Research Article
a
December
2018
Computer Science and Software Engineering
ISSN: 2277-128X (Volume-8, Issue-12)
DFT and TD-DFT Study on the Structural, Optoelectronic and
Photovoltaic Properties of Chemically Modified Donor-Acceptor
Conjugated Oligomers for Organic Solar Cells
Zakaria El Malki*
Moulay Ismaïl University, MEM, High School of Technology (ESTM), B.P 3103 Toulal, 50040 Meknes,
Morocco
Mohammed Bouachrine
Moulay Ismaïl University, FS, Faculty of Sciences, B.P. 11201 Zitoune Meknes,
Morocco
Françoise. Serein-Spirau
Molecular and Macromolecular Heterochimy, UMR, CNRS 5076, Higher National School of Chemistry,
Montpellier, France
Jean-Marc. Sotiropoulos
Pau and Adour Countries University, UMR5254 - IPREM, Chemistry-Physics Team, Helioparc –
Pau, France
.
Abstract— In this paper, a novel theoretical tool for some new low-band-gap copolymers has been developed on the basis of density functional theory (DFT) quantum chemical calculations to model their optoelectronic properties. We have designed a series of novel double organic D-π-A (electron donor-π-conjugated-acceptor) dyes employed in dye-sensitized solar cells (DSSCs). These copolymers are constituted of (Carbazole-Methylthiophene), benzothiadiazole and thiophene [(Cbz-Mth)-B-T] units essentially as well as their derivatives leading to donor (D)-acceptor (A) structure-types. The cyanoacrylic acid (A) anchoring group leads to more red shift of absorption bands. The optimized structures and optoelectronic properties of these dyes were investigated by using the Density Functional Theory DFT/B3LYP/6-31G (d, p) method and Time Dependant Density Functional Theory (TD/DFT) calculations. Firstly, we studied the insertion of thiophene as spacer unit into [(Cbz-Mth)-B]2-A backbone to reach (Carbazole-Methylthiophene), benzothiadiazole-thiophene-cyanoacrylic acid [(Cbz-Mth)-B-T]2-A copolymer, secondly, thiophene usual unit was replaced by bithiophene and three-thiophene respectively, entity to obtain
(Carbazole-Methylthiophene), benzothiadiazole-dithiophene-cyanoacrylic acid [(Cbz-Mth)-B-DT]2-A and
(Carbazole-Methylthiophene), benzothiadiazole-Three-Thiophene-cyanoacrylic acid [(Cbz-Mth)-B-TT]2-A copolymers. Later, we examined the insertion effect of bridging by C=C (CN)2 groups on the energy gaps and the electronic properties of the study copolymer [(Cbz-Mth)-B-T-C=C (CN)2-T]2-A, and finally, ethynyl spacer was added to obtain a novel oligomer model, denoted (Carbazole-Methylthiophene)–ethynyl- benzothiadiazole-dithiophene- C=C(CN)2 groups-cyanoacrylic acid [(Cbz-Mth)-E-B-T-C=C(CN)2-T]2-A and to investigate their bridging effect into the main backbone on various properties by examining structural and electronic properties. The calculated geometries indicate that these dyes are all coplanar. In order to predict the band gaps for guiding the synthesis of novel materials with low band gaps, we applied quantum-chemical techniques to calculate the band gaps in several oligomers. The analysis of microelectronic and photonic structure in one dimension program (AMPS-1D) program has been successfully used to study the compounds organic solar cells. The calculated results of these dyes demonstrate that these compounds are then blended with [6,6]-phenyl-C61- butyric acid methyl ester (PCBM) in bulk-heterojunction solar cell.
Keywords: Low band gap; Benzothiadiazole; TD/DFT calculations; Donor-Acceptor; bridging effect.
I. INTRODUCTION
ISSN(E): 2277-128X, ISSN(P): 2277-6451, pp. 38-51
© www.ijarcsse.com, All Rights Reserved Page | 39
important role for DSSCs to gain the higher solar-to-electricity conversion efficiency that have been an active research subject, recently [15–17]. The bulk-heterojunction (BHJ) represents one of the most promising architectures for all organic solar cells [18]. It is based on charge generation at the interface between two different organic semiconductors, that behave as donor and acceptor, and that are blended together. In the BHJ polymer solar cells, a conjugated polymer is employed as a donor in combination with fullerene derivatives [19] (for example [6,6]-phenyl-C61-butyric acid methyl ester or PCBM), widely used as acceptors in solution processable photovoltaic devices. The interaction between the electron donor (D) and acceptor (A) moieties in such an alternating donor-acceptor copolymer can result in the hybridization of the high-lying HOMO energy level of the donor and low-lying energy levels of the acceptor, leading to a relatively small band gap polymer semiconductor with novel electronic structure. Many organic dyes, based on the donor (π spacer) - acceptor (D-π-A) system, exhibiting relatively high DSSC performance, have so far been designed and developed. They include triarylamine dyes [20-23], hemicyanine dyes [24, 25], thiophene-based dyes [26], indoline dyes
[27-30], and phthalocyanine [31] dyes. Although remarkable progress has been made in the development of organic dyes as sensitisers for DSSCs, optimising their chemical structures is required for further improvements in performance. Based on interesting optical properties of thiophene derivative, which can be tuned efficiently by suitable modification
[32–35] and/or intramolecular charge transfer (CT) [36, 37], we are interest to D–A architecture including these varieties of polymers. Tian et al. have reported a series of donor-acceptor-π-bridge-acceptor (D-A-π-A) structural organic dyes incorporating Benzotiadiazole into the triphenylamine framework, resulting in red-shift in absorption and weakening the deprotonation effect on TiO2 film, which is beneficial for light-harvesting [38]. On the other hand, most recent papers are focused on the Carbazole (Cbz) [39-41], due to its important specific properties (photoconductivity, photoluminescence and hole transport properties).
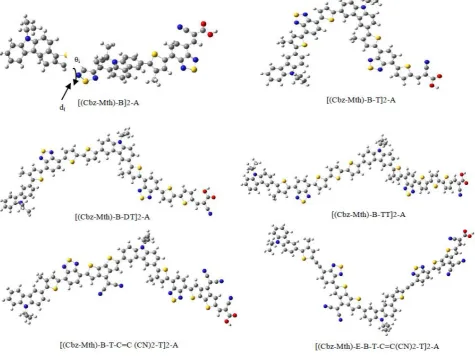
Fig 1. Chemical structure of polymers under study (a) Mth)-B]2-A, (b) Mth)-B-T]2-A, (c) [(Cbz-Mth)-B-DT]2-A, (d) [(Cbz-Mth)-B-TT]2-A, (e) [(Cbz-Mth)-B-T-C=C(CN)2-T]2-A and (f)
[(Cbz-Mth)-E-B-T-C=C(CN)2-T]2-A. N CH3 C H3 H S C H3 N S N N CH3 C H3 H S C H3 N S N C H C CN HOOC N CH3 C H3 H S C H3 N S N S N CH3 C H3 H S C H3 N S N S C H C CN HOOC
(Cbz-Mth)-B-T 2-A (Cbz-Mth)-B 2-A
N CH3 C H3 H S C H3 N S N S S N CH3 C H3 H S C H3 N S N S S C H C CN HOOC
(Cbz-Mth)-B-DT 2-A
N CH3 C H3 H S C H3 N S N S S N N N CH3 C H3 H S C H3 N S N S S N N C H C CN HOOC
(Cbz-Mth)-B-T-C=C(CN)2-T 2-A
(Cbz-Mth)-E-B-T-C=C(CN)2-T 2-A (a) (b) (c) (d) (f) Thiophene Spacer Dithiophene Spacer Bridge effect Ethynyl spacer N S N S S N N N CH3 C H3 H S C H3 N CH3 C H3 H S C H3 N S N S S N N C H C CN HOOC N CH3 C H3 H S C H3 N S N S S S N CH3 C H3 H S C H3 N S N S S S C H C CN HOOC Three-Thiophene Spacer
(Cbz-Mth)-B-TT 2-A
ISSN(E): 2277-128X, ISSN(P): 2277-6451, pp. 38-51
In the present work, we chose the benzothiadiazole (B) unit as an acceptor, targeting first alternated copolymers of Methylthiophene and Carbazole: this main backbone D–A copolymer will be compared to typical (PVK-P3HT) [42]
model structure. We report a theoretical study of the structural, electronic structures and the optical properties of (Carbazole-Methylthiophene), benzothiadiazole and thiophene based a spacer units and bridge groups essentially as well as their derivatives leading to donor (D)-acceptor (A) structure-types. The cyanoacrylic acid (A) anchoring group leads to more red shift of absorption bands. The optimized structures and optoelectronic properties of these dyes were investigated by using the Density Functional Theory DFT/B3LYP/6-31G (d,p) method and Time Dependant Density Functional Theory (TD/DFT) calculations. Firstly, we study the insertion of thiophene as spacer unit into [(Cbz-Mth)-B]2-A backbone to reach (Carbazole-Methylthiophene), benzothiadiazole-thiophene-cyanoacrylic acid [(Cbz-Mth)-B-T]2-A copolymer. Secondly, thiophene usual unit was replaced by bithiophene and three-thiophene respectively, entity to obtain (Carbazole-Methylthiophene), benzothiadiazole-dithiophene-cyanoacrylic acid [(Cbz-Mth)-B-DT]2-A and (Carbazole-Methylthiophene), benzothiadiazole-Three-Thiophene-cyanoacrylic acid [(Cbz-Mth)-B-TT]2-A copolymers. Later, we examined the insertion effect of bridging by C=C (CN)2 groups on the energy gaps and the electronic properties of the study copolymer [(Cbz-Mth)-B-T-C=C (CN)2-T]2-A, and finally, ethynyl spacer was added to obtain a novel oligomer model, denoted (Carbazole-Methylthiophene) – ethynyl- benzothiadiazole-dithiophene- C=C (CN)2 groups-cyanoacrylic acid [(Cbz-Mth)-E-B-T-C=C(CN)2-T]2-A, and to investigate their bridging effect into the main backbone on various properties by examining structural and electronic properties. The influence of the ethynyl unit on the electronic and electrochemical properties of different compounds has extensively been studied [43–46]. The molecular structures are shown in Figure. 1. The geometric structures and electronic properties were investigated by the density functional theory (DFT) at the B3LYP level and 6-31G (d, p) basis set. The calculated results were compared with the study‘s experimental data [42, 47]. The results are a useful guide for the design, synthesis, and confirmation of the experiment.
II. COMPUTATIONAL DETAILS
All molecular calculations were performed in gas phase by Density Functional Theory (DFT) level with the B3LYP hybrid functional [48-50]. All optimizations were calculated without any symmetry constraints using 6-31G (d, p) basis set on Gaussian 09 software package [51]. None of the frequency calculations generating imaginary frequencies indicate that the optimized geometries are true energy minima. The HOMO, LUMO and gap (HOMO-LUMO) energies are also deduced for the stable structures. We investigated the localization of the frontier orbitals. The spatial distributio n of the frontier orbitals (HOMO) and (LUMO) provides a strategy by which the photovoltaic properties of solar cell can be understood. The vertical excitation energy, the wave lengths, and the electronic transition energies of the oligomers were obtained by using time-dependent density functional theory TD/DFT with B3LYP calculations. The electronic absorption spectra of the dyes are calculated and simulated with TD/DFT method at the B3LYP/6-31G (d, p) level in vacuum. The analysis of microelectronic and photonic structure in one dimension program (AMPS-1D) program was successfully used to study the compounds organic solar cells.
III. RESULTS AND DISCUSSION 3.1 Structure and Geometric Properties
The geometry of our dyes has been optimized using DFT/B3LYP/6-31G (d, p) method in the gas phase and their optimized geometries are shown in Figure. 2. The selected dihedral angle (i) and bond distance (di) parameters are
collected in Table 1. After full optimization in the ground state, the results indicate that all the studied compounds are maintained completely planar (the inter-ring torsion angle is evaluated to be about -4°) (Table 1). This planarity increases the conjugation degree and demonstrates that we should observe an enhanced charge transfer. Based on this optimized structure, the access to the characteristic parameters (geometric parameters: dihedral angle (θi) (the deviation from
coplanarity between the donor and acceptor units) and inter ring bond lengths (di) (the bond length between the donor
ISSN(E): 2277-128X, ISSN(P): 2277-6451, pp. 38-51
© www.ijarcsse.com, All Rights Reserved Page | 41
structure. In order to improve the thermal stability as well, we considered using ethynyl as additional spacers between donor (Cbz-Mth) and acceptor units (Benzothiadiazole) (B) (Figure 1f) to obtain (Carbazole-Methylthiophene)-ethynyl benzothiadiazole-dithiophene-C=C(CN)2-cyanoacrylic acid [(Cbz-Mth)-E-B-T-C=C(CN)2-T]2-A. This new copolymer has the same geometric properties as [(Cbz-Mth)-B-T-C=C(CN)2-T]2-A. Their optimized structures adopt a more planar conformation as presented in Figure 2. It can be confirmed that the π-conjugated systems in these dyes should be more planar and therefore the electron can be smoothly injected from the (Cbz-Mth) donor to the cyanoacrylic acceptor.
Fig 2. Optimized structures obtained by B3LYP/6-31G (d,p) of the studied molecules.
TABLE 1 SELECTED BOND LENGTHS (Å) AND DIHEDRAL ANGLES (DEGREE) OF OPTIMIZED STRUCTURES CALCULATED
BY B3LYP/6-31G(D,P) LEVEL OF THEORY.
Dyes
(Cbz-Mth)-B
B-A
[(Cbz-Mth)-B]2-A Distance (d)
1.456 1.458
Dihedral (θ)
-5.258 6.458
(Cbz-Mth)-B
B-T T-A
[(Cbz-Mth)-B-T]2-A
Distance (d)
1.454 1.454 1.424
Dihedral (θ)
-2.036 -5.783 -0.107
(Cbz-Mth)-B
B-T T-T T-A
[(Cbz-Mth)-B-DT]2-A
Distance (d)
ISSN(E): 2277-128X, ISSN(P): 2277-6451, pp. 38-51
Dihedral (θ)
-4.464 3.296 170.39 1
-3.084
(Cbz-Mth)-B
B-T T-T T-T T-A
[(Cbz-Mth)-B-TT]2-A
Distance (d)
1.454 1.453 1.442 1.443 1.427
Dihedral (θ)
-6.655 -0.114 166.68 -166.89 -0.073
[(Cbz-Mth)-B-T-C=C (CN)2-T]2-A
Distance (d)
1.452 1.454 1.474 1.473 1.427
Dihedral (θ)
-7.240 -2.721 -0.010 2.061 0.231
(Cbz-Mth)-E
E-B B-T T- C=C (CN)2
C=C (CN)2-T
T-A
[(Cbz-Mth)-E-B-T-C=C(CN)2
-T]2-A
Distance (d)
1.399 1.408 1.453 1.474 1.473 1.424
Dihedral (θ)
-159.82 151.53 -1.356 0.159 -1.164 -0.009
Note: (Cbz-Mth) is (Carbazole-Methylthiophene), B is Benzothiadiazole, T is thiophene, E is ethynyl, C=C(CN)2 is bridge group and A is the cyanoacrylic acid.
3.2. Electronics Properties
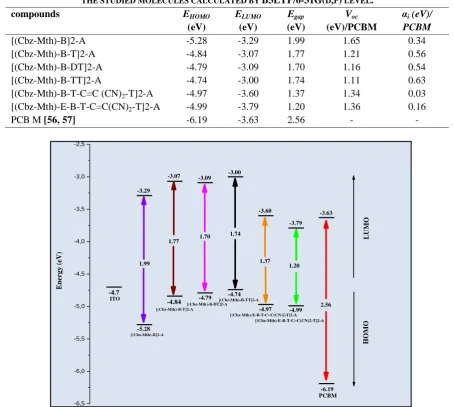
The energy gap (Egap) for [(Cbz-Mth)-B]2-A, T]2-A, DT]2-A,
[(Cbz-Mth)-B-TT]2-A, [(Cbz-Mth)-B-T-C=C(CN)2-T]2-A and [(Cbz-Mth)-E-B-T-C=C(CN)2-T]2-A, was obtained by the differences of HOMO and LUMO energy levels (ΔHOMO-LUMO) using B3LYP/6-31G(d,p). Table 2 lists the theoretical electronic
property parameters (EHOMO, ELUMO and Egap). Calculated band gaps were in the range of 1.20 eV – 1.99 eV. Through our
simulation, [(Cbz-Mth)-B]2-A have an extrapolated band gap about 1.99 eV, HOMO and LUMO energy about -5.28 and -3.29 eV respectively. Thus, and as expected, the [(Cbz-Mth)-B]2-A presents a lower band gap than pristine (Cbz-Mth)2
(4.19 eV) [42], due to the insertion of Benzothiadiazole and cyanoacrylic acid (A) acceptors units in the main backbone copolymer. Another approach to improve the properties of copolymer and to adjust the positions of HOMO and LUMO levels for an adequate application in organic solar cell (OSC) is to introduce a spacer unit in the main backbones [52]. In [(Cbz-Mth)-B]2-A compound, the thiophene spacer unit is inserted in D–A structure between Benzothiadiazole and cyanoacrylic acid (Figure 2). The insertion of thiophene as spacer unit in the backbone copolymer leads to some structural and optical changes. The band gaps in the case of the Mth)-B-T]2-A, Mth)-B-DT]2-A and [(Cbz-Mth)-B-TT]2-A copolymers are about 1.77 eV, 1.70 eV and 1.74 eV respectively which are lower by about 0.22 eV, 0.29 eV and 0.25 eV comparing to [(Cbz-Mth)-B]2-A (Figure. 3). Using the same method, as previously presented, the band gap energy calculated with DFT/B3LYP/6-31G(d,p) method of the [(Cbz-Mth)-B-T-C=C(CN)2-T]2-A copolymer is about 1.37 eV. The HOMO and LUMO energy levels are consequently located respectively at -4.97 eV and -3.60 eV. When we insert the ethynyl spacer, we notice a reduction in the band gap of 1.20 eV. The low HOMO energy level of [(Cbz-Mth)-E-B-T-C=C(CN)2-T]2-A increases the open-circuit potential (VOC) (1.36 eV), which is very sensitive to
energy levels of materials and the interfaces between the different layers [53, 54]. It is also significant in the case of [(Cbz-Mth)-B-T-C=C(CN)2-T]2-A copolymer. In fact, the HOMO energy level of the [(Cbz-Mth)-E-B-T-C=C(CN)2-T]2-A copolymer of about -4.99 eV suggests that this compound has a good stability and potential application for hole injection and transport material. Overall, as we expected, all copolymers show low-band gaps ranging from 1.99 to 1.20 eV. By increasing the electron-releasing effect, the optical band gap derived from the absorption edge of the polymers decreases in the order of [(Cbz-Mth)-B]2-A (1.99 eV) > [(Cbz-Mth)-B-T]2-A (1.77 eV) > [(Cbz-Mth)-B-TT]2-A (1.74 eV) > [(Cbz-Mth)-B-DT]2-[(Cbz-Mth)-B-TT]2-A (1.70 eV) > [(Cbz-Mth)-B-T-C=C(CN)2-T]2-[(Cbz-Mth)-B-TT]2-A (1.37 eV) > [(Cbz-Mth)-E-B-T-C=C(CN)2-T]2-A (1.20 eV) as illustrated in Figure. 3. The LUMO energy levels of all copolymers were estimated between -3.79 and -3.00 eV with small variation, which shows that the LUMO level strongly depends on the acceptor unit (B). On the contrary, HOMO level was much more modified going from -5.28 to -4.74 eV which confirms that this later is mainly determined by donor unit. The insertion of the spacer groups, namely thiophene and ethynyl, also reduces the band gap energy. Improving the efficiency of photovoltaic components (power conversion efficiency values (PCE)) necessarily requires a significant increase in the fill factor (FF) and VOC, which is a compromise for a small gap to cover
ISSN(E): 2277-128X, ISSN(P): 2277-6451, pp. 38-51
© www.ijarcsse.com, All Rights Reserved Page | 43
PCE = FF . VOC. JSC
Pin (1)
Where Pin is the incident power density, Jsc is the short-circuit current, VOC is the open-circuit voltage, and FF
denotes the fill factor. The theoretical values of VOC were calculated from the following expression [55]. The results were
1.34 eV and 1.36 eV for [(Cbz-Mth)-B-T-C=C(CN)2-T]2-A and [(Cbz-Mth)-E-B-T-C=C(CN)2-T]2-A respectively.
Donor HOMO Acceptor
LUMO
OC
E
E
V
(2)
The conjugated copolymers were consequently mixed with [6,6]-phe-nyl-C61-butyric acid methyl ester (PCBM) to generate a good interface for the transfer of charge to the electrode. Energetically, PCBM was characterized by HOMO and LUMO level in the range of -6.19 eV and -3.63 eV [56, 57]. The theoretical values of the open circuit voltage VOC of the studied molecules range from 1.11 eV to 1.65 eV in the case of PCBM (Table 2), these values are
sufficient for a possible efficient electron injection. Therefore, all the studied molecules can be used as sensitizers because the electron injection process from the excited molecule to the conduction band of the acceptor (PCBM) and the subsequent regeneration is possible in the organic sensitized solar cell.
Another parameter denoted αi determined by the difference between the LUMO energy levels of the studied
molecules and the LUMO energy level of PCBM [58]:
Donor LUMO Acceptor
LUMO
i
E
E
(3)
As shown in Table 2, the obtained values of αi were in the range of (0.03-0.63 eV). This indicates that all
LUMO level from all compounds is placed on LUMO level of PCBM. Indeed, we deduce that all compounds are likely to be injected in the excited state.
TABLE 2ENERGY VALUES OF ELUMO(EV),EHOMO(EV),EGAP(EV), THE OPEN CIRCUIT VOLTAGE VOC(EV) AND αi OF
THE STUDIED MOLECULES CALCULATED BY B3LYP/6-31G(D,P) LEVEL.
compounds EHOMO
(eV) ELUMO (eV) Egap (eV) Voc (eV)/PCBM
αi (eV)/
PCBM
[(Cbz-Mth)-B]2-A -5.28 -3.29 1.99 1.65 0.34
[(Cbz-Mth)-B-T]2-A -4.84 -3.07 1.77 1.21 0.56
[(Cbz-Mth)-B-DT]2-A -4.79 -3.09 1.70 1.16 0.54
[(Cbz-Mth)-B-TT]2-A -4.74 -3.00 1.74 1.11 0.63
[(Cbz-Mth)-B-T-C=C (CN)2-T]2-A -4.97 -3.60 1.37 1.34 0.03
[(Cbz-Mth)-E-B-T-C=C(CN)2-T]2-A -4.99 -3.79 1.20 1.36 0.16
PCB M [56, 57] -6.19 -3.63 2.56 - -
Fig 3. Sketch of B3LYP/6-31 G (d,p) calculated energies of the HOMO and LUMO level of the studied molecules.
ISSN(E): 2277-128X, ISSN(P): 2277-6451, pp. 38-51
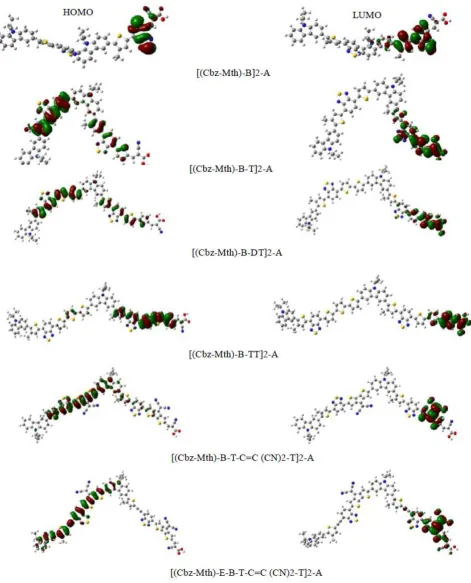
3.3. Frontier Molecular Orbitals
The excited-state properties are strongly related to the electron density in the frontier molecular orbitals (FMO); The HOMO and LUMO can provide a reasonable qualitative indication of the excitation properties and the ability of electron or hole transport [59]. The contour plot of HOMO and LUMO orbitals of the six oligomers obtained by B3LYP/6-31G (d, p) are shown in Figure.4. As shown in Figure.4, while strong localization of the HOMOs occurs on the donor subunits of the polymer backbone, especially on the thiophene units, strong delocalization of the LUMOs occurs on the bridges between the subunits proving the flow of electron density along the polymer backbone, the electron density of LUMO is mainly localized on the acceptor units (mostly in the anchoring group). So, the electronic transitions of the studied molecules from HOMO to LUMO could lead to intramolecular charge transfer from the donor units to the anchoring groups through the conjugated system.
ISSN(E): 2277-128X, ISSN(P): 2277-6451, pp. 38-51
© www.ijarcsse.com, All Rights Reserved Page | 45
3.4. Chemical Reactivity Indices
By using HOMO and LUMO energy values for a molecule, chemical potential (), chemical hardness (), electronegativity () and electrophilicity power () can be calculated as follows [60]:
2
LUMO HOMO E
E
(Chemical potential) (4)
2
HOMO LUMO E
E
(Chemical hardness) (5)
2
LUMO HOMO E
E
(electronegativity) (6)
2
2
(electrophilicity power) (7)
On the other side, we note that the PCBM has the smallest value of the chemical potential ( = - 4.91 eV) compared to six compounds (see Table 3). This is a tendency to view the electrons to escape from those compounds has a high chemical potential to PCBM which has a small chemical potential. Therefore, PCBM behaves as an acceptor of electrons and other compounds behave as a donor of electrons. As for the electronegativity, we notice that the PCBM has a higher value of electronegativity (4.91 eV) than other compounds. This indicates that the PCBM is able to attract the electrons from other studied compounds. The compound PCBM has greater than other compounds. This indicates that PCBM finds it difficult to liberate the electrons, while the other compounds are the best candidates to liberate an electron to the PCBM. Finally, we notice that the compound PCBM is more electrophilic compound than other compounds which shows the lowest values of . Therefore, these compounds are good donors of electrons.
TABLE 3CALCULATED CHEMICAL HARDNESS (), CHEMICAL POTENTIAL (), ELECTRONEGATIVITY () AND
ELECTROPHILICITY POWER () VALUES OF THE STUDIED COMPOUNDS OBTAINED BY B3LYP/6-31G(D, P) LEVEL.
compounds (eV) (eV) (eV) (eV)
[(Cbz-Mth)-B]2-A 0.99 -4.28 4.28 9.25
[(Cbz-Mth)-B-T]2-A 0.88 -3.95 3.95 8.86 [(Cbz-Mth)-B-DT]2-A 0.85 -3.94 3.94 9.13 [(Cbz-Mth)-B-TT]2-A 0.87 -3.87 3.87 8.61 [(Cbz-Mth)-B-T-C=C (CN)2-T]2-A 0.69 -4.29 4.29 13.33
[(Cbz-Mth)-E-B-T-C=C(CN)2-T]2-A 0.60 -4.39 4.39 16.06
PCB M 1.28 -4.91 4.91 9.41
3.5. Optical Properties
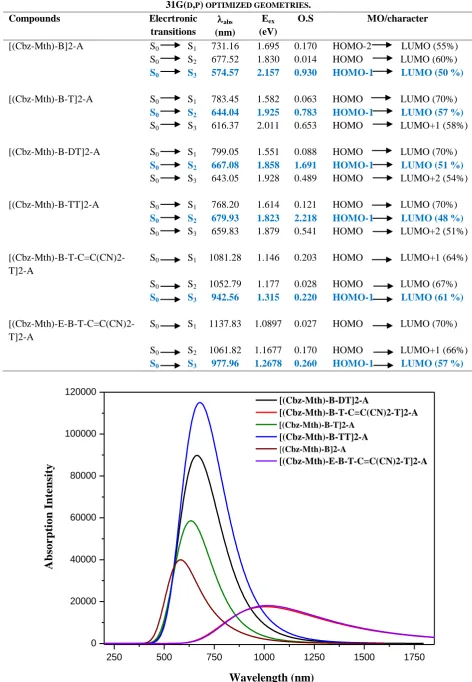
How the absorption of a new material matches with the solar spectrum is an important factor for the application as a photovoltaic material. Also, a good photovoltaic material should have broad and strong visible absorption characteristics. The optical properties of the six oligomers were investigated by UV-Vis absorption spectroscopy (Figure. 5) with TD/DFT method based on optimized ground-state geometries. We present in Table 4 the vertical excitation energy Eex (eV), calculated absorption λmax (nm), oscillator strength (O.S) and molecular orbital character (MO/character)
ISSN(E): 2277-128X, ISSN(P): 2277-6451, pp. 38-51
TABLE 4ABSORPTION SPECTRUM DATA OBTAINED BY TD/DFT METHODS FOR THE COMPOUNDS AT
B3LYP/6-31G(D,P) OPTIMIZED GEOMETRIES.
Compounds Elecrtronic
transitions
abs
(nm)
Eex
(eV)
O.S MO/character
[(Cbz-Mth)-B]2-A S0 S1 731.16 1.695 0.170 HOMO-2 LUMO (55%)
S0 S2 677.52 1.830 0.014 HOMO LUMO (60%)
S0 S3 574.57 2.157 0.930 HOMO-1 LUMO (50 %)
[(Cbz-Mth)-B-T]2-A S0 S1 783.45 1.582 0.063 HOMO LUMO (70%)
S0 S2 644.04 1.925 0.783 HOMO-1 LUMO (57 %)
S0 S3 616.37 2.011 0.653 HOMO LUMO+1 (58%)
[(Cbz-Mth)-B-DT]2-A S0 S1 799.05 1.551 0.088 HOMO LUMO (70%)
S0 S2 667.08 1.858 1.691 HOMO-1 LUMO (51 %)
S0 S3 643.05 1.928 0.489 HOMO LUMO+2 (54%)
[(Cbz-Mth)-B-TT]2-A S0 S1 768.20 1.614 0.121 HOMO LUMO (70%)
S0 S2 679.93 1.823 2.218 HOMO-1 LUMO (48 %)
S0 S3 659.83 1.879 0.541 HOMO LUMO+2 (51%)
[(Cbz-Mth)-B-T-C=C(CN)2-T]2-A
S0 S1 1081.28 1.146 0.203 HOMO LUMO+1 (64%)
S0 S2 1052.79 1.177 0.028 HOMO LUMO (67%)
S0 S3 942.56 1.315 0.220 HOMO-1 LUMO (61 %)
[(Cbz-Mth)-E-B-T-C=C(CN)2-T]2-A
S0 S1 1137.83 1.0897 0.027 HOMO LUMO (70%)
S0 S2 1061.82 1.1677 0.170 HOMO LUMO+1 (66%)
S0 S3 977.96 1.2678 0.260 HOMO-1 LUMO (57 %)
Fig 5. Simulated UV-Visible optical absorption spectra of the studied compounds calculated by TD/DFT/B3LYP/6-31G (d, p) level.
250 500 750 1000 1250 1500 1750
0 20000 40000 60000 80000 100000 120000
Wavelength (nm)
A
b
so
r
p
ti
o
n
I
n
te
n
si
ty
[(Cbz-Mth)-B-DT]2-A
[(Cbz-Mth)-B-T-C=C(CN)2-T]2-A
[(Cbz-Mth)-B-T]2-A
[(Cbz-Mth)-B-TT]2-A
[(Cbz-Mth)-B]2-A
ISSN(E): 2277-128X, ISSN(P): 2277-6451, pp. 38-51
© www.ijarcsse.com, All Rights Reserved Page | 47
3.6. Photovoltaic Properties
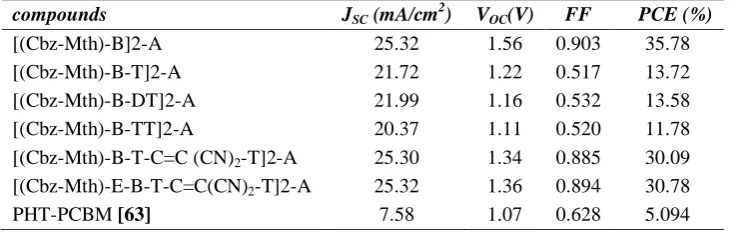
The photovoltaic properties of the six oligomers with PCBM acceptor were investigated by AMPS-1D stands for Analysis of Microelectronic and Photonic Structures – One Dimension. It was conceived as a very general and versatile computer simulation tool for the analysis of device physics and device design. It is a one-dimensional (1-D) device physics code which is applicable to any two terminal devices. It can be used for diode, sensor, photodiode, and photovoltaic device analysis [61, 62]. The current–voltage characteristics of the molecules are shown in Figure. 6, in dark and under white light illumination of 100 mW/cm2 intensity. The resulted photovoltaic parameters are summarized in Table 5. The [(Cbz-Mth)-E-B-T-C=C(CN)2-T]2-A showed a simulated value PCE (or Eff) (power conversion efficiency) of 30.78 % with a JSC of 25.32 mA/cm
2
, VOC of 1.36 V and FF of 0.894. The [(Cbz-Mth)-B-T-C=C(CN)2-T]2-A showed
a PCE of 30.09 % with JSC of 25.30 mA/cm2, VOC of 1.34 V and FF of 0.885. The slightly higher PCE and VOC for
[(Cbz-Mth)-E-B-T-C=C(CN)2-T]2-A based OSCs was observed, in comparison with that of [(Cbz-Mth)-B-T-C=C(CN)2-T]2-A and with the other studied compounds. The origin of slight enhancement in VOC for
T]2-A based device may be attributed to the deeper HOMO energy level of Mth)-E-B-T-C=C(CN)2-T]2-A (-4.99 eV) as compared to Mth)-B-T-C=C(CN)2-Mth)-E-B-T-C=C(CN)2-T]2-A (-4.97 eV), Mth)-B-TMth)-E-B-T-C=C(CN)2-T]2-A (-4.74), [(Cbz-Mth)-B-DT]2-A (-4.79) and [(Cbz-Mth)-B-TT]2-A (-4.84), due to the insertion of the spacer group ethynyl. Since VOC of
BHJ organic solar cells is proportional to the difference in the HOMO energy level of electron donor and LUMO of electron acceptor, the slightly larger energy offset between [(Cbz-Mth)-E-B-T-C=C(CN)2-T]2-A and PCBM leads to an enhanced VOC from the OSCs. In comparison, the results of the simulations, the power conversion efficiency PCE of the
studied molecules solar cells are noted to be higher than that of (Poly (3-hexylthiophene)-[6, 6]-phenyl C61-butyric acid methyl ester) P3HT-PCBM (5.094 %) [63], due to the insertion of the Carbazole, Thiophene and Benzothiadiazole units in the studied oligomers.
(a) [(Cbz-Mth)-E-B-T-C=C(CN)2-T]2-A, JSC = 25.32, Eff = 30.78%, FF = 0.89, VOC = 1.36 V
ISSN(E): 2277-128X, ISSN(P): 2277-6451, pp. 38-51
Fig 6. Simulated J-V curves (a) and (b) of the studied compounds calculated by AMPS-1D application.
TABLE 5PHOTOVOLTAIC CHARACTERISTICS OF THE COMPOUNDS SIMULATED BY AMPS-1D APPLICATION.
compounds JSC (mA/cm
2
) VOC(V) FF PCE (%)
[(Cbz-Mth)-B]2-A 25.32 1.56 0.903 35.78
[(Cbz-Mth)-B-T]2-A 21.72 1.22 0.517 13.72
[(Cbz-Mth)-B-DT]2-A 21.99 1.16 0.532 13.58 [(Cbz-Mth)-B-TT]2-A 20.37 1.11 0.520 11.78 [(Cbz-Mth)-B-T-C=C (CN)2-T]2-A 25.30 1.34 0.885 30.09
[(Cbz-Mth)-E-B-T-C=C(CN)2-T]2-A 25.32 1.36 0.894 30.78
PHT-PCBM [63] 7.58 1.07 0.628 5.094
IV. CONCLUSION
ISSN(E): 2277-128X, ISSN(P): 2277-6451, pp. 38-51
© www.ijarcsse.com, All Rights Reserved Page | 49
affects the electronic structure by donating charge carriers thereby lowering the energy band gap and raising the conjugation length. The results show that the designed compounds are possible as BHJ organic solar cell as their LUMO energy levels are much higher than that of PCBM, their calculated VOC ranges from 1.11 eV to 1.65 eV and their power
conversion efficiency PCE ranges from 11.78 % to 35.78 %. However, these values are sufficient for an efficient electron injection. Therefore, all the studied molecules can be used as organic solar cells. This theoretical approach that uses quantum calculation as DFT, TD-DFT and AMPS1D, presents a guiding tool to the synthesis process and helps to understand the structure–properties relationship of these new compounds.
ACKNOWLEDGMENTS
This work has been supported by the Volubilis project AI n°: MA/11/248 and by CNRST/CNRS cooperation (Project chimie 1009). We are grateful to the ―Association Marocaine des Chimistes Théoriciens‖ (AMCT) for its valuable help concerning the programs. I would like also to express my thanks to Zimeng Chen, Zhitao Wang and Zhaoqian Su who helped me with AMPS-1D.
REFERENCES
[1] A. Mishra, M. K. R Fischer, P. Bäuerle, Angew. Chem., Int. Ed. 48, 2474, 2009. [2] Y. Ooyama, Y. Harima, Eur. J. Org. Chem. 2903, 2009.
[3] Z. Ning, H. Tian, Chem. Commun. 5483, 2009.
[4] H. Qin, S.Wenger, M.Xu, F.Gao, X.Jing,; P.Wang, M. S. Zakeeruddin, M. Grätzel, J. Am. Chem. Soc. 130, 9202, 2008.
[5] H.-Y. Yang, Y.-S. Yen, Y.-C. Hsu, H. H. Chou, J. T. Lin, Org. Lett. 12, 16, 2010.
[6] H. Tian, X. Yang, R. Chen, R. Zhang, A. Hagfeldt, L. Sun, J. Phys. Chem. C 112, 11023, 2008. [7] Z. S. Wang, Y. Cui, Y. Dan-Oh, C. Kasada, A. Shinpo, K. Hara, J. Phys. Chem. C, 111, 7224, 2007. [8] B. O‘Regan, M. Grätzel, Nature 353, 737, 1991.
[9] A. Hagfeldt, M. Grätzel, Acc. Chem. Res. 33, 269, 2000. [10] M. Grätzel, Nature 414, 338, 2001.
[11] T. Dittrich, B. Neumann, H. Tributsch, J. Phys. Chem. C 111 2265, 2007.
[12] X.Z. Liu, Y.H. Luo, H. Li, Y.Z. Fan, Z.X. Yu, Y. Lin, L.Q. Chen, Q.B. Meng, Chem. Commun. 27, 2847, 2007. [13] J.B. Xia, F.Y. Li, H. Yang, X.H. Li, C.H. Huang, J. Mater. Sci. 42, 6412, 2007.
[14] M.X. Li, X.B. Zhou, H. Xia, H.X. Zhang, Q.J. Pan, T. Liu, H.G. Fu, C.C. Sun, Inorg. Chem. 47, 2312, 2008. [15] Z. Tian, M. Huang, B. Zhao, H. Huang, X. Feng, Y. Nie, P. Shen, S. Tan, Dyes Pigm. 87, 181, 2010.
[16] M. Matsui, A. Ito, M. Kotani, Y. Kubota, K. Funabiki, J. Jin, T. Yoshida, H. Minoura, H. Miura, Dyes Pigm. 80, 233, 2009.
[17] X. Ma, W. Wu, Q. Zhang, F. Guo, F. Meng, J. Hua, Dyes Pigm. 82, 353, 2009.
[18] W. Ma, C. Yang, X. Gong, K. Lee, A.J. Heeger, Thermally stable, efficient polymer solar cells with nanoscale control of theinterpenetrating network morphology, Adv. Funct. Mater. 15, 1617–1622, 2005.
[19] T. Kitagawa, Y. Murata, K. Komatsu, Fullerene reactivity—fullerene cations and open-cage fullerenes, in: M.M. Haley ,R.R. Tykwinski (Eds.), Carbon-Rich Compounds, Wiley-VCH Verlag GmbH& Co.KGaA,Weinheim, pp.383–420, 2006.
[20] Z. Ning, H. Tian, Triarylamine: a promising core unit for efficient photovoltaic materials. Chem Commun;:5483-95, 2009.
[21] DH. Lee, MJ. Lee, HM. Song, BJ. Song, KD. Seo, HK. Kim, et al. Organic dyes incorporating low-band-gap chromophores based on p-extended benzothiadiazole for dye-sensitized solar cells. Dyes Pigments,; 91:192-8, 2011.
[22] Z. Ning, Q. Zhang, W. Wu, H. Pei, B. Liu, H. Tian, Starburst triarylamine based dyes for efficient dye-sensitized solar cells. J Org Chem,; 73:3791-7, 2008.
[23] G. Li, KJ. Jiang, YF. Li, SL. Li, LM. Yang. Efficient structural modification of triphenylamine-based organic dyes for dye-sensitized solar cells. J Phys Chem C; 112:11591-9, 2008.
[24] ZS. Wang, FY. Li, CH. Huang, L. Wang, M. Wei, LP. Jin, et al. Photoelectric conversion properties of nanocrystalline TiO2 electrodes sensitized with hemicyanine derivatives. J Phys Chem B; 104:9676-82, 2000. [25] YS. Chen, C. Li, ZH. Zeng, WB. Wang, XS. Wang, BW. Zhang. Efficient electron injection due to a special
adsorbing group‘s combination of carboxyl and hydroxyl: dye-sensitized solar cells based on new hemicyanine dyes. J Mater Chem; 15:1654-61, 2005.
ISSN(E): 2277-128X, ISSN(P): 2277-6451, pp. 38-51
[27] T. Horiuchi, H. Miura, S. Uchida. Highly efficient metal-free organic dyes for dye-sensitized solar cells. J Photochem Photobiol A Chem; 164:29-32, 2004.
[28] T. Horiuchi, H. Miura, K. Sumioka, S. Uchida. High efficiency of dye-sensitized solar cells based on metal-free indoline dyes. J Am Chem Soc; 126: 12218-9, 2004.
[29] L. Schmidt-Mende, U. Bach, R. Humphry-Baker, T. Horiuchi, H. Miura, S. Ito, et al. Organic dye for highly efficient solid-state dye-sensitized solar cells. Adv Mater; 17:813-5, 2005.
[30] S. Ito, SM. Zakeeruddin, R. Humphry-Baker, P. Liska, R. Charvët, M. Grätzel, et al. High efficiency organic-dye-sensitized solar cells controlled by nanocrystalline- TiO2 electrode thickness. Adv Mater; 18:1202-5, 2006. [31] S. Eu, T. Katoh, T. Umeyama, Y. Matano, H. Imahori. Synthesis of sterically hindered phthalocyanines and
their applications to dye-sensitized solar cells. Dalton Trans;:5476-83, 2008.
[32] S.M. Bouzzine, A. Makayssi, M. Hamidi, M. Bouachrine, J. Mol. Struct. Theochem. 851, 254–262, 2008. [33] Y. He, J. You, L. Dou, C.-C. Chen, E. Richard, K.C. Cha, Y. Wu, G. Li, Y. Yang, Chem. Commun. 48, 7616–
7618, 2012.
[34] Z. El Malki, M. Bouachrine, M. Hamidi, F. Serein-Spirau, J. P. Lere-Porte, J. Marc Sotiropoulos, J. Mater. Environ. Sci. 7 (9), 3244, 2016.
[35] Z. El Malki, M. Bouachrine, L. Bejjit, M. Haddad, F. Serein-Spirau, J. Marc Sotiropoulos, International Journals of Advanced Research in Computer Science and Software Engineering ISSN: 2277-128X (Volume-7, Issue-6), 96-107, 2017.
[36] B.C. Thompson, L.G. Madrigal, M.R. Pinto, T.S. Kang, K.S. Schanze, J.R. Reynolds, J. Polym. Sci. Pol. Chem. 43, 1417–1431, 2005.
[37] Y. Lin, P. Cheng, Y. Li, X. Zhan, Chem. Commun. 48, 4773–4775, 2012.
[38] W. Zhu, Y. Wu, S. Wang, W. Li, Z. Wang, H. Tian, Organic DeAepeA solar cell sensitizers with improved stability and spectral response. Adv Funct Mater; 21:756-63, 2010
[39] G. Zotti, G. Schiavon, S. Zecchin, J. F. Morin, M. Leclerc, Macromolecules 35, 2122, 2002. [40] F. Garnier, G. Horowitz, X. Peng, D. Fichou, Adv.Mater 2, 562, 1990.
[41] R. E.Gill, G. G.Malliaras, J. Wildeman, G. Hadziioannou, Adv. Mater 6, 132, 1994.
[42] Z. El Malki, K. Hasnaoui, S.M. Bouzzine, L. Bejjit, M. Haddad, M. Hamidi, and M. Bouachrine, SRX Chemistry • Volume 2010 • Article ID 346843 • doi:10.3814/2010/346843, 1-8, 2010.
[43] S. Leroy-Lhez, M. Allain, J. Oberle, F. Fages, New J. Chem. 31, 1013–1021, 2007.
[44] V. Roy, Y.-G. Zhi, Z.-X. Xu, S.-C. Yu, P.W.H. Chan, C.-M. Che, Adv. Mater.17, 1258–1261, 2005.
[45] A. Marrocchi, F. Silvestri, M. Seri, A. Facchetti, A. Taticchi, T.J. Marks, Chem. Commun. 11, 1380–1382, 2009.
[46] N.S. Baek, S.K. Hau, H.-L. Yip, O. Acton, K.-S. Chen, A.K.-Y. Jen, Chem. Mater. 20, 5734–5736, 2008. [47] S. Punsay, Thesis submitted in ‗‗Synthesis and characterization of carbazole derivatives for optoelectronic
devices‘‘ for the degree of M.Sc. in Chemistry, Faculty of Science, Ubon Ratchathani University, 2011.
[48] S.H. Vosko, L. Wilk, M. Nusair, Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis, Can. J. Phys. 58, 1200–1211, 1980.
[49] A.D. Becke, Density-functional thermochemistry. 3. The role of exact exchange, J. Chem. Phys. 98, 5648–5652, 1993.
[50] A.D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A 38, 3098–3100, 1988.
[51] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H.P. Hratchian, A.F. Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J.A. Montgomery, J.J.E. Peralta, F. Ogliaro, M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J.M. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, R.L. Martin, K. Morokuma, V.G. Zakrzewski, G.A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich, A.D. Daniels, O. Farkas, J.B. Foresman, J.V. Ortiz, J. Cioslowski, D.J. Fox, Gaussian 09, Gaussian Inc., Wallingford, CT, 2009.
[52] X.-C. Huang, Li, Y.-H. Wang, Z.-H. Kang, R. Lu, E.-L. Miao, F. Wang, G.-W. Wang, H.-Z. Zhang, Opt. Mater. 35, 1373–1377, 2013.
ISSN(E): 2277-128X, ISSN(P): 2277-6451, pp. 38-51
© www.ijarcsse.com, All Rights Reserved Page | 51
[54] W.J. Potscavage, J.R.A. Sharma, B. Kippelen, Acc. Chem. Res. 42, 1758– 1767, 2009. [55] G.D. Sharma, P. BalaRaju, M.S. Roy, Sol. Energ. Mat. Sol. Cells 92, 261– 272. 2008. [56] Y.-J. Cheng, S.-H. Yang, C.-S. Hsu, Chem. Rev. 109, 5868–5923, 2009.
[57] A. Mabrouk, A. Azazi, K. Alimi, Polym. Eng. Sci. 53, 1040–1052, 2013.
[58] S. Bertho, I. Haeldermans, A. Swinnen, W. Moons, T. Martens, L. Lutsen, A. Bonfiglio, Influence of thermal ageing on the stability of polymer bulk heterojunction solar cells, Sol. Energy Mater. Sol. cells 91, (5) 385-389, 2007.
[59] N. Hergué, C. Mallet, G. Savitha, M. Allain, P. Frère and J. Roncali, Organic Letters, Vol. 13, No. 7 1762-1765, 2011.
[60] R.G. Pearson, Absolute electronegativity and hardness correlated with molecular orbital theory, Proc. Natl. Acad. Sci. 83, 8440–8841, 1986.
[61] AMPS-1D. Pennsylvania State University, http://www.empl.psu.edu/amps, 1997. [62] en.wikipedia.org/wiki/P3HT, accessed date 04/05/2012.
![Fig 1. Chemical structure of polymers under study (a) [(Cbz-Mth)-B]2-A, (b) [(Cbz-Mth)-B-T]2-A, (c) [(Cbz- Mth)-B-DT]2-A, (d) [(Cbz-Mth)-B-TT]2-A, (e) [(Cbz-Mth)-B-T-C=C(CN)2-T]2-A and (f) [(Cbz-Mth)-E-B-T-C=C(CN)2-T]2-A](https://thumb-us.123doks.com/thumbv2/123dok_us/7803529.2084614/2.595.129.478.327.746/fig-chemical-structure-polymers-study-mth-mth-cbz.webp)
![Table 5. The [(Cbz-Mth)-E-B-T-C=C(CN)2-T]2-A showed a simulated value PCE (or Eff) (power conversion efficiency) of 30.78 % with a JSC of 25.32 mA/cm2, VOC of 1.36 V and FF of 0.894](https://thumb-us.123doks.com/thumbv2/123dok_us/7803529.2084614/10.595.99.501.336.770/table-showed-simulated-value-power-conversion-efficiency-jsc.webp)