organic papers
o3210
Herreraet al. C12H19NO doi:10.1107/S1600536805027868 Acta Cryst.(2005). E61, o3210–o3211 Acta Crystallographica Section E
Structure Reports Online
ISSN 1600-5368
4-(Hexylamino)phenol
Ana M. Herrera,a‡ Sylvain Berne`sb* and Delia Lo´pezc
a
Centro de Investigaciones en Materiales y Metalurgia, Universidad Auto´noma del Estado de Hidalgo, Carretera Pachuca-Tulancingo Km. 4.5, 42074 Pachuca, Hgo., Mexico,bCentro de
Quı´mica, Instituto de Ciencias, Universidad Auto´noma de Puebla, AP 1613, 72000 Puebla, Pue., Mexico, andcFacultad de Ciencias
Quı´micas, Universidad Auto´noma de Puebla, Boulevard 14 Sur, Col. San Manuel, 72570 Puebla, Pue., Mexico
‡ Current address: Facultad de Ciencias Quı´micas Universidad Auto´noma de Puebla Boulevard 14 Sur Col. San Manuel 72570 Puebla, Pue. Mexico
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 296 K
Mean(C–C) = 0.006 A˚
Rfactor = 0.053
wRfactor = 0.170 Data-to-parameter ratio = 9.8
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography Printed in Great Britain – all rights reserved
The title compound, C12H19NO, presents a two-dimensional hydrogen-bonding pattern, where amine and hydroxyl func-tionalities serve as both donors and acceptors.
Comment
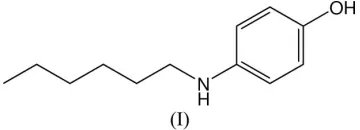
Secondary amines are common in polymer synthesis, where they are used as monomer chain extenders and crosslinking agents (Herman et al., 1990; Choi et al., 2004). The title compound, (I), is an amphiphilic monomer belonging to this class of compounds, containing a hydrophobic alkyl-chain moiety and a polar hydroxyl functionality. It was obtained by reacting 4-aminophenol with 1-bromohexane (see Experi-mental), demonstrating that, as expected, the amine func-tionality is more reactive than the hydroxy group when 4-aminophenol undergoes an SN2 reaction with primary alkyl halides.
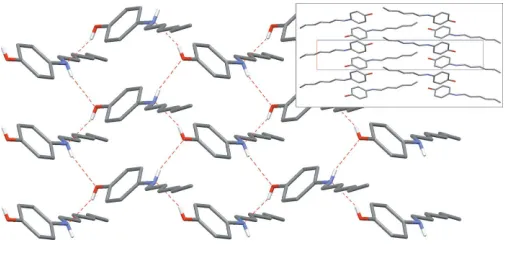
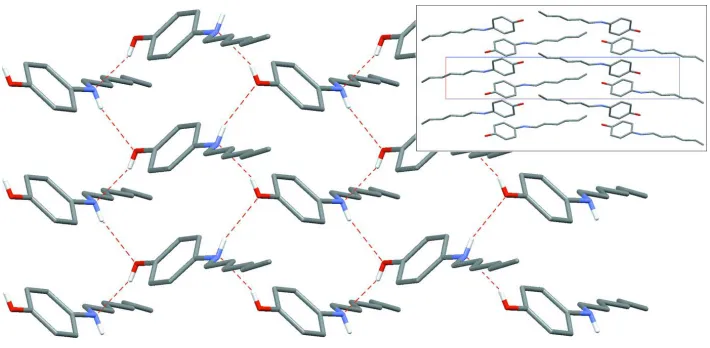
The asymmetric unit of (I) consists of one molecule in a general position with the expected geometry (Fig. 1 and Table 1). O—H and N—H bonds are involved in a network of hydrogen bonds of moderate strength (Table 2); hydroxyl and amine functionalities serve both as donor and acceptor groups,
forming a directed two-membered chain O—H N—
H O—H running along the short axis [100]. The complete network has a two-dimensional pattern (Fig. 2), giving an arrangement which avoids–interactions; the dihedral angle between two adjacent phenol rings belonging to two hydrogen-bonded molecules is 78.99 (9)(symmetry code: 1
x, 1
2+ y, 1z). The crystal structure is based on segregated stacks of aryl groups and alkyl groups, parallel to the [100] axis (Fig. 2, inset).
Experimental
To a solution of 4-aminophenol (1 g, 9 mmol) in cyclohexanone was added finely powdered K2CO3 (2.53 g, 18 mmol) under an argon
atmosphere. The mixture was refluxed for 2 h and then 1-bromo-hexane (1.82 g, 11 mmol) was added in small portions. The reaction was continued under reflux for 24 h and the mixture was then cooled and filtered to remove KBr and unreacted K2CO3. After evaporation
of the solvent, the residue was dissolved in acetone and this solution was poured into hexane in order to precipitate unreacted
[image:1.610.243.423.371.436.2]phenol and the solution reduced under reduced pressure at T < 323 K. This procedure was repeated until pure (I) was isolated as brown crystals (yield 0.82 g, 70%, m.p. 348 K). Analysis found: C 74.6, H 9.7, O 8.2, N 7.3%; calculated for C12H19NO: C 74.6, H 9.8, O 8.2, N
7.2%.1H NMR (400 MHz, CDCl3):0.89 (m, J= 8.8 Hz, 3H, CH3),
1.30 (m, J= 7.2, 6H, 3CH2), 1.58 (m, J= 6.8 Hz, 2H, CH2), 3.05 (m, J= 6.8 Hz, 2H, NCH2), 4.01 (s, 1H, OH), 6.55 (d, J= 8.4 Hz, 2H, Ph),
6.68 (d, J= 8.4 Hz, 2H, Ph).
Crystal data
C12H19NO
Mr= 193.28
Orthorhombic,P212121
a= 4.7016 (6) A˚
b= 10.1325 (14) A˚
c= 26.273 (3) A˚
V= 1251.6 (3) A˚3
Z= 4
Dx= 1.026 Mg m
3
MoKradiation Cell parameters from 55
reflections
= 4.8–11.9
= 0.07 mm1
T= 296 (1) K Prism, brown 0.550.280.20 mm
Data collection
Bruker P4 diffractometer 2/!scans
Absorption correction: none 5524 measured reflections 1327 independent reflections 833 reflections withI> 2(I)
Rint= 0.048
max= 25.0
h=5!5
k=12!12
l=31!31 3 standard reflections
every 97 reflections intensity decay: 3%
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.053
wR(F2) = 0.170
S= 1.04 1327 reflections 136 parameters
H atoms treated by a mixture of independent and constrained refinement
w= 1/[2(F
o2) + (0.1)2] whereP= (Fo2+ 2Fc2)/3 (/)max< 0.001
max= 0.11 e A˚ 3
min=0.17 e A˚ 3
Table 1
Selected geometric parameters (A˚ ,).
O1—C1 1.377 (4) O1—H1A 0.72 (5) N1—C4 1.419 (4) N1—C7 1.449 (5) N1—H1B 0.75 (5)
C7—C8 1.527 (5) C8—C9 1.486 (6) C9—C10 1.549 (7) C10—C11 1.466 (8) C11—C12 1.494 (8)
C1—O1—H1A 114 (3) C4—N1—C7 118.6 (3)
C4—N1—H1B 107 (3) C7—N1—H1B 107 (3)
Table 2
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
O1—H1A N1i 0.72 (5) 2.08 (5) 2.764 (5) 158 (5) N1—H1B O1ii
0.75 (5) 2.34 (5) 3.058 (5) 161 (4)
Symmetry codes: (i)x;y1 2;zþ
1
2; (ii)xþ1;yþ 1 2;zþ
1 2.
The H atoms bonded to heteroatoms N1 and O1 were found in a difference map and refined with free coordinates and isotropic U
parameters. H atoms bonded to C atoms were placed in idealized positions and refined as riding on their parent C atom [C—H
distances and isotropicUiso(H) parameters: methylene 0.97 A˚ and
1.2Ueq(C); methyl 0.96 A˚ and 1.5Ueq(C); aromatic 0.93 A˚ and
1.2Ueq(C)]. In the absence of significant anomalous scattering effects,
Friedel pairs were merged.
Data collection: XSCANS (Siemens, 1996); cell refinement:
XSCANS; data reduction: XSCANS; program(s) used to solve structure: SHELXTL-Plus (Sheldrick, 1998); program(s) used to refine structure:SHELXTL-Plus; molecular graphics: SHELXTL-Plus; software used to prepare material for publication: SHELXTL-Plus.
Partial support by VIEP–BUAP/CONACyT (project No. 7/ I/NAT/05) is gratefully acknowledged. AMH is indebted to PROMEP (Mexico) for providing a research grant.
References
Choi, E.-J., Ahn, J.-C., Chien, L.-C., Lee, C.-K., Zin, W.-C., Kim, D.-C. & Shin, S.-T. (2004).Macromolecules,37, 71–78.
Herman, F. M., Bikales, N. M., Ovarberger, C. G., Menges, G. & Kroschwitz, J. I. (1990).Encyclopedia of Polymer Science & Engineering, 2nd ed. New York: Wiley Interscience.
Sheldrick, G. M. (1998).SHELXTL-Plus. Release 5.10. Siemens Analytical X-ray Instruments Inc., Madison, Wisconsin, USA.
Siemens (1996).XSCANS. Version 2.21. Siemens Analytical X-ray Instru-ments Inc., Madison, Wisconsin, USA.
Figure 2
[image:2.610.315.568.67.215.2] [image:2.610.314.567.255.383.2]Part of the crystal structure of (I), showing the hydrogen-bonding scheme (dashed red lines). For clarity, H atoms bonded to C atoms have been omitted. The inset is a projection of the crystal structure along [010]. The short cell axis corresponds to theaaxis and the long axis corresponds to thecaxis.
Figure 1
supporting information
sup-1
Acta Cryst. (2005). E61, o3210–o3211supporting information
Acta Cryst. (2005). E61, o3210–o3211 [doi:10.1107/S1600536805027868]
4-(Hexylamino)phenol
Ana M. Herrera, Sylvain Bern
è
s and Delia L
ó
pez
S1. Comment
Secondary amines are ubiquitous in polymer synthesis, where they are used as monomer chain extenders and crosslinking
agents (Herman et al., 1990; Choi et al., 2004). The title compound, (I), is an amphiphilic monomer belonging to this
class of compounds, including an hydrophobic alkyl-chain moiety and a polar hydroxyl functionality. It was obtained by
reacting 4-aminophenol with 1-bromohexane (see Experimental), demonstrating that, as expected, the amine functionality
is more reactive than the hydroxy group when 4-aminophenol undergoes an SN2 reaction with primary alkyl halides.
The asymmetric unit of (I) consists of one molecule in a general position with the expected geometry (Fig. 1 and Table
1). O—H and N—H bond lengths are short [0.72 (5) and 0.75 (5) Å, respectively] compared to those found, for example,
in 4-aminophenol [0.87 and 0.90–0.96 Å, respectively; Ermer & Eling, 1994]. These groups are involved in a network of
hydrogen bonds of moderate strength (Table 2); hydroxyl and amine functionalities serve both as donor and acceptor
groups, forming a directed two-membered chain ···O—H···N—H···O—H··· running along the short axis [100]. The
complete network has a two-dimensional pattern (Fig. 2), giving an arrangement which avoids π–π interactions; the
dihedral angle between two adjacent phenol rings belonging to two hydrogen-bonded molecules is 78.99 (9)° (symmetry
code: 1 − x, 1/2 + y, 1 − z). The crystal structure is based on segregated stacks of aryl groups and alkyl groups, parallel to
the [100] axis (Fig. 2, inset).
S2. Experimental
To a solution of 4-aminophenol (1 g, 9 mmol) in cyclohexanone was added finely powdered K2CO3 (2.53 g, 18 mmol)
under an argon atmosphere. The mixture was refluxed for 2 h and then 1-bromohexane (1.82 g, 11 mmol) was added in
small portions. The reaction was continued under reflux for 24 h and the mixture was then cooled and filtered to remove
KBr and unreacted K2CO3. After evaporation of the solvent, the residue was dissolved in acetone and this solution was
poured into hexane in order to precipitate unreacted 4-aminophenol and the solution reduced under reduced pressure at T
< 323 K. This procedure was repeated until pure (I) was isolated as brown crystals (yield 0.82 g, 70%, m.p. 348 K).
Analysis found: C 74.6, H 9.7, O 8.2, N 7.3%; calculated for C12H19NO: C 74.6, H 9.8, O 8.2, N 7.2%. 1H NMR (400
MHz, CDCl3): δ 0.89 (m, J = 8.8 Hz, 3H, CH3), 1.30 (m, J = 7.2, 6H, 3 × CH2), 1.58 (m, J = 6.8 Hz, 2H, CH2), 3.05 (m, J
= 6.8 Hz, 2H, NCH2), 4.01 (s, 1H, OH), 6.55 (d, J = 8.4 Hz, 2H, Ph), 6.68 (d, J = 8.4 Hz, 2H, Ph).
S3. Refinement
The H atoms bonded to heteroatoms N1 and O1 were found in a difference map and refined with free coordinates and
isotropic U parameters. H atoms bonded to C atoms were placed in idealized positions and refined as riding on their
parent C atom [restrained C—H distances and isotropic Uiso(H) parameters: methylene 0.97 Å and 1.2Ueq(C); methyl 0.96
Å and 1.5Ueq(C); aromatic 0.93 Å and 1.2Ueq(C)]. In the absence of significant anomalous scattering effects, Friedel pairs
Figure 1
The structure of (I), with displacement ellipsoids for non-H atoms drawn at the 30% probability level.
Figure 2
Part of the crystal structure of (I), showing the hydrogen-bonding scheme (dashed red lines). For clarity, H atoms bonded
to C atoms have been omitted. The inset is a projection of the crystal structure along [010]. The short cell axis
corresponds to the a axis and the long axis corresponds to the c axis.
4-(n-Hexylamino)phenol
Crystal data
C12H19NO Mr = 193.28
Orthorhombic, P212121
Hall symbol: P 2ac 2ab a = 4.7016 (6) Å b = 10.1325 (14) Å c = 26.273 (3) Å V = 1251.6 (3) Å3 Z = 4
F(000) = 424
Dx = 1.026 Mg m−3
Melting point: 348 K
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 55 reflections θ = 4.8–11.9°
µ = 0.07 mm−1 T = 296 K Prism, brown
[image:4.610.128.483.304.478.2]supporting information
sup-3
Acta Cryst. (2005). E61, o3210–o3211Data collection
Bruker P4 diffractometer
Radiation source: fine-focus sealed tube, FN4 Graphite monochromator
2θ/ω scans
5524 measured reflections 1327 independent reflections 833 reflections with I > 2σ(I)
Rint = 0.048
θmax = 25.0°, θmin = 2.2° h = −5→5
k = −12→12 l = −31→31
3 standard reflections every 97 reflections intensity decay: 3%
Refinement
Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.053 wR(F2) = 0.170 S = 1.04 1327 reflections 136 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: See text
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(F
o2) + (0.1)2]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.11 e Å−3
Δρmin = −0.17 e Å−3
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O1 0.1149 (7) 0.5685 (3) 0.32994 (10) 0.0833 (8) H1A −0.028 (11) 0.542 (4) 0.3307 (16) 0.098 (18)* N1 0.3464 (8) 0.8998 (3) 0.16515 (12) 0.0754 (9) H1B 0.495 (10) 0.925 (4) 0.1686 (15) 0.093 (15)* C1 0.1688 (6) 0.6457 (3) 0.28791 (13) 0.0659 (9) C2 0.0336 (7) 0.6269 (4) 0.24204 (14) 0.0756 (10)
H2A −0.1008 0.5602 0.2387 0.091*
C3 0.0971 (7) 0.7073 (3) 0.20079 (13) 0.0768 (10)
H3A 0.0050 0.6933 0.1699 0.092*
C4 0.2953 (6) 0.8079 (3) 0.20483 (13) 0.0660 (9) C5 0.4315 (7) 0.8240 (4) 0.25173 (13) 0.0744 (9)
H5A 0.5675 0.8899 0.2555 0.089*
C6 0.3674 (7) 0.7438 (3) 0.29239 (14) 0.0765 (9)
H6A 0.4600 0.7564 0.3233 0.092*
C7 0.3220 (9) 0.8555 (4) 0.11289 (14) 0.0878 (11)
H7A 0.1276 0.8287 0.1061 0.105*
H7B 0.4444 0.7797 0.1075 0.105*
C8 0.4068 (10) 0.9663 (4) 0.07655 (15) 0.0992 (13)
H8A 0.2828 1.0413 0.0823 0.119*
H8B 0.5995 0.9938 0.0844 0.119*
C9 0.3925 (13) 0.9294 (5) 0.02187 (16) 0.1203 (16)
H9A 0.1998 0.9026 0.0137 0.144*
H9B 0.5166 0.8545 0.0158 0.144*
C10 0.4811 (15) 1.0442 (5) −0.01366 (18) 0.1311 (18)
H10B 0.6683 1.0750 −0.0037 0.157* C11 0.4880 (19) 1.0084 (6) −0.0677 (2) 0.168 (2)
H11A 0.2979 0.9838 −0.0785 0.202*
H11B 0.6101 0.9321 −0.0721 0.202*
C12 0.593 (2) 1.1177 (7) −0.1009 (2) 0.196 (3)
H12A 0.5281 1.1040 −0.1351 0.294*
H12B 0.7975 1.1189 −0.1004 0.294*
H12C 0.5221 1.2005 −0.0885 0.294*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.0817 (19) 0.0811 (17) 0.0870 (17) −0.0060 (15) −0.0013 (15) 0.0114 (14) N1 0.077 (2) 0.0712 (19) 0.078 (2) −0.0101 (17) 0.0024 (16) 0.0022 (16) C1 0.0611 (16) 0.0558 (18) 0.081 (2) 0.0055 (16) 0.0013 (17) 0.0081 (18) C2 0.0687 (18) 0.063 (2) 0.095 (2) −0.0132 (16) −0.0019 (19) 0.002 (2) C3 0.079 (2) 0.074 (2) 0.077 (2) −0.0062 (19) −0.0107 (19) −0.005 (2) C4 0.0651 (16) 0.0561 (17) 0.077 (2) 0.0022 (15) 0.0044 (17) −0.0031 (19) C5 0.0671 (17) 0.070 (2) 0.086 (2) −0.0114 (17) −0.005 (2) −0.0006 (18) C6 0.0724 (19) 0.075 (2) 0.082 (2) −0.0036 (19) −0.0073 (19) −0.005 (2) C7 0.102 (2) 0.079 (2) 0.082 (2) −0.005 (2) 0.011 (2) −0.007 (2) C8 0.115 (3) 0.099 (3) 0.083 (3) −0.010 (3) 0.006 (2) 0.002 (2) C9 0.151 (4) 0.115 (3) 0.095 (3) −0.004 (4) 0.016 (3) −0.003 (3) C10 0.163 (5) 0.135 (4) 0.095 (3) −0.009 (4) 0.003 (3) 0.006 (3) C11 0.226 (6) 0.150 (5) 0.127 (5) −0.003 (6) 0.019 (4) 0.012 (4) C12 0.259 (9) 0.187 (6) 0.143 (5) −0.027 (7) 0.026 (6) 0.058 (5)
Geometric parameters (Å, º)
O1—C1 1.377 (4) C7—H7B 0.9700
O1—H1A 0.72 (5) C8—C9 1.486 (6)
N1—C4 1.419 (4) C8—H8A 0.9700
N1—C7 1.449 (5) C8—H8B 0.9700
N1—H1B 0.75 (5) C9—C10 1.549 (7)
C1—C6 1.369 (4) C9—H9A 0.9700
C1—C2 1.376 (5) C9—H9B 0.9700
C2—C3 1.388 (5) C10—C11 1.466 (8)
C2—H2A 0.9300 C10—H10A 0.9700
C3—C4 1.385 (4) C10—H10B 0.9700
C3—H3A 0.9300 C11—C12 1.494 (8)
C4—C5 1.398 (4) C11—H11A 0.9700
C5—C6 1.376 (4) C11—H11B 0.9700
C5—H5A 0.9300 C12—H12A 0.9600
C6—H6A 0.9300 C12—H12B 0.9600
C7—C8 1.527 (5) C12—H12C 0.9600
C7—H7A 0.9700
supporting information
sup-5
Acta Cryst. (2005). E61, o3210–o3211C4—N1—C7 118.6 (3) C7—C8—H8A 108.7
C4—N1—H1B 107 (3) C9—C8—H8B 108.7
C7—N1—H1B 107 (3) C7—C8—H8B 108.7
C6—C1—C2 119.4 (3) H8A—C8—H8B 107.6
C6—C1—O1 118.0 (3) C8—C9—C10 112.4 (4)
C2—C1—O1 122.6 (3) C8—C9—H9A 109.1
C1—C2—C3 120.2 (3) C10—C9—H9A 109.1
C1—C2—H2A 119.9 C8—C9—H9B 109.1
C3—C2—H2A 119.9 C10—C9—H9B 109.1
C4—C3—C2 121.1 (3) H9A—C9—H9B 107.9
C4—C3—H3A 119.5 C11—C10—C9 113.8 (5)
C2—C3—H3A 119.5 C11—C10—H10A 108.8
C3—C4—C5 117.5 (3) C9—C10—H10A 108.8
C3—C4—N1 122.8 (3) C11—C10—H10B 108.8
C5—C4—N1 119.6 (3) C9—C10—H10B 108.8
C6—C5—C4 121.0 (3) H10A—C10—H10B 107.7
C6—C5—H5A 119.5 C10—C11—C12 112.9 (6)
C4—C5—H5A 119.5 C10—C11—H11A 109.0
C1—C6—C5 120.8 (3) C12—C11—H11A 109.0
C1—C6—H6A 119.6 C10—C11—H11B 109.0
C5—C6—H6A 119.6 C12—C11—H11B 109.0
N1—C7—C8 110.1 (3) H11A—C11—H11B 107.8
N1—C7—H7A 109.6 C11—C12—H12A 109.5
C8—C7—H7A 109.6 C11—C12—H12B 109.5
N1—C7—H7B 109.6 H12A—C12—H12B 109.5
C8—C7—H7B 109.6 C11—C12—H12C 109.5
H7A—C7—H7B 108.2 H12A—C12—H12C 109.5
C9—C8—C7 114.1 (4) H12B—C12—H12C 109.5
C6—C1—C2—C3 −0.3 (5) C2—C1—C6—C5 0.4 (5)
O1—C1—C2—C3 −179.7 (3) O1—C1—C6—C5 179.8 (3)
C1—C2—C3—C4 −0.3 (5) C4—C5—C6—C1 0.2 (5)
C2—C3—C4—C5 0.9 (5) C4—N1—C7—C8 −175.8 (3)
C2—C3—C4—N1 −173.8 (3) N1—C7—C8—C9 179.2 (4) C7—N1—C4—C3 −33.8 (5) C7—C8—C9—C10 −179.8 (4) C7—N1—C4—C5 151.5 (3) C8—C9—C10—C11 176.1 (7) C3—C4—C5—C6 −0.9 (5) C9—C10—C11—C12 −176.0 (6) N1—C4—C5—C6 174.1 (3)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
O1—H1A···N1i 0.72 (5) 2.08 (5) 2.764 (5) 158 (5)
N1—H1B···O1ii 0.75 (5) 2.34 (5) 3.058 (5) 161 (4)