SPECIAL ARTICLE
Sickle Cell Disease: A Question of Equity and Quality
Lauren A. Smith, MD, MPHa, Suzette O. Oyeku, MD, MPHb, Charles Homer, MD, MPHc, Barry Zuckerman, MDa
aDepartment of Pediatrics, Boston University School of Medicine, Boston Medical Center, Boston, Massachusetts;bDivision of General Pediatrics, Children’s Hospital Boston, Boston, Massachusetts;cNational Initiative for Children’s Healthcare Quality, Boston, Massachusetts
The authors have indicated they have no financial relationships relevant to this article to disclose.
ABSTRACT
Thirty years ago, the first major federal legislation concerning sickle cell disease treatment was passed, resulting in the development of comprehensive sickle cell centers. We are now at another watershed moment in the treatment of this illness with the passage in October 2004 of the Sickle Cell Treatment Act, designed to substantially expand specialized sickle cell treatment programs. This legislation offers a remarkable opportunity to significantly improve health outcomes for individuals with sickle cell disease if it is implemented with a specific focus on the distinct but related issues of equity and quality. Despite major advances in sickle cell disease treatment that have occurred over the past 3 decades, important gaps exist both in the equity of government and private philanthropic support for research and in the uniform provision of high quality clinical care. This article assesses the current gaps in funding support and in the implementation of im-provements in clinical care in order to suggest strategies for making optimal use of the opportunity that the new legislation presents to improve the health of all individuals affected by this disease.
www.pediatrics.org/cgi/doi/10.1542/ peds.2005-1611
doi:10.1542/peds.2005-1611
Abbreviations
SCD—sickle cell disease NIH—National Institutes of Health RFA—requests for application NHLBI—National Heart, Lung, and Blood Institute
AHRQ—Agency for Healthcare Research and Quality
Accepted for publication Oct 17, 2005 Address correspondence to Lauren A. Smith, MD, MPH, Department of Pediatrics, Boston Medical Center, 91 E Concord St, Maternity Building, 4th Floor, Boston, MA 02118. E-mail: [email protected]
T
HREE DECADES AFTER the publication of Robert Scott’s1,2 influential critiques of the status of re-search and clinical care for sickle cell disease (SCD), “Health Care Priority and Sickle Cell Anemia” and “Sickle Cell Anemia: High Prevalence and Low Priority,” we are at another watershed moment in the history of SCD treatment. Scott’s articles were pivotal in influenc-ing subsequent congressional hearinfluenc-ings that lead to the passage of the first major legislation concerning SCD treatment, the National Sickle Cell Anemia Control Act, in 1972, which increased funding for SCD, primarily through the development of comprehensive sickle cell centers.3Scott emphasized the relationship between the status of SCD research and civil rights and argued for “increased priority and attention by both the public and the health professions,” because, at the time, SCD re-ceived less public and professional support compared with other less prevalent diseases, such as cystic fibrosis, despite having a substantial public health impact.1,2The passage of the Sickle Cell Treatment Act in Oc-tober 2004, designed to substantially expand specialized sickle cell treatment programs, offers a remarkable op-portunity to significantly improve health outcomes for individuals with SCD if the legislation is implemented with a specific focus on the distinct but related issues of equity and quality. Important gaps exist in the equity of research funding allocation and private philanthropy and in the provision of high-quality clinical care, despite major advances in treatment.
In this article we assess the current gaps in funding support for SCD and in the full implementation of
clin-ical advances in order to suggest strategies for making optimal use of the opportunity that the newest legisla-tion presents to improve the health of all individuals affected by this disease. The issue of equity and quality in SCD care is of particular relevance to pediatricians and child health policy scholars, because pediatric providers are on the forefront of treating this disease in the first 2 decades of life.
SIGNIFICANT GAPS PERSIST IN PUBLIC AND PRIVATE SUPPORT FOR RESEARCH AND CLINICAL CARE
In the United States, there are⬎80 000 people affected with SCD.4 It affects 1 in 400 blacks and 1 in 19 000 Latinos and has a carrier rate of 1 in 12 and 1 in 100 for black and Latino populations, respectively.4,5 In 1970, Scott1highlighted a substantial difference in the research effort for sickle cell anemia compared with other chronic childhood diseases, measured by the number of National Institutes of Health (NIH) grants. Scott1noted that there were 3 times as many grants for the more highly publi-cized conditions of cystic fibrosis and muscular dystro-phy as there were for SCD. Subsequent to the passage of the 1972 SCD treatment legislation, the total number of grants for SCD increased by a factor of 10 (Table 1). Although we might now better assess research effort by overall funding rather than the number of grants, a gap in the research effort remains (Table 1). Although NIH is not the only source of funding for medical research, it is the major governmental resource, so disparities in NIH funding have important implications for research effort. For 2004, NIH reports spending $90 million on SCD
TABLE 1 NIH Research Funding and Private, Nonprofit Association Support of SCD and Cystic Fibrosis
Variable SCD Cystic Fibrosis
US prevalencea 80 000 30 000
Federal support
NIH fiscal-year 2004 funding, in millions of dollarsb 90 128
NIH funding per person with disease, $ 1125 4267
No. of federal grants
No. of grants funded in 1968c 22 65
No. of grants funded in 1972, after Sickle Cell Anemia Control Actd 215 80
No. of grants funded in 2004 331 459
Private philanthropic support, $
Cystic Fibrosis Foundation 2003 annual revenuee 152 231 000
Sickle Cell Disease Association of America 2003 annual revenue,f 498 577
Revenue per person affected with disease 6 5074
Total NIH and private support, in millions, $ 90.4 280.2
Total support per person affected with disease, $ 1130 9340
aSources: National Human Genome Research Institute. Learning about sickle cell disease. Available at: www.genome.gov/10001219; and Na-tional Institutes of Health and NaNa-tional Human Genome Research Institute. Learning about cystic fibrosis. Available at: www.genome.gov/ 10001213.
bSource: National Institutes of Health. Estimates of funding for various diseases, conditions, and research areas. Available at: www.nih.gov/news/ fundingresearchareas.htm
cSource: Scott R. Health care priority and sickle cell anemia.JAMA. 1970;214:731–734.
dSource: Office of Extramural Research and National Institutes of Health. Computer Retrieval of Information on Scientific Projects (CRISP). Available at: www.crisp.cit.nih.gov.
eSource: Cystic Fibrosis Foundation. Science People Support: 2003 Annual Report. Available at www.cff.org/publications/files/ 2003㛭Annual㛭Report.pdf.
across all of its institutes.6This funding figure includes all of the research that NIH deems either directly or periph-erally related to SCD. This contrasts with $128 million NIH spent on cystic fibrosis, which affects 30 000 indi-viduals in the United States (Table 1). Although per capita expenditures do not fully capture the differing experiences of disease by individuals, it is notable that NIH allocates almost 4 times more funding per person affected with cystic fibrosis as it does for those affected by SCD. These levels of funding have been essentially stable over the past 4 years.6
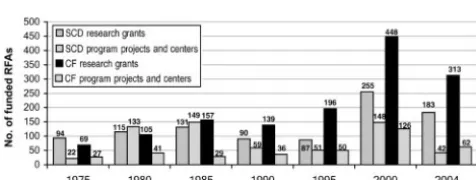
Trends since 1975 in the number of research grant requests for application (RFAs) for SCD and cystic fibro-sis are presented in Fig 1. There were not substantial differences until 1990 when the increase in the number of RFAs for cystic fibrosis research outstripped the num-ber of RFAs for SCD research. Notably, in the wake of the 1975 SCD legislation, the number of RFAs for pro-grams and centers was initially higher for SCD; however, the numbers were comparable after 1985.7 In recent years, funding has been earmarked to establish an SCD clinical research network, which is focused on translat-ing results from basic science trials to phase III clinical trials.8
In addition to the gap in federally sponsored research, there is also a substantial difference in how extensively the private sector has been mobilized to support SCD research and clinical care, which Scott2 also noted in 1970. For example, for fiscal year 2003, the Sickle Cell Disease Association of America’s total revenue was $498 577, compared with $152 million for the Cystic Fibrosis Foundation, a 300-fold difference that has sub-stantial implications for the Sickle Cell Disease Associa-tion of America’s ability to support research and advo-cacy (Table 1). For example, the Cystic Fibrosis Foundation has established the Therapeutic Develop-ment Program to provide matching research funds to stimulate the development of new treatments and the
Therapeutic Development Network to promote the co-ordination of clinical trials.9The Sickle Cell Disease As-sociation of America does not have the resources to launch such broad initiatives. When NIH and private support are combined, the funding gap between SCD and cystic fibrosis triples. Based on the combined NIH and private funding, there is⬎8 times more support per person affected by cystic fibrosis than that for those affected by SCD (Table 1).
THE ROLE OF PRIVATE ORGANIZATIONS IN RESEARCH AND CLINICAL CARE: BEYOND FUNDING
Private charities can do much to improve care and out-comes for those with particular conditions, in addition to providing direct funding for research. First, these orga-nizations provide national advocacy by creating public demand for funding for scientific and clinical advances. People with a limited knowledge of muscular dystrophy are likely familiar with the Jerry Lewis telethon, which mobilizes public support, in addition to generating fund-ing, for muscular dystrophy research. The Cystic Fibrosis Foundation has actively engaged in legislative advocacy and regularly presented testimony to Congress about NIH funding and to the Institute of Medicine regarding research efforts.10,11 Although SCD organizations made very important contributions to the passage of the recent sickle cell legislation, overall they have not been as successful in generating general public support as other organizations.12Second, private philanthropic organiza-tions serve as valuable resources for patients with the disease and can educate and empower individual pa-tients to advocate for the best care available. Third, these organizations reduce the stigma frequently associated with chronic diseases by disseminating positive images and information about the disorder. Without this effort, children with SCD are left with the 50-year-old social stigma of “bad blood” and other negative and embarrass-ing images.2,13Lastly, private philanthropic organizations support the quality of health care delivery to affected patients. The Cystic Fibrosis Foundation is a leader in this role of assuring and improving the quality of care by supporting the development and implementation of clinical practice guidelines and a formal accreditation process for cystic fibrosis care centers. These accredited care centers receive funding from the foundation to support the delivery of quality care to cystic fibrosis patients and are required to submit data to a patient registry. The registry tracks morbidity and mortality of the disease, as well as the practice patterns and outcomes at individual care centers.14 The variability in practice patterns and outcomes evident in the registry has served as a stimulus for the Cystic Fibrosis Foundation to launch an ambitious quality improvement initiative aimed at identifying and disseminating “best practices” throughout their care center network. All of these efforts
FIGURE 1
require financial and organizational resources, and, in this arena, SCD patients have been left behind.
CLINICAL AND QUALITY IMPROVEMENTS IN SICKLE CELL CARE
In addition to the ongoing lack of equity in public re-search funding and private sector involvement, the cur-rent status of SCD can be further evaluated in the con-text of equity in the quality of clinical care. In the 3 decades since the Scott articles,1,2 despite continued funding gaps, there have, nevertheless, been substantial improvements in SCD treatments, leading to an increase in life expectancy from 14 years in 1973 to the mid- to late 40s now (Table 2).15–17Recent survival data indicate an 85.6% overall survival and an 88.5% stroke-free survival at age 18, substantially higher than the 50% overall survival when Scott wrote his commentaries.1,17 From 1968 to 1992, mortality has decreased 40 –50% in children with SCD.18The reduction in mortality is attrib-utable to several factors. First, all but 2 states have uni-versal newborn screening for SCD that allows for the initiation of secondary and tertiary prevention and man-agement strategies from birth, such as penicillin prophy-laxis, which was proven effective at preventing invasive bacterial infections in 1986.19 Recent cohort data indi-cate that only 20% of deaths before adulthood are be-cause of infection, compared with much higher propor-tions in prior cohorts of SCD patients.17 Second, the institution of hydroxyurea as a treatment has decreased painful crises and reduced mortality by 40% in adults with SCD.20,21Third, the Stroke Prevention Trial in Sickle Cell Anemia demonstrated the effectiveness of prevent-ing this potentially devastatprevent-ing complication by screen-ing patients with transcranial Doppler studies, followed by appropriate initiation of transfusion therapy.22A
re-cent study demonstrated a 75% decline in stroke rates in children in the 3 years after the Stroke Prevention Trial findings were published.23 Although it has not been widely used, bone marrow transplantation has been demonstrated to successfully treat SCD in selected pa-tients.24
Simultaneously, during the past decade, there has seen substantial growth in the field of quality improve-ment, with efforts aimed at improving systems of clinical care. The Institute of Medicine reports,Crossing the Qual-ity ChasmandTo Err is Humanhighlight the rationale for these efforts.25–29 Quality improvement strategies have been used extensively to assess and enhance care for adult and pediatric patients with other chronic diseases, such as asthma, diabetes, heart disease, and renal dis-ease, among others.30A recent study of Medicare bene-ficiaries needing dialysis showed that quality improved for all patients and, importantly, that the quality gap between white and minority patients narrowed when quality improvement strategies were instituted.31 Al-though quality improvement has been embraced by leaders in medicine as an indispensable part of health care, these effective tools have not been incorporated routinely and effectively in the clinical care of patients with SCD.
EQUITY IN THE EMPHASIS ON AND AVAILABILITY OF QUALITY CARE
Given the notable advances in SCD care, the gap in ensuring widespread adoption of effective practices and their delivery in a context that meets child and family needs is of the gravest concern.30,32Consensus reports on SCD management, such as the National Heart, Lung, and Blood Institute (NHLBI) guidelines, The Management of Sickle Cell Diseaseand the American Academy of
Pediat-TABLE 2 SCD Life Expectancy and Highlights in Advances in SCD Treatment Year Life Expectancy,
y
Advances
1949 Sickling of red blood cells attributed to hemoglobinopathy
1970 10–12 National Association for Sickle Cell Disease established (presently, Sickle Cell Disease Association of America)
1972 National Sickle Cell Anemia Control Act passed, funding 15 comprehensive sickle cell centers
1975 First statewide newborn screening implemented
1978 Cooperative Study of Sickle Cell Disease begun
1980 20–22
1986 Prophylactic penicillin proven effective in preventing pneumococcal infection
1990 30–35
1995 Hydroxyurea treatment found to decrease complications in adults 1996 Bone marrow transplantation found effective at curing sickle cell disease
1997 Stroke Prevention Trial study demonstrates effectiveness of screening and transfusion therapy 2000 40–45 Heptavalent pneumococcal vaccine introduced
2001 Corrected SCD in transgenic mouse model by gene therapy
2004 Sickle Cell Treatment Act47passed, providing funding for 40 sickle cell disease treatment centers, the establishment of a National Coordinating Center, and federal matched funds for genetic counseling and education
rics Policy Statement “Health Supervision for Children With Sickle Cell Disease” outline for both primary care and specialty physicians what high quality, comprehen-sive sickle cell care should include, such as: the provision and coordination of care through a medical home, the integration of primary and specialty care, family and patient education regarding symptoms of serious com-plications, genetic counseling, prevention of infection through antibiotic prophylaxis and vaccination, screen-ing for stroke risk and neurocognitive testscreen-ing, judicious use of properly screened blood transfusions, appropriate pain management, treatment of renal complications, and regular ophthalmologic evaluations.5,16,33–35
However, there is no coordinated process to ensure the widespread adoption of treatment guidelines and no requirement for NHLBI-funded comprehensive sickle cell centers to track their implementation or assess their effectiveness, such as occurs through the cystic fibrosis registry.36Because there are effective treatments, mean-ingful access to these treatments can improve patient outcomes. In many areas of the country, pediatricians share responsibility with hematologists for caring for children with SCD. It is the minority of sickle cell pa-tients who receive care in one of the comprehensive sickle cell centers.8 Many primary care providers who practice in settings outside comprehensive sickle cell centers may not be fully aware of treatment guidelines. There is also a crucial need to increase the workforce capacity to care for adult patients with SCD to provide appropriate continuity of care for adolescents transition-ing to adult care.8
The limited evidence on the quality of SCD care sug-gests that the significant gains in clinical care for SCD have not been uniformly distributed. For example, al-though penicillin prophylaxis is effective in preventing morbidity and mortality because of invasive pneumo-coccal disease, recent data demonstrate inadequate pro-phylaxis rates among publicly insured children com-pared with their privately insured counterparts.37,38 Studies have also noted rural-urban differences in func-tioning and health care use among patients with SCD.39 Quinn et al,17in a discussion of the survival outcome for their Dallas cohort, state that the encouraging “survival estimates from our study also reflect the availability of specialized sickle cell disease-related care, which does not, unfortunately, apply to every child with sickle cell disease in the United States.” In support of this assertion, geographic differences exist in the mortality of young children with SCD. Mortality varies 16-fold between those states with the lowest and highest rates; even wider disparities are apparent at the county level.40 Be-cause it is unlikely that these differences in health out-comes are related to geographic differences in underly-ing disease severity, such disparities are more likely attributed to variation in factors, such as the adoption of high-quality care practices. The lack of systematic
re-search on quality and access issues is striking in this context.39
Thus, the promise of quality improvement remains incompletely used within SCD care. The diffusion of medical knowledge is often slow and uneven.41 The manner in which innovations, such as specific clinical advances or quality improvement strategies, spread can serve either to improve or exacerbate racial disparities. The more effective the technology is, the more likely unequal access to it will worsen disparities.42The widely different geographic rates of child mortality indicate that this dynamic is likely at work in the case of SCD. We know very little about what proportion of patients with SCD actually get the type of care recommended in pub-lished guidelines, what the barriers are to uniform pro-vision of this care, and what needs to be done to over-come them. For example, currently there is no way of knowing how many patients are offered hydroxyurea treatment when appropriate. A predominant focus on research rather than also promoting the effective incor-poration of important clinical advances into actual SCD care delivered within and outside comprehensive treat-ment centers misses the opportunity to improve out-comes for children and adults with this disease.
A CONSIDERATION OF RACE IN THE TREATMENT OF SCD
There is no denying that race matters in the United States: as a country, we continue to struggle with the implication of past and present racial bias for health. Racial and ethnic disparities in health and health care are described in the Institute of Medicine’s report, Un-equal Treatmentand the Secretary of Health and Human ServicesNational Health Care Disparities Report.43–45In gen-eral, these and other reports focus on racial and ethnic differences in diagnosis, treatment, and outcome within the same disease or condition. SCD is largely absent from discussions of disparities precisely because it predomi-nantly affects patients of 1 race; hence, there are no racial differences in care or outcome within the condi-tion. However, the focus on intracondition differences obscures important considerations of differences in out-comes between conditions that may disproportionately affect different sectors of the population.
RECOMMENDATIONS
Thirty years after Scott’s1,2 influential commentary, we can create another watershed moment in the history of sickle disease care by thoughtful implementation of the important recent legislation, the Sickle Cell Treatment Act. Just as basic and clinical research and early identi-fication benefited after the passage of the SCD legislation in 1972, equity and quality in SCD care will occur if it is explicitly funded. The recent legislation, signed into law in October 2004, was designed to expand specialized sickle cell treatment programs.47The law provides fund-ing for genetic counselfund-ing, community outreach and education, and the establishment of 40 SCD treatment centers. Specific details on which entities will be eligible to receive funding from the legislation have not been fully outlined. However, it seems that community health centers will be encouraged to partner with comprehen-sive SCD treatment centers to increase access to high-quality care in areas where there is currently no NHLBI-funded SCD center. In 2004, Congress appropriated $200 000 for the establishment of a demonstration pro-gram and a National Coordinating Center to coordinate and distribute data, best practices, and results from the activities of the 40 sickle cell treatment centers, as well as to develop educational materials and model protocols for the prevention and treatment of SCD. In December 2005, the Senate approved $2.2 million to fund the new law.48
This period after the Sickle Cell Treatment Act has been signed into law offers a unique opportunity for all of the agencies responsible for funding SCD research, clinical care, and quality improvement to make consid-erations of equity and quality an explicit priority in both the specific implementation of the law and in overall funding priorities more generally. The following recom-mendations could facilitate this effort.
First, the Health Resources and Services Administra-tion, charged with overseeing the National Coordinating Center, should ensure that the center emphasizes the diffusion of clinical advances by requiring that all clinical programs use the clinical patient data collection registry already being developed for the 10 existing NIH-funded comprehensive sickle cell centers and that these data at the organizational level be available to the public.49This registry can serve as a model for collecting and dissem-inating data on clinical care and outcomes that would be adopted by all 40 of the proposed clinical centers, as well as other nonfederally supported SCD care centers. Im-plementation of such a disease registry will provide ro-bust data on provider practices, use, and guideline im-plementation. Such data are lacking for health services and outcomes research related to SCD.
Second, the Center for Medicare and Medicaid Ser-vices, responsible for reimbursing clinical serSer-vices, ge-netic counseling, and community education, should
mandate reporting of quality of care data before federal matching funds will be disbursed.
Third, NIH and the Agency for Healthcare Research and Quality (AHRQ) should make the consistent imple-mentation of scientific advances in clinical care for SCD an explicit funding priority. Both AHRQ and NIH have highlighted the importance of focusing on improving knowledge diffusion, AHRQ through its Translating Re-search into Practice initiative and NIH through its initia-tive on “reengineering the clinical research enter-prise.”50,51NIH is appropriately beginning to shift more attention to expanding the idea of “translational search” to include translating findings from clinical re-search into daily, routine clinical care in all practice settings.50,52
Fourth, an explicit focus on equity and quality should be included in considerations of the potential clinical advances generated by the new tools and techniques of genomics. The implications of therapy in the genome era was addressed in a recent conference hosted by NIH and are being considered by the Trans-NIH Sickle Cell Dis-ease Therapies Working Group representing staff from 8 NIH institutes.8
These recommendations are relevant to both federal and state governments, because both levels of govern-ment have a substantial financial interest in ensuring the diffusion of quality care. A recent analysis indicates that there are ⬃75 000 hospitalizations for SCD annually, resulting in direct costs of more than $475 million, two thirds of these costs are borne by government funded programs.53 This underestimates the total health care costs of SCD, because it does not include emergency department visits or other ambulatory care. The uniform provision of excellent care should reduce costs by sub-stantially lowering the complications that result in emer-gency department visits and hospitalizations.54
The focus on federal support of SCD research and clinical care is warranted, although some private philan-thropy has been extraordinarily effective in creating or-ganizations that stimulate research, improve quality, and educate and support families. By its very nature, such philanthropic efforts have the potential to accentuate disparities based on income, race, or degree of public sympathy rather than based on more objective measures of need. Although greater philanthropic support for SCD would be highly desirable, it would be unrealistic to expect private organizations to completely fill the re-search and quality of care gap in SCD, because those affected by it are more likely to be economically disen-franchised because of their racial or ethnic minority status. Because private funding is not always linked to disease prevalence, government has a legitimate role in balancing private efforts with its own funding and pri-ority setting based on agreed-on measures of the overall burden of different diseases.55
to suffer avoidable complications and even death be-cause effective new therapies have not been uniformly implemented. Although this is not unique to SCD, the severity of the disease and the nature of who suffers from it make the impact of this failure both severe and disparate. It is shortsighted to continue to invest in pducing new knowledge without making a similarly ro-bust commitment to ensuring the universal and equita-ble diffusion of this knowledge so that all patients will reap the full benefit of our investments in research. In the wake of the signing of the Sickle Cell Treatment Act, now is the time to focus on equity and quality in SCD care to demonstrate unequivocally our ability to provide the best health care regardless of the specific disease or ethnic group affected.
ACKNOWLEDGMENTS
Dr Oyeku was supported by grant T32 HP10018-08 from the Health Resources and Services Administration, De-partment of Health and Human Services, to Harvard Pediatric Health Services Research Fellowship Program, Children’s Hospital Boston, at the time of the submission of the article.
We thank Michael Silverstein, MD, Paul Wise, MD, MPH, Lee Pachter, DO, and Sharon Muret-Wagstaff, PhD, MPA, for their helpful review of the article and Sarah Reich, BA, and Jennifer Kreslake, MPH, for their assistance in preparing the article.
REFERENCES
1. Scott R. Health care priority and sickle cell anemia. JAMA. 1970;214:731–734
2. Scott R. Sickle cell anemia: high prevalence and low priority. N Engl J Med.1970;282:164 –165
3. National Heart, Lung, and Blood Institute.Sickle Cell Research for Treatment and Cure. Bethesda, MD: National Institutes of Health; 2002. NIH Publication 02–5214
4. National Human Genome Research Institute. Learning about sickle cell disease. Available at: www.genome.gov/10001219. Accessed November 17, 2004
5. National Heart, Lung, and Blood Institute.The Management of Sickle Cell Disease. Bethesda, MD: National Institutes of Health; 2002. NIH Publication 02–2117
6. National Institutes of Health. Estimates of funding for various diseases, conditions, and research areas. Available at: www.nih. gov/news/fundingresearchareas.htm. Accessed March 21, 2005
7. National Institutes of Health and US Department of Health and Human Services. Computer Retrieval of Information on Scien-tific Projects (CRISP). Available at: http://crisp.cit.nih.gov. Ac-cessed September 22, 2005
8. National Institutes of Health.New Directions for Sickle Cell Ther-apy in the Genome Era. Conference proceedings. Bethesda, MD: November 19 –21, 2003
9. Cystic Fibrosis Foundation. Science People Support: 2003 An-nual Report. Available at www.cff.org/publications/files/ 2003㛭Annual㛭Report.pdf. Accessed March 21, 2005
10. Cystic Fibrosis Foundation. Legislative Activities and Updates. Available at: www.cff.org/legislative㛭action. Accessed June 1, 2005
11. National Heart, Lung, and Blood Institute. Public Interest
Or-ganization Meeting. Available at: www.nhlbi.nih.gov/public/ liaison.htm. Accessed February 9, 2000
12. Tapper M.In the Blood: Sickle Cell Anemia and the Politics of Race. Philadelphia, PA: University of Pennsylvania Press; 1999 13. Wailoo K.Dying in the City of Blues: Sickle Cell Anemia and the
Politics of Race and Health. Chapel Hill, NC: University of North Carolina Press; 2001
14. Cystic Fibrosis Foundation.Patient Registry 2003 Annual Report. Bethesda, MD: Cystic Fibrosis Foundation; 2004
15. Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease: Life expectancy and risk factors for early death.N Engl J Med.1994;330:1639 –1644
16. Claster S, Vichinsky EP. Managing sickle cell disease. BMJ. 2003;327:1151–1155
17. Quinn CT, Rogers ZR, Buchanan GR. Survival of children with sickle cell disease.Blood.2004;103;4023– 4027
18. Davis H, Schoendorf KC, Gergen PJ, Moore RM. National trends in the mortality of children with sickle cell disease, 1968 through 1992.Am J Public Health.2004;87;1317–1322 19. Gaston MH, Verter JI, Woods G, et al. Prophylaxis with oral
penicillin in children with sickle cell anemia. A randomized trial.N Engl J Med.1986;314:1593–1599
20. Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia.N Engl J Med.1995;332:1317–1322
21. Steinberg MH, Barton F, Castro O, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA. 2003;289: 1645–1651
22. Adams RJ, McKie VC, Hsu L, al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial doppler ultrasonography.N Engl J Med. 1998;339:5–11
23. Fullerton H, Adams RJ, Shoujun Z, Johnson SC. Declining stroke rates in Californian children with sickle cell disease. Blood.2004;104;336 –339
24. Walters MC, Patience M, Leisenring W, et al. Bone marrow transplantation for sickle cell disease.N Engl J Med.1996;335: 369 –376
25. Institute of Medicine, Board on Health Care Services, Commit-tee on Identifying Priority Areas for Quality Improvement. Priority Areas for National Action: Transforming Health Care Qual-ity. Washington, DC: National Academy Press; 2003
26. Chassin MR, Galvin RW, and the National Roundtable on Health Care Quality. The urgent need to improve health care quality: Institute of Medicine National Roundtable on Health Care Quality.JAMA.1998;280:1000 –1005
27. Berwick DM. A user’s manual for the IOM’s “Quality Chasm” report.Health Aff (Millwood).2002;21;80 –90
28. Institute of Medicine, Committee on Quality Health Care in America.Crossing the Quality Chasm: A New Health System for the 21st Century. Washington, DC: National Academy Press; 2001 29. Institute of Medicine.To Err Is Human: Building a Safer Health
System. Washington, DC: National Academy Press; 2000 30. Bodenheimer T, Wagner EH, Grumbach K. Improving primary
care for patients with chronic illness: the chronic care model, part 2.JAMA.2002;288;1909 –1914
31. Sehgal AR. Impact of quality improvement efforts on race and sex disparities in hemodialysis.JAMA.2003;289;996 –1000 32. Wagner E. Quality improvement can’t be optional. Eff Clin
Pract.2001;4;239 –249
33. Vichinsky EP. Comprehensive care in sickle cell disease: Its impact on morbidity and mortality. Semin Hematol.1991;28: 220 –226
34. Wood AJ. Management of sickle cell disease.Drug Ther.2004; 340;1021–1030
Oncology CoG. Health supervision for children with sickle cell disease.Pediatrics.2002;109:526 –535
36. National Institutes of Health and National Heart, Lung and Blood Institute.Comprehensive Sickle Cell Centers. RFA HL-01-015. Bethesda, MD: National Institutes of Health and National Heart, Lung and Blood Institute; 2000
37. Benjamin LJ, Dampier CD, Jacox A, et al. Guideline for the Management of Acute and Chronic Pain in Sickle Cell Disease. Glen-view, IL: American Pain Society; 1999
38. Sox CM, Cooper WO, Koepsell TD, DiGiuseppe DL, Christakis DA. Provision of pneumococcal prophylaxis for publicly in-sured children with sickle cell disease. JAMA. 2003;290; 1057–1061
39. Telfair J, Haque A, Etienne M, Tang S, Strasser S. Rural/urban differences in access to and utilization of services among people in Alabama with sickle cell disease.Public Health Rep.2003;118: 27–36
40. Davis H, Gergen PJ, Moore RM. Geographic differences in mortality of young children with sickle cell disease in the United States.Public Health Rep.1997;112:52–58
41. Rogers E.Diffusion of Innovations. 4th ed. New York, NY: Free Press; 1995
42. Wise PH. The transformation of child health in the United States.Health Aff (Millwood).2004;23;9 –25
43. Institute of Medicine.Unequal Treatment: Confronting Racial and Ethnic Disparities in Healthcare. Washington, DC: National Acad-emy Press; 2003
44. Department of Health and Human Services and Agency for Healthcare Research and Quality.National Healthcare Disparities Report. Rockville, MD: Department of Health and Human Ser-vices and Agency for Healthcare Research and Quality; 2003 45. US Department of Health and Human Services and Agency for
Healthcare Research and Quality.National Healthcare Disparities Report. Rockville, MD: US Department of Health and Human
Services and Agency for Healthcare Research and Quality; 2004. Report 05-0014
46. Telfair J, Myers J, Drezner S. Does race influence the provision of care to persons with sickle cell disease? Perceptions of mul-tidisciplinary providers.J Health Care Poor Underserved.1998;9: 184 –195
47. US House of Representatives and Committee on Ways and Means. Inclusion of Primary and Secondary Medical Strategies for Children and Adults With Sickle Cell Disease as Medical Assistance Under the Medicaid Program. Washington, DC: House of Representatives and Committee on Ways and Means; 2004. R4520, Section 712 48. Senate approves Talent-Cochran request for $2.2 million to fund sickle cell treatment law. Available at: www. sicklecelldisease.org/pdf/senateapprovestalentcochran. Ac-cessed March 21, 2006
49. Gawande A. The bell curve.The New Yorker. December 6, 2004. Available at: www.newyorker.com/fact/content/?041206fa_ fact. Accessed September 27, 2005
50. National Institutes of Health. NIH roadmap initiatives. Avail-able at: http://nihroadmap.nih.gov/initiatives.asp. Accessed October 18, 2004
51. Agency for Health Care Policy and Research.Translating Re-search Into Practice. Rockville, MD: Agency for Health Care Policy and Research; 2004. Report HS-99-003
52. Zerhouni E. MEDICINE: the NIH roadmap.Science.2003;302; 63–72
53. Davis H, Moore RM, Gergen PJ. Cost of hospitalizations asso-ciated with sickle cell disease in the United States.Public Health Rep.1997;112:40 – 43
54. Woods K, Karrison T, Koshy M, Patel A, Friedmann P, Cassel C. Hospital utilizaiton patterns and costs for adult sickle cell pa-tients in Illinois.Public Health Rep.1997;11:44 –51
55. Gross CP, Anderson GF, Powe NR. The relation between fund-ing by the National Institutes of Health and the burden of disease.N Engl J Med.1999;340:1881–1887
ESTIMATED COST-EFFECTIVENESS OF GROWTH HORMONE THERAPY FOR IDIOPATHIC SHORT STATURE
Objective:To estimate the cost-effectiveness of growth hormone (GH) ther-apy for idiopathic short stature (ISS). . . .
Patients: A cohort of 10-year-old pre-pubertal boys with ISS treated with GH.
Interventions: Comparison of children treated for 5 years with GH therapy vs children receiving no intervention. . . .
Conclusions:Targeted treatment of children with ISS with the greatest potential for growth appears critical for maximizing cost-effectiveness of GH treatment. However, the significance of the cost per inch is difficult to judge until the utility gains associated with height gain after GH therapy for ISS can be ascertained.
DOI: 10.1542/peds.2005-1611
2006;117;1763
Pediatrics
Lauren A. Smith, Suzette O. Oyeku, Charles Homer and Barry Zuckerman
Sickle Cell Disease: A Question of Equity and Quality
Services
Updated Information &
http://pediatrics.aappublications.org/content/117/5/1763 including high resolution figures, can be found at:
References
http://pediatrics.aappublications.org/content/117/5/1763#BIBL This article cites 29 articles, 7 of which you can access for free at:
Subspecialty Collections
sub
http://www.aappublications.org/cgi/collection/hematology:oncology_
Hematology/Oncology
following collection(s):
This article, along with others on similar topics, appears in the
Permissions & Licensing
http://www.aappublications.org/site/misc/Permissions.xhtml in its entirety can be found online at:
Information about reproducing this article in parts (figures, tables) or
Reprints
DOI: 10.1542/peds.2005-1611
2006;117;1763
Pediatrics
Lauren A. Smith, Suzette O. Oyeku, Charles Homer and Barry Zuckerman
Sickle Cell Disease: A Question of Equity and Quality
http://pediatrics.aappublications.org/content/117/5/1763
located on the World Wide Web at:
The online version of this article, along with updated information and services, is
by the American Academy of Pediatrics. All rights reserved. Print ISSN: 1073-0397.