and Li-Ya Wang
Department of Chemistry, Luoyang Normal University, Luoyang 471022, People’s Republic of China
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 295 K
Mean(C–C) = 0.003 A˚
Rfactor = 0.039
wRfactor = 0.110
Data-to-parameter ratio = 10.8
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography
Printed in Great Britain – all rights reserved
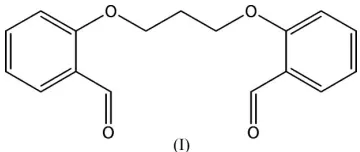
Molecules of the title compound, C17H16O4, form a chain
along the c axis via – stacking interactions involving neighboring aromatic rings. The chains are linked through intermolecular C—H O hydrogen bonds, resulting in a zigzag packing arrangement.
Comment
Much attention has been paid to the synthesis, coordination chemistry and catalytic application of salicylaldehyde, its Schiff bases and metal complexes (Hataet al., 2004; Scherhag & Spicer, 2000; Mukherjeeet al., 2001; Liet al., 2000), leading to the characterization of many derivatives. Meanwhile, interactions, such as C—H , – and weak hydrogen-bonding interactions (C—H X:X= O, N, Cl, Br), are known to play crucial roles in molecular self-assembly and crystal symmetry in biology, chemistry and materials science (Leiningeret al., 2000; Mu¨ller-Dethlefs & Hobza, 2000; Conn & Rebek, 1997; Hunteret al., 1991; Desiraju, 2005).
In the title molecule, (I), four intramolecular C—H O hydrogen bonds are observed (Table 1). The planes of the aromatic rings form a dihedral angle of 99.88 (5), with the aldehyde groups oriented in opposite directions (Fig. 1). Interestingly, the C1–C6 benzene ring is offset-stacked along
[image:1.610.242.422.393.469.2] [image:1.610.207.457.549.723.2]Received 11 July 2005 Accepted 26 July 2005 Online 6 August 2005
Figure 1
thec axis with respect to the corresponding rings of neigh-boring molecules at (x, 1
2y,
1
2+ z) and (x, 1 2 y,
1 2+ z)
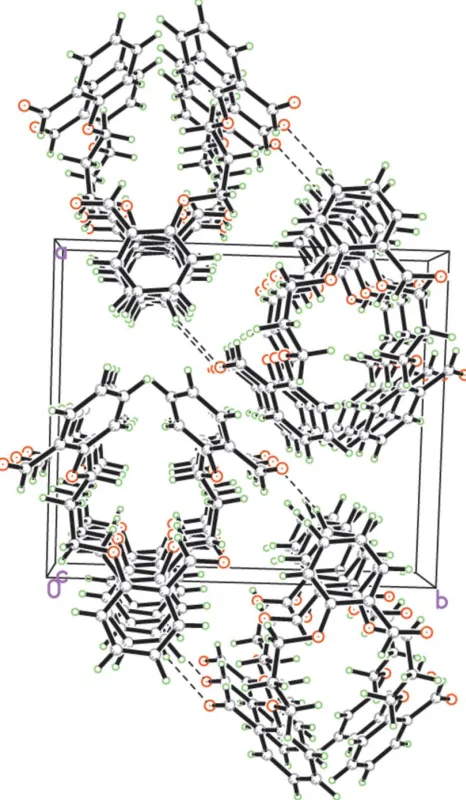
(Fig. 2). The dihedral angle formed by adjacent rings is 3.53 (7). The centroid–centroid distance between two neigh-bouring ring centers is 3.915 (3) A˚ and the perpendicular distances of the center of a benzene ring to two neighboring rings are 3.532 (4) and 3.539 (4) A˚ . These values indicate the presence of reasonable–aromatic interactions (Hunter & Sanders, 1990), which greatly contribute to the stabilization of the one-dimensional chain structure. There is a weak C— H O hydrogen bond between adjacent chains (Table 1), resulting in a two-dimensional zigzag packing arrangement parallel to theabplane.
Experimental
The title compound was prepared according to the literature method Atkins et al.(1994). A mixture of salicylaldehyde (60 mmol), 1,3-dibromopropane (30 mmol) and anhydrous K2CO3(60 mmol) was
stirred under N2in anhydrous dimethylformamide (60 ml) for 20 h,
then cooled to room temperature in an ice-water bath to ensure precipitation. After filtration, the precipitate was recrystallized from 95% ethanol to give the title compound (70% yield). A small amount of the precipitate was dissolved in 95% ethanol to make a clear solution and kept at room temperature for a week to give light-yellow pillar-shaped crystals suitable for X-ray diffraction analysis.
Crystal data
C17H16O4
Mr= 284.30 Monoclinic,P21=c a= 12.8275 (12) A˚ b= 14.8431 (13) A˚ c= 7.8272 (7) A˚
= 95.334 (2) V= 1483.8 (2) A˚3 Z= 4
Dx= 1.273 Mg m
3
MoKradiation Cell parameters from 1628
reflections
= 3.0–19.1
= 0.09 mm1 T= 295 (2) K Pillar, light yellow 0.210.180.13 mm
organic papers
o2776
Huet al. C [image:2.610.48.297.71.270.2]17H16O4 Acta Cryst.(2005). E61, o2775–o2777
Figure 2
[image:2.610.320.558.75.290.2]The one-dimensional chain structure of (I) formed by–interactions along thecaxis (dashed lines represent the–interactions). H atoms have been omitted.
Figure 3
A packing diagram of (I), viewed down thecaxis. Dashed lines denote C—H O hydrogen bonds.
Figure 4
[image:2.610.53.286.322.722.2]Data collection
Bruker SMART APEX-II CCD diffractometer
’and!scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996) Tmin= 0.981,Tmax= 0.988
10969 measured reflections
2758 independent reflections 1562 reflections withI> 2s˘I) Rint= 0.030
max= 25.5
h=15!15 k=17!17 l=9!9
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.039
wR(F2) = 0.110
S= 1.01 2758 reflections 255 parameters
All H-atom parameters refined
w= 1/[2(F
o2) + (0.0437P)2
+ 0.1205P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.11 e A˚
3
min=0.11 e A˚
3
Extinction correction:SHELXL97 Extinction coefficient: 0.0093 (16)
Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
C3—H3 O1 0.93 (2) 2.51 (2) 2.871 (3) 103 (1) C11—H11 O2 0.95 (2) 2.50 (2) 2.828 (3) 100 (1) C13—H13 O3 0.98 (2) 2.36 (2) 2.737 (3) 102 (2) C14—H14 O4 0.98 (2) 2.43 (2) 2.786 (3) 101 (1) C4—H4 O2i
1.04 (2) 2.48 (2) 3.454 (3) 156 (2)
Symmetry code: (i)x;yþ1 2;z
1 2.
The H atoms were located in a difference Fourier map and refined isotropically [C—H = 0.93–(2)–1.04–(2)–A˚ ].
Data collection:SMART(Bruker, 1997); cell refinement:SAINT
(Bruker, 1997); data reduction:SAINT; program(s) used to solve structure:SHELXS97(Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics:
SHELXTL (Bruker, 1997); software used to prepare material for publication:SHELXTL.
The support of this work by the Natural Science Fundation of Henan Province (grants Nos. 2004601012, 0511020100 and 234) is gratefully acknowledged.
References
Atkins, I. M., Lindoy, L. F., Matthew, O. A., Meehan, G. V., Sobolev, A. N. & White, A. H. (1994).Aust. J. Chem.47, 1155–1162.
Bruker (1997).SMART,SAINTandSHELXTL. Bruker AXS Inc., Madison, Wisconsin, USA.
Conn, M. M. & Rebek, J. Jr (1997).Chem. Rev.97, 1647–1668. Desiraju, G. R. (2005).Chem. Commun.pp. 2995–3001.
Hata, M., Akutsu, H., Yamada, J. & Nakatsuji, S.(2004).Molecules,9, 746–756. Hunter, C. A. & Sanders, J. K. M. (1990).J. Am. Chem. Soc.112, 5525–5534. Hunter, C. A., Singh, J. & Thornton, J. M. (1991).J. Mol. Biol.218, 837–846. Leininger, S., Olenyuk, B. & Stang, P. J. (2000).Chem. Rev.100, 853–908. Li, Z. N., Liu, G. & Zheng, Z. (2000).Tetrahedron,56, 7787–7791.
Mukherjee, P. S., Dalai, S., Mostafa, G., Lu, T.-H., Rentschler, E. & Chaudhuri, N. R. (2001).New J. Chem.25, 1203–1207.
Mu¨ller-Dethlefs, K. & Hobza, P. (2000).Chem. Rev.100, 143–167.
Scherhag, G. & Spicer, M. D. (2000).J. Chem. Soc. Dalton Trans.pp. 1237– 1238.
Sheldrick, G. M. (1996).SADABS. University of Go¨ttingen, Germany. Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of
Go¨ttingen, Germany.
organic papers
Acta Cryst.(2005). E61, o2775–o2777 Huet al. C
supporting information
sup-1 Acta Cryst. (2005). E61, o2775–o2777
supporting information
Acta Cryst. (2005). E61, o2775–o2777 [https://doi.org/10.1107/S1600536805023925]
2,2
′
-(Propane-1,3-diyldioxy)dibenzaldehyde
Pu-Zhou Hu, Lu-Fang Ma, Jian-Ge Wang, Bang-Tun Zhao and Li-Ya Wang
2,2′-(Propane-1,3-diyldioxy)dibenzaldehyde
Crystal data
C17H16O4
Mr = 284.30
Monoclinic, P21/c
Hall symbol: -P 2ybc
a = 12.8275 (12) Å
b = 14.8431 (13) Å
c = 7.8272 (7) Å
β = 95.334 (2)°
V = 1483.8 (2) Å3
Z = 4
F(000) = 600
Dx = 1.273 Mg m−3
Melting point: 98 K
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 1628 reflections
θ = 3.0–19.1°
µ = 0.09 mm−1
T = 295 K
Pillar, light yellow 0.21 × 0.18 × 0.13 mm
Data collection
Bruker SMART APEX-II CCD diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ and ω scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996)
Tmin = 0.981, Tmax = 0.988
10969 measured reflections 2758 independent reflections 1562 reflections with I > 2s˘I)
Rint = 0.030
θmax = 25.5°, θmin = 2.7°
h = −15→15
k = −17→17
l = −9→9
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.039
wR(F2) = 0.110
S = 1.02 2758 reflections 255 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
All H-atom parameters refined
w = 1/[σ2(F
o2) + (0.0437P)2 + 0.1205P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.11 e Å−3
Δρmin = −0.11 e Å−3
Extinction correction: SHELXL97, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4
supporting information
sup-2 Acta Cryst. (2005). E61, o2775–o2777
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O1 0.09389 (13) 0.44218 (11) 0.2032 (2) 0.1170 (6)
O2 0.36143 (14) −0.07326 (11) −0.3507 (2) 0.1090 (6)
O3 0.11249 (10) 0.17765 (8) 0.23069 (17) 0.0769 (4)
O4 0.32562 (10) 0.07694 (9) 0.06363 (17) 0.0769 (4)
C1 0.02200 (15) 0.20877 (13) 0.1464 (2) 0.0645 (5)
C2 0.01279 (15) 0.30219 (12) 0.1336 (2) 0.0665 (5)
C3 −0.07773 (19) 0.33811 (18) 0.0461 (3) 0.0833 (7)
C4 −0.1563 (2) 0.2836 (2) −0.0232 (3) 0.0918 (7)
C5 −0.14618 (19) 0.1923 (2) −0.0065 (3) 0.0898 (7)
C6 −0.05814 (17) 0.15385 (17) 0.0772 (3) 0.0778 (6)
C7 0.40320 (14) 0.10281 (12) −0.0332 (2) 0.0624 (5)
C8 0.46916 (17) 0.17475 (14) 0.0072 (3) 0.0774 (6)
C9 0.54887 (18) 0.19447 (17) −0.0924 (3) 0.0876 (7)
C10 0.56466 (19) 0.14414 (17) −0.2338 (3) 0.0850 (7)
C11 0.49788 (18) 0.07428 (16) −0.2777 (3) 0.0772 (6)
C12 0.41631 (14) 0.05178 (12) −0.1799 (2) 0.0636 (5)
C13 0.0952 (2) 0.36121 (17) 0.2106 (3) 0.0869 (7)
C14 0.3490 (2) −0.02504 (15) −0.2292 (3) 0.0824 (6)
C15 0.13203 (18) 0.08225 (14) 0.2319 (4) 0.0806 (7)
C16 0.24032 (19) 0.0698 (2) 0.3191 (3) 0.0898 (7)
C17 0.32353 (18) 0.11701 (17) 0.2311 (3) 0.0783 (6)
H3 −0.0767 (16) 0.4009 (15) 0.039 (2) 0.093 (7)*
H4 −0.2227 (19) 0.3118 (15) −0.087 (3) 0.117 (8)*
H5 −0.1984 (17) 0.1508 (15) −0.056 (3) 0.104 (8)*
H6 −0.0507 (15) 0.0913 (14) 0.092 (2) 0.087 (7)*
H8 0.4634 (15) 0.2068 (14) 0.108 (3) 0.094 (7)*
H9 0.5970 (18) 0.2470 (15) −0.060 (3) 0.118 (8)*
H10 0.6244 (16) 0.1574 (13) −0.307 (3) 0.094 (7)*
H11 0.5067 (15) 0.0368 (13) −0.374 (3) 0.089 (7)*
H13 0.1564 (18) 0.3309 (14) 0.270 (3) 0.100 (7)*
H14 0.2910 (15) −0.0366 (12) −0.158 (2) 0.077 (6)*
H15A 0.0808 (16) 0.0524 (13) 0.299 (3) 0.096 (7)*
H16A 0.2414 (16) 0.0938 (14) 0.437 (3) 0.105 (8)*
H17A 0.3084 (14) 0.1845 (14) 0.216 (2) 0.087 (6)*
supporting information
sup-3 Acta Cryst. (2005). E61, o2775–o2777
H16B 0.2564 (16) 0.0035 (16) 0.322 (2) 0.103 (7)*
H17B 0.3968 (16) 0.1103 (13) 0.293 (2) 0.091 (6)*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.1279 (14) 0.0659 (10) 0.1588 (16) −0.0041 (9) 0.0215 (12) −0.0057 (10)
O2 0.1277 (14) 0.0965 (11) 0.1008 (12) −0.0033 (9) −0.0002 (10) −0.0328 (10)
O3 0.0671 (9) 0.0640 (8) 0.0990 (10) 0.0110 (6) 0.0049 (7) 0.0026 (7)
O4 0.0704 (9) 0.0852 (9) 0.0756 (9) −0.0039 (7) 0.0106 (7) −0.0084 (7)
C1 0.0597 (12) 0.0685 (12) 0.0673 (12) 0.0067 (10) 0.0161 (9) 0.0042 (10)
C2 0.0691 (13) 0.0633 (12) 0.0700 (12) 0.0072 (10) 0.0225 (10) 0.0028 (10)
C3 0.0848 (17) 0.0795 (16) 0.0892 (16) 0.0193 (14) 0.0273 (13) 0.0165 (13)
C4 0.0724 (16) 0.117 (2) 0.0871 (17) 0.0154 (16) 0.0122 (13) 0.0184 (15)
C5 0.0717 (16) 0.110 (2) 0.0881 (16) −0.0036 (15) 0.0071 (13) 0.0017 (15)
C6 0.0745 (15) 0.0733 (15) 0.0871 (15) −0.0001 (12) 0.0148 (12) 0.0017 (12)
C7 0.0560 (11) 0.0646 (11) 0.0652 (12) 0.0087 (9) −0.0009 (9) 0.0026 (10)
C8 0.0713 (14) 0.0796 (14) 0.0805 (15) −0.0040 (11) 0.0030 (12) −0.0111 (12) C9 0.0757 (16) 0.0858 (16) 0.1007 (19) −0.0065 (13) 0.0045 (14) 0.0037 (14) C10 0.0782 (16) 0.0899 (17) 0.0882 (17) 0.0042 (13) 0.0138 (13) 0.0162 (14) C11 0.0871 (16) 0.0824 (15) 0.0621 (13) 0.0204 (13) 0.0078 (12) 0.0058 (12) C12 0.0667 (12) 0.0598 (11) 0.0622 (12) 0.0102 (9) −0.0059 (10) 0.0037 (9) C13 0.0910 (17) 0.0678 (15) 0.1042 (18) 0.0059 (13) 0.0209 (14) −0.0002 (13) C14 0.0896 (17) 0.0768 (15) 0.0789 (16) 0.0048 (12) −0.0021 (13) −0.0068 (12) C15 0.0795 (16) 0.0646 (14) 0.1006 (18) 0.0114 (11) 0.0232 (14) 0.0188 (13) C16 0.0911 (17) 0.0954 (18) 0.0847 (17) 0.0299 (14) 0.0187 (13) 0.0210 (14) C17 0.0732 (15) 0.0912 (17) 0.0701 (14) 0.0162 (12) 0.0053 (12) −0.0041 (12)
Geometric parameters (Å, º)
O1—C13 1.203 (2) C8—C9 1.374 (3)
O2—C14 1.213 (2) C8—H8 0.93 (2)
O3—C1 1.361 (2) C9—C10 1.366 (3)
O3—C15 1.438 (2) C9—H9 1.01 (2)
O4—C7 1.361 (2) C10—C11 1.368 (3)
O4—C17 1.442 (2) C10—H10 1.02 (2)
C1—C6 1.382 (3) C11—C12 1.393 (3)
C1—C2 1.395 (2) C11—H11 0.95 (2)
C2—C3 1.398 (3) C12—C14 1.460 (3)
C2—C13 1.459 (3) C13—H13 0.98 (2)
C3—C4 1.365 (3) C14—H14 0.984 (19)
C3—H3 0.93 (2) C15—C16 1.501 (3)
C4—C5 1.366 (3) C15—H15A 0.98 (2)
C4—H4 1.04 (2) C15—H15B 1.01 (2)
C5—C6 1.376 (3) C16—C17 1.497 (3)
C5—H5 0.97 (2) C16—H16A 0.99 (2)
C6—H6 0.94 (2) C16—H16B 1.01 (2)
supporting information
sup-4 Acta Cryst. (2005). E61, o2775–o2777
C7—C12 1.399 (2) C17—H17B 1.02 (2)
C1—O3—C15 118.53 (16) C11—C10—H10 119.5 (11)
C7—O4—C17 117.79 (15) C10—C11—C12 121.9 (2)
O3—C1—C6 124.01 (18) C10—C11—H11 121.6 (12)
O3—C1—C2 115.78 (17) C12—C11—H11 116.4 (12)
C6—C1—C2 120.21 (19) C11—C12—C7 118.35 (19)
C1—C2—C3 118.4 (2) C11—C12—C14 119.9 (2)
C1—C2—C13 120.93 (19) C7—C12—C14 121.8 (2)
C3—C2—C13 120.7 (2) O1—C13—C2 125.0 (2)
C4—C3—C2 121.2 (2) O1—C13—H13 119.0 (13)
C4—C3—H3 125.6 (13) C2—C13—H13 115.9 (12)
C2—C3—H3 113.2 (13) O2—C14—C12 123.4 (3)
C3—C4—C5 119.3 (2) O2—C14—H14 120.1 (11)
C3—C4—H4 119.8 (13) C12—C14—H14 116.5 (11)
C5—C4—H4 120.9 (12) O3—C15—C16 106.1 (2)
C4—C5—C6 121.6 (3) O3—C15—H15A 108.8 (12)
C4—C5—H5 122.5 (13) C16—C15—H15A 109.7 (12)
C6—C5—H5 115.8 (13) O3—C15—H15B 109.4 (12)
C5—C6—C1 119.3 (2) C16—C15—H15B 110.6 (12)
C5—C6—H6 122.6 (12) H15A—C15—H15B 112.0 (17)
C1—C6—H6 118.1 (12) C17—C16—C15 113.8 (2)
O4—C7—C8 123.70 (18) C17—C16—H16A 108.2 (13)
O4—C7—C12 117.03 (17) C15—C16—H16A 108.2 (12)
C8—C7—C12 119.27 (19) C17—C16—H16B 108.4 (12)
C9—C8—C7 120.5 (2) C15—C16—H16B 108.1 (12)
C9—C8—H8 119.6 (13) H16A—C16—H16B 110.2 (17)
C7—C8—H8 119.7 (13) O4—C17—C16 107.4 (2)
C10—C9—C8 121.2 (2) O4—C17—H17A 108.6 (11)
C10—C9—H9 119.3 (13) C16—C17—H17A 112.0 (10)
C8—C9—H9 119.5 (13) O4—C17—H17B 107.3 (10)
C9—C10—C11 118.8 (2) C16—C17—H17B 113.6 (10)
C9—C10—H10 121.7 (11) H17A—C17—H17B 107.8 (15)
C15—O3—C1—C6 7.4 (3) C7—C8—C9—C10 0.4 (3)
C15—O3—C1—C2 −173.19 (18) C8—C9—C10—C11 1.5 (3)
O3—C1—C2—C3 179.04 (16) C9—C10—C11—C12 −1.7 (3)
C6—C1—C2—C3 −1.5 (3) C10—C11—C12—C7 0.1 (3)
O3—C1—C2—C13 −1.4 (2) C10—C11—C12—C14 −178.19 (18)
C6—C1—C2—C13 178.05 (18) O4—C7—C12—C11 −177.34 (15)
C1—C2—C3—C4 1.0 (3) C8—C7—C12—C11 1.7 (3)
C13—C2—C3—C4 −178.5 (2) O4—C7—C12—C14 0.9 (2)
C2—C3—C4—C5 0.0 (3) C8—C7—C12—C14 180.00 (18)
C3—C4—C5—C6 −0.6 (3) C1—C2—C13—O1 178.9 (2)
C4—C5—C6—C1 0.1 (3) C3—C2—C13—O1 −1.6 (3)
O3—C1—C6—C5 −179.63 (18) C11—C12—C14—O2 2.0 (3)
C2—C1—C6—C5 0.9 (3) C7—C12—C14—O2 −176.30 (18)
supporting information
sup-5 Acta Cryst. (2005). E61, o2775–o2777
C17—O4—C7—C12 168.27 (16) O3—C15—C16—C17 −59.6 (3)
O4—C7—C8—C9 177.01 (18) C7—O4—C17—C16 −173.18 (16)
C12—C7—C8—C9 −2.0 (3) C15—C16—C17—O4 −63.6 (3)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
C3—H3···O1 0.93 (2) 2.51 (2) 2.871 (3) 103.3 (14)
C11—H11···O2 0.95 (2) 2.50 (2) 2.828 (3) 100.4 (14)
C13—H13···O3 0.98 (2) 2.36 (2) 2.737 (3) 102.1 (15)
C14—H14···O4 0.98 (2) 2.43 (2) 2.786 (3) 100.7 (12)
C4—H4···O2i 1.04 (2) 2.48 (2) 3.454 (3) 156.4 (17)