Full Terms & Conditions of access and use can be found at
https://www.tandfonline.com/action/journalInformation?journalCode=imbc20
Molecular Membrane Biology
ISSN: 0968-7688 (Print) 1464-5203 (Online) Journal homepage: https://www.tandfonline.com/loi/imbc20
Structure-function relationships of a membrane
pore forming toxin revealed by reversion
mutagenesis
Boonhiang Promdonkoy & David J. Ellar
To cite this article: Boonhiang Promdonkoy & David J. Ellar (2005) Structure-function relationships of a membrane pore forming toxin revealed by reversion mutagenesis, Molecular Membrane Biology, 22:4, 327-337, DOI: 10.1080/09687860500166192
To link to this article: https://doi.org/10.1080/09687860500166192
Published online: 09 Jul 2009.
Submit your article to this journal
Article views: 137
View related articles
Structure-function relationships of a membrane pore forming toxin
revealed by reversion mutagenesis
BOONHIANG PROMDONKOY
1, & DAVID J. ELLAR
21
National Center for Genetic Engineering and Biotechnology, National Science and Technology Development Agency, Klong
Luang, Pathumthani, Thailand, and2Department of Biochemistry, University of Cambridge, Cambridge, UK
(Received 13 December 2004; and in revised form 20 April 2005)
Abstract
Cyt2Aa1 is a haemolytic membrane pore forming toxin produced byBacillus thuringiensissubsp.kyushuensis. To investigate membrane pore formation by this toxin,second-site revertants of an inactive mutant toxin Cyt2Aa1-I150A were generated by random mutagenesis using error-prone PCR. The decrease in side chain length caused by the replacement of isoleucine by alanine at position 150 in theaD-b4 loop results in the loss of important van der Waals contacts that exist in the native protein between I150 and K199 and L203 onaE. 28 independent revertants of I150A were obtained and their relative toxicity can be explained by the position of the residue in the structure and the effect of the mutation on side-chain interactions. Analysis of these revertants revealed that residues onaA,aB,aC,aD and the loops betweenaA andaB,aD andb5,b6 andb7 are important in pore formation. These residues are on the surface of the molecule suggesting that they may participate in membrane binding and toxin oligomerization. Changing the properties of the amino acid side-chains of these residues could affect the conformational changes required to transform the water-soluble toxin into the membrane insertion competent state.
Keywords:Bacillus thuringiensis, cytolyticd-endotoxin, Cyt2Aa1 toxin, reversion mutagenesis, transmembrane pore
Introduction
Bacillus thuringiensis (Bt) insecticidal protein toxins
are divided into two families (Cry and Cyt) with unrelated amino acid sequences [1,2]. Although the X-ray crystallographic structures of Cry and Cyt toxins are entirely different [3,4], the mechanism of action of these two families is thought to be the same. Both Cry and Cyt toxins form small pores approximately 1/2 nm in diameter in the larval midgut epithelial cell membranes leading to colloid-osmotic lysis [5]. Cyt2Aa1 which is the subject of this study is produced as a 29 kDa protoxin which is processed at both ends by gut proteases to generate the active 21/23 kDa toxin [6].
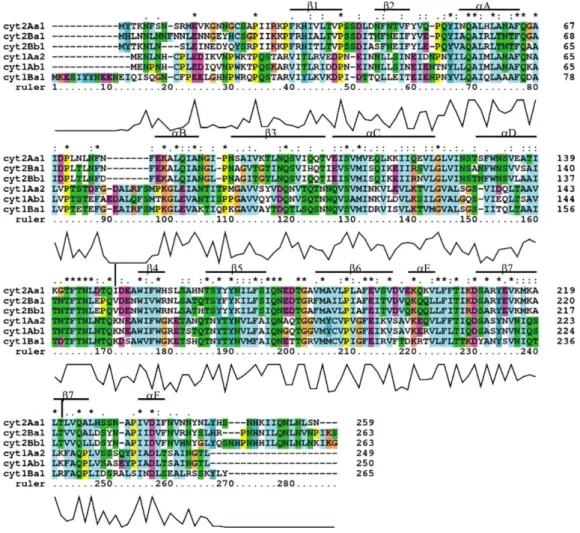
Cyt2Aa1 exists as an inactive protoxin dimer linked by intertwined N-terminal strands in a con-tinuous 12-strandedb-sheet [4]. The high sequence similarity between members of the Cyt toxin family (see Figure 1) indicates that they have the same structural fold as Cyt2Aa1 [4].
Protease processing by insect gut enzymes cleaves the intertwined N-terminal b1 strands responsible for dimerization to release the active toxin as a
monomer and simultaneously eliminate several structural barriers to pore formation [4,6]. Activated toxin molecules can form pores in cell membranes [7], phospholipid vesicles and bilayers [8/10]. Lytic pore formation is believed to require toxin mono-mers to assemble into an oligomeric complex [4,11,12].
The Cyt toxin pore is not likely to be formed by the six a-helices in the structure, as they are too short to span the 30 A˚ width of the lipid bilayer [4]. In contrast, strands b5, b6 and b7 are sufficiently long to span the bilayer. Over these three strands the sheet shows an amphiphilic or hydrophobic char-acter and therefore they could oligomerise to form a
b-barrel pore [4].
The aim of this study was to investigate structure-function relationships associated with membrane pore formation by Cyt2Aa1. A non-haemolytic mutant of Cyt2Aa1 was generated and used as a template for reversion mutagenesis. Analysis of the revertant proteins should provide information about residues that are important in pore formation. This method proved valuable in the Correspondence: Boonhiang Promdonkoy, National Center for Genetic Engineering and Biotechnology, National Science and Technology Development Agency, 113 Paholyothin Road, Klong 1, Klong Luang, Pathumthani 12120, Thailand. Tel: /662 5646700, ext. 3340. Fax:/662 5646707. E-mail: [email protected]
Molecular Membrane Biology, JulyAugust 2005; 22(4): 327337
ISSN 0968-7688 print/ISSN 1464-5203 online#2005 Taylor & Francis DOI: 10.1080/09687860500166192
study of Staphylococcal a-hemolysin [13]. Cyt2Aa1 was chosen in preference to other Cyt d-endotoxins because the availability of its crystal structure [4] enables the position of mutated residues to be mapped on the structure of the molecule.
Materials and methods
Plasmids and oligonucleotides
Plasmid pMSV1Cyt2Aa1 (wild type) is based on pALTER (Promega) and carries the full-length
cyt2Aa1 gene from Bt subsp. kyushuensis [14].
pUC18 was described by Yanisch-Perron et al. [15]. Two oligonucleotides were designed to change the sequence of Cyt2Aa1 from isoleucine at position 150 and from threonine at position 221 to alanine (I150A and T221A). To facilitate identification of the mutants, the oligonucleotide for I150A was designed so as to introduce a new Hae III
restriction site and the T221A oligonucleotide was designed to abolish aHind III site. DNA sequences of oligonucleotides for I150A and T221A were
5?-TTTAGACACTCAGGCCGATGAAGCATG-3?
and 5?
-TAAAATGAAAGCTCTTGCTTTAGTT-CAAG-3?, respectively. Oligonucleotides for error-prone PCR were universal M13 forward and reverse sequencing primers. All oligonucleotides were pro-duced by the Oligonucleotide Synthesis Facility, Department of Biochemistry, University of Cam-bridge, UK.
Site-directed mutagenesis and cloning
Site-directed mutagenesis was performed using the Altered Sites II In Vitro Mutagenesis System (Promega) as described by the manufacturer.
Sin-gle-stranded template was prepared from
pMSV1Cyt2Aa1 using R408 helper phage. Muta-genesis reactions were used to transform the Figure 1. Multiple sequence alignment of amino acids from Cytd-endotoxins. The alignment was performed using Clustal X. The number on the right of each sequence indicates the residue number at the end of each line. The symbols (*), (:) and (.) represent identical, highly conserved and less conserved amino acids, respectively. The graph under the sequences indicates the degree of amino acid identity for all toxins (from 0% at the bottom to 100% at the top). The secondary structures of Cyt2Aa1 are shown on top of the sequence. The ruler number under the sequences is shown for indication of the alignment position only. The residues in Cyt2Aa1 which were selected for mutagenesis are marked by½. This figure is reproduced in colour inMolecular Membrane Biologyonline.
mismatch repair deficient E. coli strain ES1301
mutS by electroporation [16]. Transformed cells were grown overnight in 5 ml LB (Luria-Bertani) medium containing 100 mg/ml ampicillin. Plasmid DNA was then isolated and re-transformed into
E. coli JM109 or TG1 cells. Transformants were
analysed for introduced mutations by restriction endonuclease digestion and DNA sequencing. Wild type and mutant genes were also sub-cloned into plasmid pUC18 for high efficient expression in
E. coli.
Error-prone PCR
Error-prone PCR was performed as described by Panchal and Bayley [13]. The template for PCR was
the cyt2Aa1-I150A gene in plasmid pUC18. The
3?- and 5?-universal primers were as described above. Random mutations were introduced using a large number of PCR cycles (50) and a high concentration of dNTPs (0.4 mM each). PCR reactions were carried out in standard PCR buffer (Pharmacia) plus 1.0 ng of plasmid template, 0.8 mM of each primer, 3.0 mM MgCl2and 2.5 units of Taq DNA polymerase (Pharmacia) in 50ml reaction volume for 50 cycles at 948C, 30 sec; 508C, 30 sec; 728C, 2 min (10 min for the last cycle). PCR products were then cloned into plasmid pUC18 and transformed into
E. coli JM109 or TG1.
Screening for revertants
The screening method was modified from the method described by Panchal and Bayley [13]. Small colonies that appeared after 8 h growth at 378C were replicated onto nitrocellulose membranes (Gelman Sciences), which were then placed colony side up onto a second LB-ampicillin plate previously spread with 50 ml of 0.1 M IPTG and 50ml of 2% X-gal, and incubated overnight at 37oC. The replicate colonies were lysed by removing the nitrocellulose membranes and placing them (colony side up) for 1 h on Whatman paper (no. 3) soaked with 0.1 M NaHCO3, 1% Triton X-100 (v/v) containing 2 mg/ml lysozyme. The toxin inclusions were solubilized and processed by removing the mem-branes and placing them on Whatman paper (no. 3) soaked with 50 mM Na2CO3 pH 10.5 containing 0.1 mg/ml proteinase K for 1 h at 378C. The membranes were then washed with PBS buffer (8 mM Na2HPO4, 1.5 mM KH2PO4, 140 mM NaCl, 2.7 mM KCl pH 7.4) plus 1 mg/ml BSA by gently floating the membranes (colony side up) on the surface of the wash buffer for 15 min. The mem-branes were then inverted onto blood agar plates prepared by adding 2.5 ml of washed human blood
to 100 ml of tryptone soya agar (Oxoid) held at 508C. Colonies showing distinct zones of haemolysis within 24 h at room temperature were individually picked from the master plates for further analysis.
Protein preparation and processing
Cyt2Aa1 inclusions were released fromE. coli cells by sonication as described previously [17]. Prior to use, the inclusions were solubilized at 1/2 mg/ml at 378C for 1 h in 50 mM Na2CO3 pH 10.5 with occasional shaking. Solubilized toxin was separated from insoluble materials by centrifugation at 12,000 g for 5 min and stored at /208C in 1 ml aliquots. Concentrations of solubilized toxin were determined by the method of Bradford [18] using the BioRad protein assay kit and BSA as standard. For proteo-lytic processing, the solubilized material was mixed with 0.1/10% (w/w) proteinase K and incubated at 378C for 10 min to 3 h.
Circular dichroism analysis
Protein samples in 50 mM Na2CO3 pH 10.5 in a 0.02 cm cylindrical quartz cuvette were analysed using a Jasco J-715 spectropolarimeter at 25oC. The instrument was calibrated with camphorsulfonic acid (CSA) and continuously purged with nitrogen gas. Scanning was set to accumulate for 4 averaged spectra in the 180/320 nm region and subtracted with baseline spectra from buffer. All spectra were analysed and compared at the same protein con-centration.
Intrinsic fluorescence spectroscopy
Cyt toxin fluorescence spectra were obtained by emission scanning of protein solution in a 10 mm path length square quartz cell. The protein samples were excited at 280 nm using a Jasco FP-6500 spectrofluorometer. The emission spectra were recorded from 300/500 nm with medium scanning speed using 3 nm of excitation and emission slit width. All spectra were subtracted with baseline spectra from buffer.
In vitro haemolysis assay
Haemolysis assays in microtitre plates were as described by Thomas and Ellar [19] except that a 4% human blood (National Blood Transfusion Service) suspension in haemolysis buffer (50% PBS, 2.25% glucose, 0.05% gelatine) was used. Processed toxin samples were diluted in two-fold serial dilutions with haemolysis buffer. 0.1 ml aliquots of the diluted toxin samples were mixed with 0.1 ml of diluted blood cells and left at
room temperature. The haemolytic end-point was recorded after 24 h of incubation.
Mosquito larvicidal assay
Mosquito larvicidal assays were performed as described by Koni and Ellar [6] with modification. Solubilized toxin samples in 50 mM Na2CO3 pH 10.5 were precipitated by adding 0.1 volume of 11% (w/v) citric acid to lower the pH to approximately 4.5 and the samples stored at /20oC for at least 2 h until required. The material was then used directly without further manipulation. The assays were carried out 3 times in duplicate using 10 larvae of 3-day-old Aedes aegypti (reared from eggs supplied by Zeneca Agrochemical, UK and the Institute of Molecular Biology and Genetics, Mahidol Univer-sity, Thailand) in 1.5 ml microtubes containing 0.9 ml of water in which the larvae were reared and 0.1 ml of precipitated toxin samples. The final concentration of toxin in each tube was a 2-fold serial dilution from 250 mg/ml to 0.25 mg/ml. The negative control contained 90ml of 50 mM Na2CO3 pH 10.5 and 10ml of 11% citric acid. Mortality was recorded after 24 h at room temperature.
Results
Expression and activity of primary mutants
Ward et al. [20] demonstrated that replacing K154 or K225 with alanine in the closely related toxin Cyt1Aa (Figure 1) caused a loss of activity without affecting expression. Therefore, it was decided to substitute alanine at the equivalent residues I150 and T221 in Cyt2Aa1. Both mutant proteins expressed at levels similar to that of the wild type and were soluble in 50 mM Na2CO3 pH 10.5 without the addition of a reducing agent suggesting that the mutations did not affect dimerization and crystal packing. No differences between solubilized mutant proteins and wild type protein were detect-able on SDS-PAGE (data not shown). Proteinase K treatment of the mutant proteins gave identical products to the wild type (data not shown) indicat-ing that the processindicat-ing sites in the mutants had not changed. The haemolytic activity of T221A was similar to the wild type, but Cyt2Aa1-I150A was not haemolytic even at very high concentrations (250mg/ml; Table I). Finally analysis of both proteins using Circular Dichroism (CD) and intrinsic fluorescence revealed no significant differ-ences in structure between the inactive mutant and the wild type (data not shown).
The Cyt2Aa1-I150A mutant was therefore
selected as the template for reversion mutagenesis. The equivalent mutation in Cyt1Aa (K154A) was
one of 6 mutations showing low toxicity and reduced phospholipid binding, but no change in expression levels [20]. All these mutations mapped to loops lying along the edge of the b-sheet adjacent to the helix pair aC-aD [4]. Because alanine replaced a charged residue in 4 of these mutants and because these charged residues are not conserved between Cyt1Aa1 and Cyt2Aa1, Li et al. [4] proposed that this region is required to remain exposed to the aqueous phase when b5, b6 andb7 insert into the membrane. In the remaining two low toxicity mutants, alanine was replaced with proline suggest-ing that main chain flexibility as well as the appro-priate hydrophobicity is needed in this region. In the model for toxin insertion proposed by Li et al. [4] these properties were important in allowing helix Table I. Activity of wt, T221A, Cyt2Aa1-I150A and revertants of Cyt2Aa1-Cyt2Aa1-I150A. The revertants are indicated in square bracket [ ].
Cyt2Aa1 toxin Haemolysis end-point (mg/ml)a Mosquito larvicidal activity LC50 (mg/ml)b wt 0.25 1/2 T221A 0.50 2/4 I150A /250 /250 [Q50R] 250 /250 [N74D] 4 4/8 [F77S] 250* 128/256 [F77L] 2 4/8 [I83T] 250* /250 [V105A] 2 8/16 [T129A] 32 32/64 [T148I] 1 1/2 [A150V] 0.25 1/2 [S166G] 2 2/4 [N170S] 250* 128/256 [E178G] 250 128/256 [V195A] 2 8/16 [N8S,F47S] 250* /250 [F47L,N259H] 16 16/32 [S9F,N74D] 4 4/8 [F77L,S210N] 16 64/128 [I68M,S166G] 16* 64/128 [D69A,I171T] 250* /250 [K79R,I83T] 250* /250 [V109A,A185V] 16 4/8 [V111I,K218R] 250 /250 [V125A,A185T] 1 4/8 [S166G,I171V] 2 2/4 [A185V,F205L] 4 2/4 [K14R,V49A,S130N] 250 128/256 [Q119R,T180A,I207T] 250* 128/256 [D39G,S108P,I171T,N259D] 16* 64/128 a
Haemolysis end-point of toxin activated with 1% (w/w) protei-nase K at 37oC for 1 h (*activated with 0.5% (w/w) proteinase K at 37oC for 30 min).bConcentration of precipitated toxin that cause 50% mortality (LC50) of 3-day-oldAedes aegypti laevae.
Probit analysis was not applied because the change in toxicity from one dose to the next was dramatic.
pair aC-aD to peel away from theb-sheet to lie on the membrane surface while the sheet itself rear-ranges to form the oligomeric trans-membrane pore. On the basis of these suggestions the loss of toxicity when the highly hydrophobic isoleucine is replaced by alanine in Cyt2Aa1-I150A could result from a disruption of the hydrophobicity which positions the surface-located aC-aD helix pair adjacent to the edge of the inserted b-sheet. Because monomer interaction following membrane insertion is crucial to the assembly of the oligomeric pore it is important to keep in mind the possibility that interference with monomer recognition could also result in loss of toxicity. If the activity of the Cyt2Aa1-I150A mutant could be recovered by substitution at other residue positions in the mole-cule, the location of these substitutions could there-fore reveal regions important in the steps of membrane recognition, toxin insertion and pore formation.
Reversion mutagenesis
Reversion mutagenesis of the inactive Cyt2Aa1-I150A was performed using error-prone PCR. Ran-dom mutations were introduced into PCR products by using Taq polymerase which lacks 3?-5? exonu-clease activities [21] and has an error rate of 2/10
4 errors/base [22]. This error rate was further increased by using a large number of PCR cycles and a high concentration of dNTPs. PCR products were then cloned into pUC18, expressed in
E. coli and colonies screened on blood agar plates.
This screen is a powerful method to identify a few haemolytic revertants among thousands of non-haemolytic colonies [13]. In the work reported here, 40 positive colonies were obtained after screen-ing approximately 10,000 colonies. This reversion frequency of 0.4% is very similar to that reported for
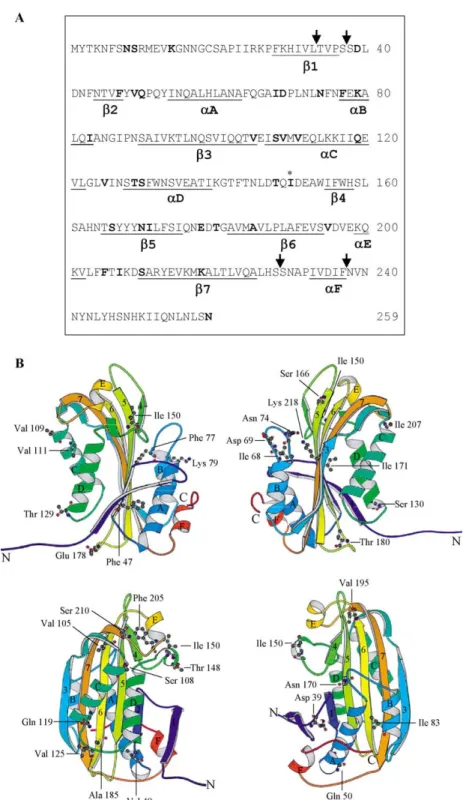
a-haemolysin (0.5%) [13]. DNA sequencing of the positive clones revealed 28 different revertants. Of these, 13 revertants were single mutations, 12 were double mutations, 2 were triple mutations and 1 was a quadruple mutation (see Table I). Four single revertants were found repeatedly: [V105A] (7 colo-nies), [N170S] (4 colocolo-nies), [A150V] (3 colonies) and [T148I] (2 colonies). The positions of revertant residues in the primary sequence and in the toxin structure are shown in Figures 2A and 2B, respec-tively.
Solubilization and proteolytic processing of revertant toxins
All revertant proteins expressed inE. coliat the same level as the wild type and formed typical inclusion
bodies that could be solubilized in 50 mM Na2CO3 pH 10.5 without the need for a reducing agent (data not shown). This result was not unexpected since none of the revertants has a residue changed to cysteine. Cyt2Aa1 contains a single cysteine residue located near the N-terminus which does not appear to play an important role in crystal packing [4]. After solubilization, all revertant proteins showed SDS-PAGE profiles similar to the wild type (data not shown).
Most revertants gave similar products to the wild type when treated with proteinase K to simulate protoxin activation (data not shown). However, 9 revertants: [F77S], [I83T], [N170S], [N8S,F47S],
[I68M,S166G], [D69A,I171T], [K79R,I83T],
[Q119R,T180A,I207T] and [D39G,S108P,I171T, N259D] were unusually sensitive to proteinase K digestion and were almost completely digested by 0.5% (w/w) proteinase K at 37oC for 1 h. These proteins were only partially digested by 0.1% (w/w) proteinase K under the same conditions (data not shown). Most of the mutated residues in the proteinase K-sensitive revertants were located on the surface of the molecule (see Figure 2B). The remaining two mutated residues in this group (N170S and I171T) are located in the middle of b5. Interestingly 8 of these 9 protease sensitive revertants which all showed much reduced mosqui-tocidal and haemolytic activities compared to the wild type toxin, have a residue mutated to serine or threonine. The protease sensitivity of the rela-tively toxic revertant [I68M,S166G] is likely to be the result of I68M because the active form of revertant [S166G] is not sensitive to proteinase K digestion.
Activity of the revertants
The results of haemolysis assays (see Table I) show that none of the revertant toxins was as active as the wild type, except [A150V]. Larvicidal assays using Aedes aegypti generally showed very similar results to the haemolytic assays, but there were some revertants (e.g., [T148I] and [A185V, F205L]) with significantly less haemolytic activity but comparable larvicidal activity to the wild type (Table I).
The revertants could be divided into three groups according to their activity. The first group, [N74D], [F77L], [V105A], [T148I], [S166G], [V195A], [S9F,N74D], [V109A,A185V], [V125A,A185T], [S166G,I171V], and [A185V,F205L], had relatively high activity similar to wild type. A second group,
[T129A], [F47L,N259H], [I68M,S166G], and
[F77L,S210N], had moderate activity, while a third group, containing the remaining revertants all had
low activity. The revertant [D39G,S108P, I171T,N259D] may be classified into the moderate or high activity group, although its activity is very difficult to quantitate because of its protease sensi-tivity.
Positions of revertant residues in the structure of molecule
Two revertants ([A150V] and [T148I]) contain mutations close to or at the position occupied by A150 in the inactive primary mutant. Both of these return a hydrophobic residue to the loop at the edge Figure 2. Amino acid sequence and structure of wild type Cyt2Aa1. (A) The position of the original inactive mutant (I150A) is marked by * and the positions of revertant residues are shown in bold. Proteinase K processing sites are indicated by arrows. (B) 3-D structure of Cyt2Aa1 from different views showing the positions of wild type residues that were replaced in the revertants. The images were produced by MolScript v2.0.2 [33]. This figure is reproduced in colour inMolecular Membrane Biologyonline.
of theb-sheet adjacent to the helix pairaC-aD from which isoleucine was removed in the primary inactive mutant. The fact that both revertants showed very high activity (Table I) indicated that hydrophobicity is required in this segment.
Seven of the revertants ([N74D], [F77S], [F77L],
[S9F,N74D], [F77L,S210N], [I68M,S166G],
[D69A,I171T]) contained mutations in the loop separating helices aA-aB. (Revertant [F77L] appears to be more active than revertants [F77S] and the double revertant [F77L,S210N], but the intrinsic activities of these three mutants are difficult to measure because their low activities are undoubt-edly partly due to their increased protease sensitiv-ity.) In the protoxin, residues in this loop play a role in stabilizing the packing of helix pairaA-aB against the b-sheet [4]. It was suggested above that an impairment of the hinge function of the loop linking helixaD to theb-sheet causes the loss of activity in the Cyt2Aa1-I150A mutant. The location of these seven revertants suggests that mutations in the helix
aA-aB loop can compensate for this. Since the report by Du et al. [23] indicates that helices aA and aB are exposed on the membrane surface, it is possible that disruption to the connectivity between helix pair aA-aB and the b-sheet may compensate for the change in the connectivity between helix pair
aC-aD and the sheet that has occurred in the Cyt2Aa1-I150A mutant. The two revertants located inb2 ([N8S,F47S], [F47L,N259H], or the adjacent
b2-aA loop ([K14R,V49A,S130N], [Q50R]) may compensate by a similar mechanism.
Three revertants ([T129A], [V125A,A185T] and [K14R,V49A,S130N]) have replacements in the loop joining helices C and D. This may indicate that disruption to the connectivity between this region and the sheet can compensate for the inactivating I150A change in the segment between helix aD and the sheet. It is noteworthy that the addition of the A185T replacement in the double revertant [V125A,A185T] dramatically increases the activity. If as suggested by Li et al. [4] and Du et al. [23]b5,b6 andb7 from several monomers span the membrane in the oligomeric pore, these strands would have alternate residues facing the membrane lipid or the channel lumen. A185 in b6 faces the membrane lipid [4] and a change to a more hydrophobic residue may favour sheet insertion. This may be a partial explanation for the activity of revertant [A185V,F205L]. However A185 is hydro-gen bonded to I175 inb5 and a strengthening of this interaction in A185T may also promote b5,b6 and
b7 insertion. The hydrophobicity of the residue at position 205 in the loop between helix aE andb7 is highly conserved (see Figure 1) and any restoration
of activity caused by the F205L change should be due to other reasons.
Four revertants ([V109A,A185V], [V111I, K218R], [Q119R,T180A,I207T], [D39G,S108P, I171T,N259D]) are found with replacements in helix aC and one in helix aB ([I83T]). The important intra-molecular salt bridge between E192 and K218 [4] is preserved in [V111I,K218R] and therefore restoration of activity is attributable to the V111I change. The finding that these helices are not membrane-protected [23] suggests that in addi-tion to their importance in connectivity to the sheet, their surface location may also be important in the monomer recognition that precedes oligomerization. This is supported by studies using synthetic peptides [24,25], reporting thataA andaC had the ability to bind the membrane and self-associate. Thus changes to both the amphipathic character and sequence in the surface exposed helices may also be compen-satory.
The revertants [S166G] and [S166G,I171V] show similar levels of activity, suggesting that the conservative change from isoleucine to valine in the double revertant may have no effect on the toxin activity. [S166G] is located at the N-terminal end of
b5 where it forms a hydrogen bond with H158 inb4. Du et al. [23] have shown that b2 remains on the membrane surface after insertion of b5. Therefore one suggestion to explain restoration of activity in this revertant would be a decrease in b2/b5 con-nectivity caused by substitution of glycine for serine. A similar explanation could be proposed for restora-tion of activity in the mutant [N170S] since N170 forms a hydrogen bond with N44 in b2. (The increased protease sensitivity of N170S makes it difficult to compare its intrinsic activity with that of S166G.).
The revertant [E178G] is located in the b5/b6 loop which is proposed to be oriented towards the cell interior during sheet insertion [4]. Although 5 of the loop residues are highly conserved (Figure 1) the lack of conservation at this position suggests that the negative charge on E178 is not important during insertion. The ability of a glycine substitution at this position to compensate for the distal I150A sub-stitution may suggest that loop flexibility favours membrane insertion.
Discussion
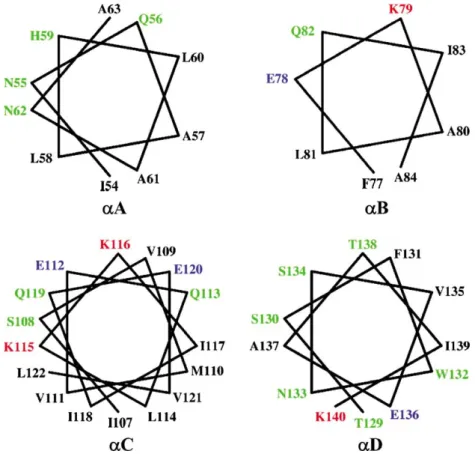
Thea/barchitecture of the Cyt2Aa1 toxin monomer is unusual in that the two outer layers sandwiching the b-sheet consist of helical hairpins that can lift upwards and away from the sheet without unraveling it [4]. Helices aA,aB,aC andaD are amphipathic (see Figure 3) with hydrophobic residues packed
against the b-sheet. The model for membrane insertion and pore formation proposes that upon membrane contact, helix pair aC-aD swings away from the sheet to lie on the membrane surface allowingb5,b6 andb7 to move into the lipid bilayer with the loop between b5 and b6 oriented towards the cell interior. Subsequent association of these inserted strands from several monomers creates an oligomeric pore [4]. The appearance of new proteo-lytic sites upon toxin binding to the membrane and the location of these sites [23] are consistent with the model. In a variation on their proposal Li et al. [4] suggested that helix hairpin aA-aB might remain packed against the sheet during insertion rather than remaining on the surface. This appears to be ruled out by the recent finding that only the segment containing b4, b5, b6, aE and b7 remains tightly associated with the membrane after proteolysis of membrane bound Cyt2Aa toxin [23].
As measured by their solubility, protease sensitiv-ity, expression levels and reaction with wild-type antibody (data not shown), the non-toxic Cyt2Aa1-I150A and the wild type toxin were identical. In addition no significant structural differences between the proteins were detectable using Circular Dichroism (CD) and intrinsic fluorescence. The dramatic difference in toxicity between the two
proteins despite this similarity in properties and structure, suggests that replacement of isoleucine by alanine may affect the conformational changes required for transforming the water-soluble toxin into the membrane-bound pore form. Li et al. [4] proposed a key role in membrane insertion for the hinge region between helix pair aC-aD and the
b-sheet both in terms of surface-seeking ability and flexibility. Because residue 150 is located in this region, the loss of activity in the Cyt2Aa1-I150A mutant may originate in changes in one or both of these properties. By reversion mutagenesis we have located residues proximal and distal from this mutation that can restore toxin activity in this mutant. Comparative analysis of these revertants may therefore help us to understand the insertion mechanism.
Proteolytic activation of the Cyt2Aa1 protoxin removes 33/37 amino acids at the N-terminus and 22-31 amino acids at the C-terminus [6] to generate either the fully-activated (S38 to S228) or the partially activated toxin (T34 to F237 or S38
to F237 or T34 to S228). Mutation of
residues upstream or downstream of these regions should have no effect on toxin activity. Therefore, residues N8S, S9F, K14R, N259H and N259D contained in revertants [N8S,F47S], [S9F,N74D], Figure 3. Helical wheels of helicesaA,aB,aC andaD. All helices show amphipathic character. Residues on the hydrophobic face could interact with the lipid bilayer of the membrane while residues on the hydrophylic side may interact with residues from other monomers to form the oligomer. This figure is reproduced in colour inMolecular Membrane Biologyonline.
[K14R,V49A,S130N], [F47L,N259H] and [D39G, S108P,I171T,N259D] are not considered to be responsible for restoring the activity of these rever-tants.
Combining our results with the findings of Du et al. [23] lead us to refine the model proposed by Li et al. [4] to describe the events taking place during pore formation (see Figure 4). The refined model proposes that upon membrane contact both helix hairpins aA-aB,aC-aD swing away from the sheet. These helices together with aF,b2,b3, b4 and the loops between them locate on the membrane surface with b5, b6 and b7 inserting into the membrane. After insertion, this places alanine in the primary inactive Cyt2Aa1-I150A mutant on the membrane surface.
Experiments to investigate the effect of tempera-ture on pore formation of Cyt2Aa1 and studies on membrane insertion and pore formation of acrylo-dan-labelled mutants of Cyt2Aa1 [17,26] show that the toxin binds and inserts into the membrane as a monomer. Oligomerization occurs when toxin molecules bind in close proximity to each other and the pores are formed when oligomers reach a critical number. Cyt2Aa thus exists in three different configurations (water-soluble monomer, membrane inserted monomer and membrane inserted oligo-mer) in any or all of which a residue may have an important role. Toxicity may therefore be restored in a revertant because of compensatory changes in the steps of membrane insertion or oligomerization or both. Using the refined model, it could be suggested that in addition to a possible impairment of the
hinge region between helix pair aC-aD and the sheet, the loss of toxicity in Cyt2Aa1-I150A could be due to the reduction of binding between the toxin molecule and the membrane surface.
If the monomer-monomer recognition that pre-cedes pore formation involves recognition/associa-tion between the surface exposed elements, then clearly toxicity may be affected by a broad range of residue changes in the elements. For example aA,
aB, andaC are amphipathic (see Figure 3) and the model proposes that residues on the hydrophobic face interact with the lipid bilayer while residues on the other side of the helix are available to recognize residues from other monomers to form the oligomer. V109 is the only hydrophobic residue located on the hydrophilic face of helix C (Figure 3) which is known to be capable of self-association in a mem-brane [24,25]. Substitution of V109 by alanine at this position in revertant [V109A,A185V] may therefore further stabilize the surface location of this element and/or increase its ability to oligomerize with other monomers. Many of the other high activity revertants that map to the loops or helices exposed on the surface in the model e.g., N74D, F77L, V105A, V109A, V125A, and T148I, may compensate by similar mechanisms.
The model shows b4 to be on the membrane surface rather than inserted in the bilayer. Proteo-lysis of membrane-bound Cyt2Aa1 occurred at a number of cleavage sites in theaD-b4 loop to leave
b4 associated with b5,b6 andb7 in the membrane protected fragment [23]. However proteolysis of membrane bound Cyt1Aa revealed cleavage sites in
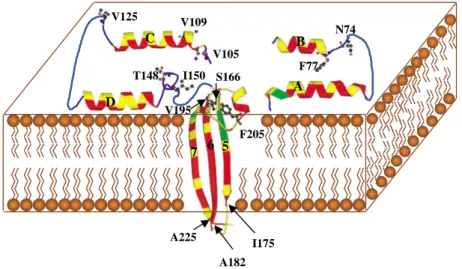
A B C D 5 6 7 N74 F77 V109 V105 T148 I150 F205 S166 V195 V125 A225 A182 I175
Figure 4. Schematic diagram of a possible model for insertion of Cyt2Aa1 into the cell membrane. The helicesaA,aB,aC andaD bind the membrane and form an oligomer with other protomers. The regions betweenaD andb5 and betweenb6 andb7 act as hinges during the conformational changes following membrane binding. Sheetsb5,b6 andb7 insert into the membrane and form an oligomer with similar sheets from other protomers to form ab-barrel pore. Residues in helices and sheets are coloured according to their hydrophobicity (red for hydrophobic, yellow for polar or charged and green for tyrosine). Sheetb3 which links helicesaB andaC is not shown. The positions of the primary inactive mutant (I150A) and the high activity revertants are shown in ball and stick as the original residues in the wild type. I175, A182 and A225 are the residues at the C or N termini ofb5,b6 andb7, respectively.
b4 including one at the C-terminal end. This indicates that in Cyt1Aa, b4 is membrane bound but not inserted and we suggest this is also likely to be true for Cyt2Aa1.
Li et al. [4] proposed that b5, b6 and b7 from several toxins form a pore based on a b-barrel structure. Experiments using polarity sensitive acry-lodan-labeled toxin have confirmed that the pore is oligomeric with each monomer contributing three strands (b5,b6,b7) [17]. Strandsb5 andb7 show a pattern of alternating hydrophobic and hydrophilic residues similar to that of the known membrane spanning regions of a-hemolysin [27] and anthrax toxin protective antigen [28]. Strand b6 is highly hydrophobic and the pattern is less obvious than in
b5 andb7. The amino acid sequences ofb5,b6 and
b7 are not conserved but the alternating hydropho-bic and hydrophilic pattern is highly conserved in all Cytd-endotoxins (Figure 1).
The crystal structures of the transmembrane domains of several proteins have been determined. In many of these structures, bands of aromatic residues (Tyr, Phe and Trp) lie at the hydrophobic/ hydrophilic interface of the lipid bilayer and are considered to be a general characteristic of trans-membrane proteins such as a-hemolysin [27], OmpA [29] and porin from Rhodobacter capsulatus
[30]. Although the three b-barrel structures are diverse in terms of subunit stoichiometry and architecture, each shows the band of aromatic residues (Tyr, Phe and Trp) at the hydrophobic/ hydrophilic interface of the lipid bilayer. These residues have intermediate polarity and are consid-ered to interact favorably with the membrane at the hydrophobic/hydrophilic interface. In Cyt2Aa1, there are three tyrosine residues near the N-terminus of b5 and b7 (Y167, Y169 in b5 and Y213 in b7) which are conserved in all Cytd-endotoxins (Figure 1). After the Cyt toxin monomers have assembled and formed a pore in the membrane, these residues could form a band around theb-barrel at a point that the model proposes to be the point of entry ofb5 and
b7 (Figure 4). This band would be very similar in position to the band in a-hemolysin.
The recent solution of the crystal structure of a fungal pore forming toxin, volvatoxin A2 (VVA2), has shown it to be almost identical to Cyt2Aa1 [31]. Electron microscopy of this toxin suggested that it could form an oligomer with 18 subunits [31]. Investigation of truncated fragments of VVA2 sug-gested that the N-terminal fragment domain which consists mostly of a-helices is responsible for oligo-merization and the C-terminal fragment domain consists of three b-sheets inserted into the mem-brane [32]. In Cyt2Aa1, sheets b5,b6 andb7 were reported to insert into the membrane and are
protected from protease digestion [23]. Experiments using synthetic peptides corresponding to helicesaA and aC [25] demonstrated that these helices could form oligomers. We therefore propose that Cyt2Aa1 forms a b-barrel transmembrane pore by inserting
b5, b6 and b7 from 18 toxin molecules into the membrane with helicesaA andaC playing a role in the oligomerization step.
Acknowledgements
We thank Dr Jade Li for valuable suggestions and discussion and Dr Paul Davis for assistance with graphics and multiple sequence alignment and Zeneca Agrochemicals, UK and the Institute of Molecular Biology and Genetics, Mahidol Univer-sity, Thailand, for supplying mosquito eggs. Support to B.P. from the Thailand National Center for Genetic Engineering and Biotechnology is gratefully acknowledged.
References
[1] Schnepf HE, Crickmore N, Van Rie J, Lereclus D, Baum J, Feitelson J, Zeigler DR, Dean DH.Bacillus thuringiensisand its pesticidal proteins. Microbiol Mol Biol Rev 1998;62: 775/806.
[2] Crickmore N, Zeigler DR, Feitelson J, Schnepf E, Van Rie J, Lereclus D, Baum J, Dean DH. Revision of the nomencla-ture for theBacillus thuringiensis pesticidal crystal proteins. Microbiol Mol Biol Rev 1998;62:807/813.
[3] Li J, Carroll J, Ellar DJ. Crystal structure of insecticidal d-endotoxin from Bacillus thuringiensis at 2.5A˚ resolution. Nature 1991;353:815/821.
[4] Li J, Koni PA, Ellar DJ. Structure of the mosquitocidal d-endotoxin CytB fromBacillus thuringiensissp.kyushuensis
and implications for membrane pore formation. J Mol Biol 1996;257:129/152.
[5] Knowles BH, Ellar DJ. Colloid-osmotic lysis is a general feature of the mechanism of action ofBacillus thuringiensis d-endotoxins with different specificity. Biochim Biophys Acta 1987;924:509/518.
[6] Koni PA, Ellar DJ. Biochemical characterisation ofBacillus thuringiensiscytolytic delta-endotoxins. Microbiol 1994;140: 1869/1880.
[7] Chow E, Singh FJP, Gill SS. Binding and aggregation of the 25 kilodalton toxin ofBacillus thuringiensissubsp.israelensis
to insect cell membranes and alteration by monoclonal antibodies and amino acid modifiers. Appl Environ Micro-biol 1989;55:2779/2788.
[8] Drobniewski FA, Ellar DJ. Purification and properties of a 28-kilodalton hemolytic and mosquitocidal protein toxin of
Bacillus thuringiensis subsp.darmstadiensis 73-E10-2. J Bac-teriol 1989;171:3060/3067.
[9] Knowles BH, Blatt MR, Tester M, Horsnell JM, Carroll J, Menestrina G, Ellar DJ. A cytolytic d-endotoxin from
Bacillus thuringiensis var. israelensis forms cation-selective channels in planar lipid bilayers. FEBS Lett 1989;244:259/ 262.
[10] Knowles BH, White PJ, Nicholls CN, Ellar DJ. A broad spectrum cytolytic toxin from Bacillus thuringiensis var.
[11] Maddrell SHP, Lane NJ, Harrison JB, Overton JA, Moreton RB. The initial stages in the action of an insecticidal d -endotoxin of Bacillus thuringiensis var. israelensis on the epithelial cells of the malpighian tubules of the insect,
Rhodnius prolixus. J Cell Sci 1988;90:131/144.
[12] Maddrell SHP, Overton JA, Ellar DJ, Knowles BH. Action of activated 27000 Mrtoxin fromBacillus thuringiensis var. israelensis on malpighian tubules of the insect, Rhodnius prolixus. J Cell Sci 1989;94:601/608.
[13] Panchal RG, Bayley H. Interactions between residues in
Staphylococcal a-hemolysin revealed by reversion mutagen-esis. J Biol Chem 1995;270:23072/23076.
[14] Koni PA, Ellar DJ. Cloning and characterization of a novel
Bacillus thuringiensis cytolytic delta-endotoxin. J Mol Biol 1993;229:319/327.
[15] Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: Nucleotide sequences of the M13MP18 and pUC19 vectors. Gene 1985;33:103/119. [16] Dower WJ, Miller JF, Ragsdale CW. High efficiency
trans-formation ofE. coliby high voltage electroporation. Nucleic Acids Res 1988;16:6127/6145.
[17] Promdonkoy B, Ellar DJ. Membrane pore architecture of a cytolytic toxin from Bacillus thuringiensis. Biochem J 2000;350:275/282.
[18] Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248/254.
[19] Thomas WE, Ellar DJ. Bacillus thuringiensis var.israelensis
crystald-endotoxin: Effects on insect and mammalian cells
in vitroandin vivo. J Cell Sci 1983;60:181/197.
[20] Ward ES, Ellar DJ, Chilcott CN. Single amino acid changes in theBacillus thuringiensisvar.israelensis d-endotoxin affect the toxicity and expression of the protein. J Mol Biol 1988;202:527/535.
[21] Tindall KR, Kunkel TA. Fidelity of DNA synthesis by the Thermus aquaticus DNA polymerase. Biochemistry 1988;27:6008/6013.
[22] Cha RS, Thilly WG. Specificity, efficiency, and fidelity of PCR. PCR Methods Applic 1993;3:18/29.
[23] Du J, Knowles BH, Li J, Ellar DJ. Biochemical characteriza-tion of Bacillus thuringiensis cytolytic toxins in association with a phospholipid bilayer. Biochem J 1999;338:185/193. [24] Gazit E, Shai Y. Structural characterization, membrane interaction, and specific assembly within phospholipid membranes of hydrophobic segments from Bacillus thurin-giensis var.israelensiscytolytic toxin. Biochemistry 1993;32: 12363/12371.
[25] Gazit E, Burshtein N, Ellar DJ, Sawyer T, Shai Y.Bacillus thuringiensis cytolytic toxin associates specifically with its synthetic helices A and C in the membrane bound state: Implication for the assembly of oligomeric transmembrane pores. Biochemistry 1997;36:15546/15554.
[26] Promdonkoy B, Ellar DJ. Investigation of the pore forming mechanism of a cytolytic d-endotoxin from Bacillus thur-ingiensis. Biochem J 2003;374:255/259.
[27] Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. Structure of Staphylococcal a-hemolysin, a heptameric transmembrane pore. Science 1996;274:1859/ 1866.
[28] Petosa C, Collier RJ, Klimpel KR, Leppla SH, Liddington RC. Crystal structure of the anthrax toxin protective antigen. Nature 1997;385:833/838.
[29] Pautsch A, Schulz GE. Structure of the outer membrane protein A transmembrane domain. Nature Struct Biol 1998;5:1013/1017.
[30] Weiss MS, Schulz GE. Structure of porin refined at 1.8 A˚ resolution. J Mol Biol 1992;227:493/509.
[31] Lin SC, Lo YC, Lin JY, Liaw YC. Crystal structures and electron micrographs of fungal volvatoxin A2. J Mol Biol 2004;343:477/491.
[32] Weng YP, Lin YP, Hsu CI, Lin JY. Functional domains of a pore-forming cardiotoxic protein, volvatoxin A2. J Biol Chem 2004;279:6805/6814.
[33] Kraulis P. MolScript: A program to produce both detailed and schematic plots of protein structures. J Appl Crystallog 1991;24:946/950.