R E V I E W
Open Access
Targeting T cell malignancies using
CAR-based immunotherapy: challenges and
potential solutions
Lauren C. Fleischer
1,2, H. Trent Spencer
1,2and Sunil S. Raikar
2*Abstract
Chimeric antigen receptor (CAR) T cell therapy has been successful in treating B cell malignancies in clinical trials; however, fewer studies have evaluated CAR T cell therapy for the treatment of T cell malignancies. There are many challenges in translating this therapy for T cell disease, including fratricide, T cell aplasia, and product
contamination. To the best of our knowledge, no tumor-specific antigen has been identified with universal expression on cancerous T cells, hindering CAR T cell therapy for these malignancies. Numerous approaches have been assessed to address each of these challenges, such as (i) disrupting target antigen expression on CAR-modified T cells, (ii) targeting antigens with limited expression on T cells, and (iii) using third party donor cells that are either non-alloreactive or have been genome edited at the T cell receptorαconstant (TRAC) locus. In this review, we discuss CAR approaches that have been explored both in preclinical and clinical studies targeting T cell antigens, as well as examine other potential strategies that can be used to successfully translate this therapy for T cell disease.
Keywords:CAR, Immunotherapy, T-ALL, T cell lymphoma
Introduction
T cell malignancies encompass a heterogeneous group of diseases, each reflecting a clonal evolution of dysfunc-tional T cells at various stages of development. T cell acute lymphoblastic leukemia (T-ALL) accounts for 15% and 25% of childhood and adult ALL cases respectively, and is the most common form of T cell cancer seen in children [1, 2]. T-lymphoblastic lymphoma (T-LLy) is a non-Hodgkin lymphoma with similar biology to T-ALL. Adult T cell leukemia/lymphoma (ATLL) is an ex-tremely aggressive form of blood cancer driven by the human T cell lymphocytic virus type 1 (HTLV1) [3–5]. Other rare forms of T cell leukemia include T cell large
granular lymphocytic leukemia (T-LGL) and
T-prolymphocytic leukemia (T-PLL) [6]. T cell lymphomas
are broadly divided into two categories, cutaneous T cell lymphoma (CTCL) and peripheral T cell lymphoma
(PTCL) [7]. Mycosis fungoides (MF) and Sezary
syn-drome (SS) represent the two most common subtypes of
CTCL, accounting for the majority of cases [8]. PTCL
can be classified into several different subtypes, among which include anaplastic large cell lymphoma (ALCL), angioimmunoblastic T cell lymphoma (AITL), extrano-dal natural killer (NK)-T cell lymphoma (ENKTL), enteropathy-associated T cell lymphoma (EATL), hepa-tosplenic T cell lymphoma (HSTCL), and PTCL-not otherwise specified (PTCL-NOS) which is the most common of the group [9,10].
The overall prognosis for T cell malignancies varies depending on the type of disease, but in general is much poorer when compared to B cell malignancies. While the survival in T-ALL/LLy has significantly improved with the intensification of chemotherapy, there still re-main very limited options for patients with
relapsed/re-fractory disease [11–13]. ATLL remains a very
challenging disease to treat, with a median survival of less than 12 months for the acute form of this disease [3–5]. Advanced stage CTCL has a median overall sur-vival of 5 years [14,15], whereas outcomes of PTCL vary
© The Author(s). 2019Open AccessThis article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. * Correspondence:[email protected]
2Cell and Gene Therapy Program, Department of Pediatrics, Aflac Cancer and
Blood Disorders Center, Children’s Healthcare of Atlanta and Emory University School of Medicine, Atlanta, GA, USA
depending upon the subtype, with ENKTL, EATL, and HSTCL having the poorest prognosis [9, 10]. While im-munotherapy has revolutionized the treatment landscape of various cancers with the use of monoclonal anti-bodies, checkpoint inhibitors, bispecific T cell engagers, and chimeric antigen receptor (CAR) T cell therapy, only limited responses have been seen in T cell disease [15]. Some promising results have been seen with use of brentuximab vedotin, a CD30-directed immunotoxin, in
CD30-positive PTCL and CTCL [16, 17] and the use of
pembroluzimab, a programmed cell death receptor 1 (PD-1) inhibitor, in the treatment of ENKTL [18]; how-ever, these positive results have been limited to very specific subsets of T cell disease. One form of immuno-therapy that has not yet been successfully translated to T cell malignancies is that of chimeric antigen receptor (CAR)-based immunotherapy. CAR T cell therapy has been extremely successful in relapsed/refractory B cell malignancies as evidenced by the recent Food and Drug Administration (FDA) approval of two CAR T cell thera-peutics for this disease [19–23]. However, implementing this technology to treat T cell malignancies has been dif-ficult, primarily due to the lack of a tumor-specific sur-face antigen in cancerous T cells. In this review, we will discuss the challenges involved in translating this novel technology to T cell disease, review all the preclinical and clinical progress made in adapting this therapy for this challenging disease, and examine potential solutions for the future development of this innovative therapy.
CAR T cell therapy
Genetic engineering of primary T cells was first pre-sented in the late 1980s [24]. Since then, chimeric anti-gen receptor T cells have emerged as a promising technique for the treatment of relapsed/refractory malig-nancies. CAR therapy brings together numerous fields including immunology, tumor biology, genetic engineer-ing, synthetic biology, and pharmacology. CARs are comprised of the intracellular signaling domain from the natural T cell receptor (TCR), CD3ζ, linked to a single-chain variable fragment (scFv) which serves as the anti-gen recognition domain. The scFv sequence is derived from a monoclonal antibody by combining the variable heavy (VH) and light (VL) domains using a small peptide
linker. Commonly used CARs also include one or two costimulatory domains, such as CD28, 4-1BB, ICOS, and/or OX40. Although the kinetics have yet to be fully elucidated, it is essential that CAR T cells have mecha-nisms of trafficking to the tumor site where they can recognize their cognate antigen. This results in CAR T cell activation and expansion, and ultimately cytolytic activity against cells expressing the target antigen. CAR-based ligand recognition is advantageous compared to TCR-based ligand recognition because CAR-targeting is
not restricted by major histocompatibility complex (MHC) interactions. Therefore, CARs can recognize cell surface proteins that have not been processed and pre-sented by antigen presenting cells (APCs). Importantly, the interactions between scFvs and ligands have much higher affinity and avidity compared to that of TCR-ligand interactions [25]. Furthermore, the immune syn-apse formed from the interaction between a CAR and its ligand likely results in a much greater functional avidity than is observed using a targeted antibody approach with the same antibody (25).
CARs targeting the B cell antigen CD19 have been studied extensively for the treatment of B cell malignan-cies. In 2017, the FDA approved the first CAR T cell therapy, Kymriah, a CD19-directed CAR therapy for the treatment of relapsed/refractory B cell acute lympho-blastic leukemia (B-ALL) and in 2018, Yescarta was ap-proved to treat relapsed diffuse large B cell lymphoma (DLBCL). These therapies, including others in clinical trials, have been widely successful in eliminating malig-nant cells and re-inducing remission in patients who were otherwise treatment-refractory [19–21, 26, 27]. Pa-tients receiving CAR therapy undergo leukapheresis resulting in the collection of T cells, which are subse-quently modified using a lentiviral or retroviral vector to express the CAR. These cells are expanded ex vivo while the patient undergoes lymphodepletion, a process in-volving chemotherapeutic agents. Finally, the CAR T cells are re-infused into the patient [28]. Lymphodeple-tion prior to re-infusion of the autologous T cells has been shown to augment both CAR T cell proliferation as well as persistence [29–31]. The administered dose of CAR T cells and the pre-existing tumor burden do not appear to be the sole determinants of the degree of T cell expansion, engraftment, and overall response. Other factors may be involved, such as the density of cognate
antigen expression on the cancer cells [32]. However,
the optimal degree of persistence of CAR T cells re-quired to prevent leukemic relapse has not been deter-mined [25,33].
One of the mechanisms of relapse post-CD19
CAR T cell therapy is due to surface antigen escape with relapsed leukemia cells being CD19-negative. The mech-anism may be due to the expansion of a small subset of CD19-negative cancer cells or alternatively, the cells may downregulate CD19 from the cell surface in order to evade detection by CAR T cells, rendering them resist-ant [19, 21, 34–37]. Additionally, it was recently shown that a phenomenon referred to as trogocytosis is a mechanism of antigen escape whereby the antigen is
transferred to the CAR T cell [38]. It has also been
patient [39]. Transduction of the leukemic cell resulted in masking of the target antigen through interactions be-tween the CAR and the cognate antigen on the same cell. Clonal expansion of this population resulted in re-sistance to CAR therapy. This report emphasized the im-portance of strict and perfect isolation of normal, healthy T cells for modification with the CAR construct. As we discuss below, this is particularly challenging in T cell leukemia patients who are more likely to have circu-lating cancerous T cells, and therefore have a higher probability of these cells being inadvertently isolated, transduced, and re-infused.
Of note, there are severe toxicities that have been as-sociated with CAR therapy. Cytokine release syndrome (CRS) is a systemic inflammatory response directly resulting from robust T cell activation following infu-sion. IL-6 is one pro-inflammatory cytokine that is se-creted at high levels during CRS. During a particularly severe CRS condition, tocilizumab, an IL-6R antagonist monoclonal antibody, was used to rapidly and effectively reverse the symptoms of a pediatric patient [27]. Toci-lizumab has since been FDA approved for treatment of
CAR T cell-induced life-threatening CRS [40].
Neuro-logical toxicities have been reported following CAR T cell infusion as well; however, preventative approaches
remain elusive [36, 41–44]. Compared to CRS and
neurotoxicity, a much more manageable consequence of CAR T cell therapy targeting B cell malignancies is the resulting B cell aplasia. This is a potentially lifelong out-come due to memory cell formation against a B cell anti-gen; but currently is managed by periodic infusions of intravenous immunoglobulins. Unfortunately, this is an extremely problematic outcome for T cell malignancies, as persistent T cell aplasia would be life threatening. There are currently > 200 clinical trials using CAR T cells registered at clinicaltrials.gov being carried out in the USA. However, the majority of these trials are enrol-ling patients with B cell malignancies. Advances are be-ing made to expand CAR T cell therapy to the treatment of other cancers, and to minimize toxicities associated with treatment while reducing difficulty and cost of production.
Translating CAR T cell therapy for treatment of T cell malignancies
Harnessing and redirecting the cytotoxicity of T cells to malignant B cells has been established, but reprogram-ming T cells to kill malignant T cells, while sparing nor-mal T cells, is much more complex and challenging. This requires aberrant expression of an antigen on ma-lignant T cells that is absent or expressed at very low levels on normal T cells. CAR therapy requires isolation of healthy T cells from malignant T cells, a complicated procedure that can result in product contamination and
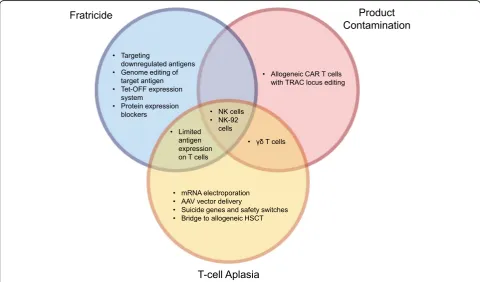
subsequent CAR-modification of tumor cells. Addition-ally, expression of the targeted antigen on CAR T cells results in fratricide and limited expansion of the CAR T cells. Furthermore, targeting of an antigen regularly expressed on normal T cells would result in T cell apla-sia, leading to profound immunosuppression, likely to be associated with high rates of morbidity and mortality (Fig.1).
Various approaches have been used to overcome these challenges, including CRISPR-Cas9 genome editing to remove the antigen from the CAR T cells [45–47], Tet-OFF expression system to limit fratricide during ex vivo expansion [48], protein expression blocker (PEBL) to re-tain the antigen in the ER/Golgi to prevent cell surface expression [49,50], or using CAR-modified natural killer cells instead of T cells [47,51–54]. Additionally, to date, four targets have been investigated as targets for CAR T cell therapy for the treatment of T cell malignancies with limited to no expression in the normal population of T
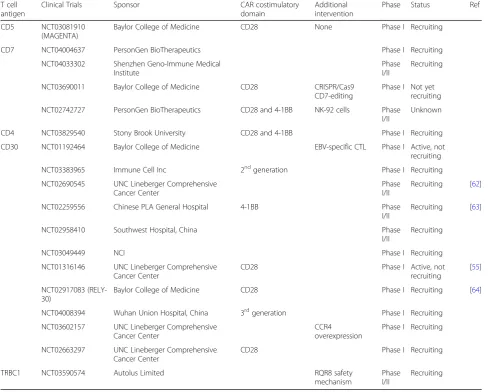
cells, CD30, CD37, TRBC1, and CD1a [55–58]. Table1
provides a summary of potential solutions to the three main challenges seen in adapting CAR technology for T cell malignancies—fratricide, T cell aplasia, and product contamination. A list of all current CAR-based clinical trials targeting T cell disease is presented in Table 2. Below, we review all preclinical and clinical CAR studies targeting T cell malignancies categorized according to the target antigen of interest.
CD5
CD5 expression is limited to normal T cells and a small subpopulation of B cells, called B-1a cells [65–69]. CD5 acts as a negative regulator of TCR signaling and has a role in protecting against autoimmunity [70, 71]. CD5 is highly expressed on many T cell malignancies, particu-larly T-ALL and PTCLs, rendering it a good target for CAR T cell therapy [72–74]. Since CD5 expression on T cells is approximately ten times that on B cells [75], a low-affinity, high-avidity CAR targeting CD5 may steer clear of CD5-positive B cells while selectively killing T cells [76,77]. Furthermore, CD8+tumor-infiltrating lym-phocytes (TILs) express lower levels of CD5 compared to that of peripheral blood T cells, and one study showed downregulation of CD5 improves the ability of T cells to lyse malignant cells [78]. CD5 was previously targeted as a tumor antigen in clinical trials using immunotoxin-conjugated CD5 monoclonal antibodies, with responses seen in patients with cutaneous T cell lymphoma and T-ALL [79,80].
Table 1Strategies to overcome challenges in translating CAR therapy to treat T cell malignancies
Challenge Strategy Reference
Fratricide Targeting downregulated antigens (e.g., CD5) [59]
Genome editing of target antigen [45–47]
Targeting antigens with limited expression on T cells (e.g., CD30, CD37, TRBC1, CD1a)
[55–58]
Tet-OFF expression system [48]
Protein expression blockers (PEBLs) [49]
Using NK cells or NK-92 cells [47,51–54,60]
T cell aplasia Targeting antigens with limited expression on T cells (e.g., CD30, CD37, TRBC1, CD1a)
[55–58]
mRNA electroporation
Adeno-associated viral (AAV) vector delivery
Using NK cells or NK-92 cells [47,51–54,60]
UsingγδT cells
Suicide genes and safety switches
Bridge to allogeneic hematopoietic stem cell transplant (HSCT)
Product contamination Allogeneic CAR T cells with TRAC locus editing [46,61]
Using NK cells or NK-92 cells [47,51–54,60]
UsingγδT cells Fratricide
Anti-tumor cytolytic activity
Product Contamination
T-cell aplasia
in vitro cytotoxicity against two T-ALL cell lines and primary T-ALL cells and delayed leukemia progression in two different CD5-positive T-ALL models [59]. Based on these results, CD5-CAR T cells with a CD28 costi-mulatory domain are being tested in patients with relapsed or refractory T cell disease (MAGENTA trial, NCT03081910). Our group used CRISPR-Cas9 to knockout CD5 expression in primary T cells prior to transduction with the CD5-CAR. We showed that gene editing of CD5 in effector CAR T cells increased CAR surface expression and decreased self-activation [47]. The increased CAR surface expression is predicted to enhance CAR T cell anti-tumor efficacy. We also showed antagonism of vasoactive intestinal peptide (VIP) signaling in conjunction with inhibition of the
PI3Kδ pathway increased expansion of
CD5-CAR-modified T cells as well as their cytotoxicity against CD5-specific tumor cell lines. This combination of com-pounds was also demonstrated to prolong in vivo
persistence of treated T cells in NOD scid IL2Rγ-chain knockout (NSG) mice [81].
Interestingly, use of 4-1BB as the costimulatory do-main in a CD5-CAR resulted in a significant fratricidal
effect [48]. It was shown that tumor necrosis factor
(TNF) receptor-associated factor (TRAF) signaling from the 4-1BB endodomain upregulated the intercellular ad-hesion molecule 1 (ICAM1), which subsequently stabi-lized the fratricidal immunological synapse between CD5-CAR T cells containing the 4-1BB costimulatory domain. To limit and control the effects of fratricide, a Tet-OFF expression system was used, which allowed for controlled transgene expression using the small mol-ecule inhibitor, doxycycline. In the presence of doxycyc-line, CD5-41BB-CAR T cells expanded ex vivo without evidence of fratricide, while maintaining a more naïve genotype. Doxycycline was removed from the culture prior to injecting the CD5-41BB-CAR T cells into mice, resulting in CD5-CAR expression and improved survival Table 2Clinical CAR trials targeting T cell malignancies
T cell antigen
Clinical Trials Sponsor CAR costimulatory
domain
Additional intervention
Phase Status Ref
CD5 NCT03081910 (MAGENTA)
Baylor College of Medicine CD28 None Phase I Recruiting
CD7 NCT04004637 PersonGen BioTherapeutics Phase I Recruiting
NCT04033302 Shenzhen Geno-Immune Medical Institute
Phase I/II
Recruiting
NCT03690011 Baylor College of Medicine CD28 CRISPR/Cas9
CD7-editing
Phase I Not yet recruiting
NCT02742727 PersonGen BioTherapeutics CD28 and 4-1BB NK-92 cells Phase I/II
Unknown
CD4 NCT03829540 Stony Brook University CD28 and 4-1BB Phase I Recruiting
CD30 NCT01192464 Baylor College of Medicine EBV-specific CTL Phase I Active, not
recruiting
NCT03383965 Immune Cell Inc 2ndgeneration Phase I Recruiting
NCT02690545 UNC Lineberger Comprehensive Cancer Center
Phase I/II
Recruiting [62]
NCT02259556 Chinese PLA General Hospital 4-1BB Phase
I/II
Recruiting [63]
NCT02958410 Southwest Hospital, China Phase
I/II
Recruiting
NCT03049449 NCI Phase I Recruiting
NCT01316146 UNC Lineberger Comprehensive Cancer Center
CD28 Phase I Active, not
recruiting
[55]
NCT02917083 (RELY-30)
Baylor College of Medicine CD28 Phase I Recruiting [64]
NCT04008394 Wuhan Union Hospital, China 3rdgeneration Phase I Recruiting
NCT03602157 UNC Lineberger Comprehensive Cancer Center
CCR4 overexpression
Phase I Recruiting
NCT02663297 UNC Lineberger Comprehensive Cancer Center
CD28 Phase I Recruiting
TRBC1 NCT03590574 Autolus Limited RQR8 safety
mechanism
Phase I/II
outcomes in a T-ALL mouse model. Furthermore, there was a survival advantage in mice treated with Tet-OFF CD5-41BB-CAR T cells compared to survival of mice treated with CD5-CD28-CAR T cells without the Tet-OFF expression system [48].
Alternatively, we expressed the CD5-CAR in NK-92 cells, an interleukin-2 (IL-2) dependent natural killer cell line, which are inherently CD5-negative. Our data demon-strates that CD5-CAR-modified NK-92 cells have in-creased cytotoxicity against T cell leukemia cell lines compared to the cytotoxicity of naïve NK-92 cells [47,51], and there is a significant improvement in survival of T-ALL xenograft mouse models compared to survival of mice treated with naïve NK-92 cells [47]. This data con-firms previously published data illustrating significantly improved survival and enhanced tumor reduction in irra-diated T-ALL mouse models treated with CD5-CAR-modified NK-92 cells compared to that of mice treated with control NK-92 cells [53]. Recently, another group tested CD5-CAR-modified 92 cells, using a NK-specific costimulatory domain 2B4 in their CAR con-structs [82]. Interestingly, the CD5-2B4-CAR NK-92 cells displayed superiority to CD5-41BB-CAR NK-92 cells, in both in vitro and in vivo experiments [82].
CD7
CD7 is a transmembrane glycoprotein with expression on
T cells and NK cells [83]. The majority of T-ALLs are
CD7-positive, despite some populations lacking expres-sion of other common markers, such as the TCR [74,84]. Additionally, early T cell precursor acute lymphoblastic leukemia (ETP-ALL), a high-risk subset of T-ALL, highly express CD7 [84–86]. Two clinical trials have been initi-ated in China studying CD7-CAR-modified T cells for the treatment of CD7-positive malignancies (NCT04033302 and NCT04004637). However, preclinical studies showed significantly reduced expansion of CD7-CAR T cells com-pared to control T cells, as a result of fratricide [45, 49]. Fratricide appears to be observed to a greater extent in
CD7-CAR T cells compared to CD5-CAR T cells [45]. It
is hypothesized that this is due to a more incomplete in-ternalization mechanism of CD7 from the cell surface fol-lowing ligation of the antigen with an anti-CD7 scFv. CRISPR-Cas9 editing of CD7 from the cell surface of T cells prior to CAR expression demonstrated a superior method of developing CD7-CAR T cells. These cells ex-hibited limited fratricide, expanded in vitro, and showed no evidence of impaired cytotoxicity in vitro nor in vivo. Investigations in a T-ALL mouse xenograft model re-vealed a statistically significant prolonged survival of CD7-edited CD7-CAR-treated mice compared to survival of control mice [45]. Based on these results, a phase I clinical trial has been initiated testing CD7-CD28-CAR T cells in T-ALL patients (NCT03690011). Additionally, a UCART7
was generated using CRISPR-Cas9 genome editing to
dis-rupt the CD7 and TCRαconstant (TRAC) loci. This study
demonstrated that NSG mice engrafted with primary T-ALL blasts and treated with UCART7 donor cells exhib-ited tumor clearance from the peripheral blood, and, did not develop graft versus host disease (GvHD) or other se-vere side effects [46].
A new technique using protein expression blockers (PEBLs) has been established as an alternative to gen-ome editing. This strategy couples an scFv with a reten-tion peptide to maintain the protein of interest in the ER/Golgi preventing cell surface expression of the anti-gen. PEBL-CD7-CAR T cells exhibited superior cytotox-icity against primary T-ALL cells in vitro compared to non-PEBL CD7-CAR T cells. Using a patient-derived xenograft (PDX) model of ETP-ALL, upon detection of leukemic cell expansion in peripheral blood, PEBL-CD7-CAR T cells were injected. PEBL-CD7-PEBL-CD7-CAR T cell-treated mice had a significant survival advantage over control mice. However, CD7-positive relapse did occur in all PEBL-CD7-CAR T cell-treated mice [49].
Despite CD7 expression on NK-92MI cells (IL-2 produ-cing NK-92 cells), they have been used for CD7-CAR ther-apy demonstrating only a small percentage of cells are CD7-positive, and upon CD7-CAR expression, fewer than
1% CD7-positive NK-92MI cells remain [60]. Two
CD7-CAR constructs, a monovalent and bivalent construct, were generated using a humanized CD7 nanobody sequence that had been previously developed in the laboratory. Both CAR constructs demonstrated enhanced CD7-specific cytotox-icity against T-ALL cell lines and primary patient cells ex vivo when expressed in NK-92MI cells. The bivalent
CD7-CAR-modified-NK-92MI cells exhibited slightly
greater cytotoxicity compared to that of the monovalent CAR-modified cells, and significantly inhibited disease pro-gression in a T-ALL PDX model when compared to naïve unmodified NK-MI cells.
CD4
administered 24 h post-CAR T cell injection. A > 95% depletion of CD4-CAR-modified T cells was observed within 6 h following injection signifying the use of alem-tuzumab as a safety mechanism for CAR T cell therapy [87]. Additionally, a phase I clinical trial to assess the safety and feasibility of CD4-CAR T cell infusions in pa-tients with relapsed/refractory T cell lymphoma and T cell leukemia has been initiated (NCT03829540).
However, expression of CD4 on T cells can complicate CD4-CAR T cell therapy as previously described. NK-92 cells are inherently CD4-negative, and therefore the use of NK-92 cells as opposed to T cells reduces risk of frat-ricide and avoids the need for further modifications. Additionally, it abrogates the risk of aplasia of CD4-positive cells that can occur with long-term engraftment of CAR T cells. Anti-CD4-CAR NK-92 cells have shown in vitro success eliminating PTCL cell lines and both adult and pediatric primary cells. Using a xenograft model in NSG mice, CD4-CAR NK-92 cell-treated mice demonstrate significantly prolonged survival compared to control-modified NK-92 cell-treated mice [54].
CD37
CD37 is a member of the tetraspanin superfamily with ex-pression limited to lymphoid tissues, particularly B cells [88,89]. CD37 expression in cancer cells is typically char-acteristic of B cell malignancies; however, its expression
can be found in some cases CTCL and PTCL [90, 91].
Since CD37 is not expressed in T cells, there is no evi-dence of fratricide occurring in anti-CD37 CAR T cells. However, in the presence of CD37-positive PTCL cell lines, CD37-CAR T cells exhibit increased activation and degranulation as well as specific cytolytic activity in vitro [56]. The restricted expression of CD37 makes it a safer target for CAR T cell therapy, given there would be no concern of T cell aplasia. Additionally, CD37 is not expressed in NK cells, providing an opportunity to utilize NK cells as effector cells in place of T cells. The versatility of CD37-CARs to treat B cell and T cell lymphomas sug-gests that this may be an important target for further
in-vestigations. While CD37 is predominantly being
examined for dual targeting for B cell malignancies, the target has potential for CAR therapy against T cell malignancies.
CD30
CD30, a member of the tumor necrosis factor receptor (TNFR) superfamily, promotes T cell proliferation and cytokine production following TCR stimulation, while also having an opposing role in promoting apoptosis [92]. Expression is limited to a subset of activated lym-phocytes found around the follicular regions of lymphoid tissues [93–95]. While CD30 is well known for its strong expression in virtually all classical Hodgkin lymphoma,
expression of CD30 can also be found on a subset of PTCLs, including ALCL [92–94,96]. One study demon-strated that CD30 expression is upregulated during chemotherapy regimens in ALL patients. Of 34 ALL patients, approximately 38% had CD30-positive
T-ALL [96]. Therefore, some T-ALL patients who relapse
following chemotherapy may still respond to CD30-directed CAR therapy.
Preclinical studies have previously demonstrated
CD30-CAR T cell capacity for lysing tumor cells [97,98] and numerous clinical investigations into CD30-CAR T cell therapy have been launched with encouraging re-sults. Eleven phase I/II trials treating patients with
CD30-positive malignancies are currently active
(NCT01316146 [55], NCT01192464, NCT03049449,
NCT02690545 [62], NCT02958410, NCT02663297,
NCT03383965, NCT02917083 [64], NCT04008394,
NCT02259556 [63], and NCT03602157). To date, no
toxicities related to CAR T cell infusion nor impaired immunity against common viruses has been reported from these trials. However, one trial reported that the in vivo CAR T cell expansion and persistence was re-duced following subsequent infusions compared to those following initial doses [55]. The decreased persistence of the CAR T cells may have prevented the development of severe adverse events such as CRS and neurotoxicity that are commonly observed following CAR T cell infusion. Of the two ALCL patients in this trial, one patient was non-responsive to the therapy, while the other entered complete remission lasting 9 months [55]. Results from another phase I trial in China for patients with relapsed/ refractory CD30-positive lymphomas (NCT02259556) corroborate the limited toxicity and anti-tumor activity of CD30-CAR T cells [63].
TRBC1
T cells express the αβ TCR; the β-chain can either be
encoded by the T cell receptor beta constant 1 (TRBC1) gene or TRBC2 gene [99,100]. Therefore, expression of TRBC1 and TRBC2 is mutually exclusive. Additionally, CD4- and CD8-positive T cell populations express both subsets and CD8-positive T cell populations specific for common viruses also contain both TRBC1 and TRBC2 cells [58]. However, as malignant T cells develop from a single cell, the entire population of cancerous cells will be either TRBC1- or TRBC2-positive. Numerous T cell malignancy cell lines and primary samples have been an-alyzed by flow cytometry to validate the homogeneity of
β-chain expression in a malignant cell population [58].
Many cancer cells downregulate theαβTCR; however, it
is expressed on > 95% of PTCLs [101] and > 30% of T-ALLs [102].
with TRBC1, but not against non-transduced cells or cells transduced with TRBC2, even in a mixed popula-tion. Furthermore, in primary samples from patients with T cell malignancies, the anti-TRBC1 CAR T cells preserved a significant fraction of healthy T cells (TRBC2 cells), thereby circumventing a limitation of CAR T cell therapy for the treatment of T cell
malignan-cies [58]. In an NSG mouse model using
TRBC1-positive Jurkat T cells to establish cancer, mice treated with the anti-TRBC1 CAR T cells exhibited reduced tumor burden and elongated survival. In additional pre-clinical studies, NSG mice were injected with both TRBC1 and TRBC2 cancer cells, and then treated with either naïve T cells or anti-TRBC1 CAR T cells. TRBC1-positive Jurkat T cells could not be detected in mice treated with anti-TRBC1 CAR T cells; however, TRBC2-positive cells were identified. This is in contrast to mice treated with naïve T cells, whose bone marrow con-firmed the presence of both TRBC1-positive and
TRBC2-positive cells [58]. Thus, targeting
TRBC1-positive malignant cells offers a unique approach to avoiding T cell aplasia, a consequence of many proposed CAR T cell therapies for the treatment of T cell malignancies.
CD3
CD3 is a pan T cell marker comprised of four distinct polypeptide chains, epsilon, gamma, delta, and zeta, which form pairs of dimers, transmitting T cell activa-tion signals. As CD3 is exclusively expressed on T cells, it has been a popular target in preclinical CAR T cell therapies for the treatment of T cell malignancies. As expected, due to fratricidal issues, manufacturing of anti-CD3 CAR T cells does not yield a viable cellular
product [61]. Various approaches using an anti-CD3
CAR have been investigated including the use of
tran-scription activator-like effector nuclease (TALEN)
mRNA to disrupt the TRAC locus and using NK-92 cells in place of T cells as the effector cell type. Disruption of
the TRAC locus prevents assembly of the TCRαβ/CD3
complex, allowing for anti-CD3-CAR expression without compromising cellular proliferation and viability. Enrich-ment of the CAR-positive, CD3-negative population was observed. In patient T-ALL samples, anti-CD3 CAR T cells demonstrated specific cytotoxicity against CD3-positive cells. In a T-ALL NSG model, anti-CD3 CAR T cells were shown to clear luciferase-expressing CD3-positive Jurkat cells, but showed no effect in NSG mice engrafted with CD3-negative Jurkat cells [61]. To cir-cumvent the need for additional modifications, NK-92 cells can also be used to express the anti-CD3-CAR, since they are CD3-negative cells. CD3-CAR NK-92 cells demonstrated efficient ex vivo lysis of PTCL primary samples, resulting in less than 0.5% lymphoma cells
remaining at 5:1 effector to target ratios. Furthermore, CD3-CAR NK-92-treated T-ALL NSG mice exhibited prolonged survival with ~ 87% reduced tumor burden through day 23 [52].
CD1a
CD1a is a lipid-presenting molecule whose expression is restricted to developing cortical thymocytes, skin Lang-erhans cells, and some circulating myeloid dendritic cells
[103, 104]. Neither T cells nor CD34+ hematopoietic
progenitors express CD1a, making it a fratricide-resistant target, while limiting the risk of on-target/off-tumor toxicity. Expression in T cell malignancies is only limited to cortical T-ALL, a major subset of T-ALL ac-counting for ~ 35–40% of all T-ALL cases [105, 106]. A study showed that CD1a-CAR T cells expanded without fratricide, and had long-term persistence in an in vivo model [57]. Additionally, these cells demonstrated spe-cific cytotoxic activity against CD1a-positive T-ALL cell lines and primary blasts in vitro, and exhibited potent anti-leukemic activity in a PDX model of cortical T-ALL. Thus, while not applicable to all T cell malignan-cies, targeting CD1a with CAR T cells may be successful in the specific subset of cortical T-ALL cases.
“Off-the-shelf”CAR T cell therapy
One of the greatest challenges in utilizing autologous CAR T cell therapy for the treatment of T cell malignan-cies is the separation of healthy T cells from malignant T cells, in order to generate a CAR T cell product that is not contaminated with cancerous T cells. To date, there has been one reported case from the University of Penn-sylvania of CD19-CAR modification of a single leukemic B cell, resulting in CD19-positive relapse and ultimately death of the patient [39]. This task of isolating healthy T cells is even more difficult when a proportion of the pa-tient’s T cells are malignant, especially in cases of T cell leukemia where there is a high likelihood of circulating cancerous T cells. Thus, manufacturing of autologous CAR T cells for the treatment of T cell malignancies has a very high likelihood of resulting in CAR-modified leukemic cells. This would likely result in relapse as these cells would likely escape recognition by normal CAR-T cells.
Additionally, there remain numerous challenges to using a patient’s own cells to manufacture CAR T cells. Patients with advanced disease undergoing CAR T cell therapy typically are heavily pre-treated, having previ-ously undergone numerous rounds of chemotherapy, which can result in low T cell counts and/or T cells that may not be healthy enough to expand well making it very difficult to manufacture an efficacious CAR T cell
product [107]. This issue is much more prevalent in
T cells associated with aging [107–110]. Additionally, given that many of these patients have advanced disease, a patient may experience disease progression, co-morbidities, or even death in the time it takes to manu-facture autologous CAR T cells. This is especially true in most relapsed T cell malignancies, which tend to be ag-gressive and chemo-resistant in nature. Lastly, each starting autologous T cell product is different—variable function, maturation, CD4/CD8 ratios, and phenotypic ratios—and the heterogeneity of each individual product has led to unpredictable results and variable potency of the therapy.
An alternative to autologous CAR T cell manufactur-ing is the use of allogeneic T cells as the cell source. In order to make this approach feasible, expression of the
endogenous αβTCR in allogeneic CAR T cells must be
blocked as it would likely result in GvHD, unless the donor is a human leukocyte antigen (HLA) match. This process involves leukapheresis from a healthy donor, followed by isolation of the donor’s T cells. Following transduction of the T cells with a CAR-encoding retro-viral vector, subsequent genome editing of the TRAC locus is required to prevent expression of the endogen-ous TCR. Cells that remain TCR-positive are then de-pleted from the expanded CAR T cell product prior to cryopreservation. This creates an “off the shelf” cellular product that can be banked until it is needed for ther-apy. This approach resulted in successful remission in two infant B-ALL cases treated with allogeneic CD19-CAR T cells modified at the TRAC and CD52 loci. The allogeneic CAR T cells persisted until conditioning for
stem cell transplant [111]. Another group utilized
shRNA to knock downβ2-microglobulin in conjunction
with a knock-in strategy to insert a CD19-CAR into the
TRAC locus. Knock down of β2-microglobulin reduces
the ability of class I HLA molecules to form heterodi-mers on the cell surface. Reducing expression of both
β2-microglobulin and TRAC resulted in decreased
allo-geneic attack by CD8 T cells and NK cells [112]. This
strategy may be useful to reduce allo-recognition in pa-tients receiving CAR T cell therapy. Other groups have exploited similar approaches in preclinical CAR T cell investigations targeting CD7 and CD3, as previously de-scribed [46,61].
CRISPR-Cas9 genome editing has become a popular technique to prevent gene expression or to correct gene
expression. One study targeting CD7 generated
“fratri-cide resistant, allo-tolerant”CAR T cells using CRISPR-Cas9 to disrupt both CD7 and the TRAC loci (UCAR T7). NSG mice engrafted with primary T-ALL blasts de-veloped GvHD when treated with wildtype donor T cells; however, mice treated with UCART7 donor cells were able to clear the tumor cells from the peripheral blood, and, furthermore, did not develop GvHD or other
severe side effects [46]. TALENs, an alternative genome editing technique, have also been used to prevent ex-pression of the TRAC locus in order to limit fratricide of anti-CD3-CAR T cells and prevent MHC-recognition of foreign host cells. Genome editing the TRAC locus
pre-vents stable assembly of the TCRαβ/CD3 complex.
Dis-ruption of the TRAC locus using TALEN mRNA prior to transduction with an anti-CD3-CAR lentiviral vector yielded CAR T cells that proliferated well and greatly re-duced tumor burden in an NSG mouse model of human leukemia [61].
As described above, PEBLs have been recently devel-oped to selectively prevent expression of individual pro-teins. PEBLs have been shown to effectively retain CD3ε in the ER/Golgi to prevent MHC recognition of host cells during allogeneic use of anti-CD19 CAR T cells [50]. Disruption of TCRαβsignaling had no effect on T cell proliferation. There was no evidence of GvHD in an NSG mouse model of leukemia treated with the PEBL-CD19-CAR T cells, whereas 60% of the mice treated
with CAR T cells that were not expressing the CD3ε
PEBL developed GvHD. Furthermore, both PEBL and CAR can be expressed from the same vector using a 2A sequence, resulting in only one transduction of the cells [50]. While this study utilized PEBL in conjunction with an anti-CD19-CAR, this system can potentially be ap-plied with other CAR constructs to target T cell antigens.
Alternative effector cell types
While CAR-modified αβ T cells can have a memory
phenotype resulting in T cell aplasia, NK cells and gamma delta (γδ) T cells will not. Utilizing these innate cells for CAR therapy is a viable alternative that groups are exploring. One disadvantage to preventing memory cell formation and using effector cells with limited per-sistence is reduced tumor control. However, this limita-tion can potentially be overcome by utilizing these cells in multiple dosing regimens. Repeated dosing of short-lived CAR-expressing cells can be used to induce
remis-sion; thus, providing a bridge to an allogeneic
hematopoietic stem cell transplant (HSCT) if needed. Since these products would be utilized in an allogeneic setting, they can be cryopreserved and would be readily available when needed for use.
Natural killer cells and NK-92 cells
Ex vivo-expanded NK cells are short-lived, and do not persist for extended periods of time in vivo compared to that ofαβ T cells [113]. CAR-modified NK cells have a turnover time of 1–2 weeks; therefore, there is reduced concern of aplasia of antigen-expressing cells [114]. Cur-rently, there are two active clinical trials using
NCT01974479). Additionally, some studies use NK-92 cells, an IL-2-dependent NK-lymphoma-derived cell line. NK-92 cells are often used as an alternative to primary NK cells due to their ease of expansion under current
good manufacturing process (cGMP) conditions [115]
and transfection with CAR mRNA [116]. CAR-modified
NK or NK-92 cell infusion can result in tumor cell clear-ance without the risk of GvHD. Therefore, these cells typically only require one genetic modification. Add-itionally, with the exception of CD7, NK cells do not
ex-press antigens targeted in T cell malignancies.
Therefore, neither fratricide nor T cell aplasia are of pri-mary concern.
CAR-expressing NK-92 cells have been extensively assessed in preclinical studies targeting various cancers such as B cell malignancies [117–119], multiple myeloma [120],
acute myeloid leukemia (AML) [121], breast carcinoma
[122,123], neuroblastoma [124], and glioblastoma [125]. As previously discussed, multiple groups have initiated preclin-ical studies using CAR-modified NK-92 cells for the treat-ment of T cell malignancies, targeting antigens such as CD5, CD7, CD4, and CD3, demonstrating reduced tumor burden and an overall survival benefit in NSG mouse models of T cell leukemia [47,52–54,60]. The safety and efficacy of NK-92 cells has been evaluated in clinical trials displaying a good safety profile with few mild to moderate adverse events
[126–128] (NCT00900809, NCT00990717). To date, five
clinical trials have been initiated involving infusion of CAR-modified NK-92 cells targeting a variety of antigens, includ-ing CD33 [129], human epidermal growth factor receptor 2 (HER2), B cell maturation antigen (BCMA), CD19, and the T cell antigen, CD7 (NCT02944162, NCT03383978, NCT03940833, NCT02892695, and NCT02742727).
Inherent NK-cell cytotoxicity is dependent on the
bal-ance of activating and inhibitory killer-cell
immunoglobulin-like receptor (KIR) signals. Inhibitory and activating KIRs on NK cells form a balance, as there are often signals from both inhibitory and activating re-ceptors. The inhibitory signals predominate, typically through higher affinity for their ligands; however, strong activating signals can override the inhibitory signals, li-censing NK cells to kill. If donor inhibitory KIRs do not recognize patient HLA, there is reduced inhibitory sig-naling to counteract the activating sigsig-naling [130, 131]. While NK-92 cells lack many of the inhibitory KIRs expressed on primary NK cells, they have a wide range of activating receptors [132]. Similar to NK cells, NK-92 cells have the capability to produce perforin and granzyme upon activation, as well as display cytotoxic activity through upregulation of TNF-related
apoptosis-inducing ligand (TRAIL), Fas ligand (FasL), and TNFα
[133]. Additionally, NK-92 cells have demonstrated evi-dence of serial killing, with each cell killing numerous target cells [134]. However, as NK-92 cells were derived
from a NK cell lymphoma, they require irradiation prior to infusion into a patient to prevent expansion, resulting in persistence for about 1 week in vivo and potentially exhibiting reduced cytotoxicity. Alternatively, suicide mechanisms can be engineered into the cells to ate the risk of NK-92-cell persistence in vivo and elimin-ate the need for irradiation, thereby resulting in greelimin-ater cytotoxicity of the infused cells.
NK cells exhibit their cytotoxic activity through numer-ous means, including expression of FasL or TRAIL, secre-tion of perforin and granzyme, as well as through antibody-dependent cellular cytotoxicity (ADCC) mecha-nisms [131, 135, 136]. A major limitation to the use of CAR T cells is antigen escape; however, as NK cells can kill through other mechanisms, downregulation of the cognate antigen on tumor cells may not halt anti-tumor activity. NK cells also express the natural killer group 2D (NKG2D) receptor, which recognizes cellular stress li-gands such as MHC class I chain-related protein A/B (MICA/B) and UL16 binding proteins (ULBPs) [137,138], resulting in cytotoxicity against exceedingly stressed cells. As NK cells do not recognize targets on healthy cells, they have limited off-target toxicity [131]. Additionally, their serial killing capability allows each individual NK cell to kill, on average, four tumor cells [139]. However, NK cells are notoriously difficult to expand ex vivo, transduce with viral vectors, cryopreserve, and they have limited life span in vivo [128,140]. While autologous NK cells can be ob-tained by leukapheresis followed by selection of CD56-positive cells, allogeneic NK cells derived from a third party donor requires an additional step for depletion of alloreactive T cells from the donor product [141].
Purification and expansion of NK cells from peripheral blood mononuclear cells (PBMCs) have been optimized in cGMP protocols to clinically relevant numbers [142–144]. This is a time-consuming process as only 10% of PBMCs are NK cells [145]. However, recently developed methods are being used to enhance NK-cell expansion, such as through K562-feeder cell expression of OX40 ligand [146]. As mentioned above, a limitation to CAR-NK ther-apy is the extreme sensitivity of NK cells to
cryopreserva-tion. They have demonstrated poor viability and
diminished cytotoxicity after cryopreservation. While cytotoxicity can be restored to normal levels after a few days in culture with exogenous IL-2, the low viability post-cryopreservation remains a concern [141].
Gamma delta T cells
While αβ T cells function as a part of the adaptive
im-mune system,γδT cells play roles in both the innate and
the adaptive immune systems. γδT cells and αβT cells
recognition [148, 149]. Lack of MHCI- and MHCII-restriction makeγδT cells optimal candidates for allogen-eic cell therapy. The peripheral blood subset ofγδT cells known as Vγ9Vδ2 T cells represents the most commonly studied subset in this context. Studies by our group have demonstrated that similar transduction efficiencies can be
achieved in Vγ9Vδ2 T cells grown under cGMP
serum-free conditions as are achieved inαβT cells using lenti-viral vectors. Additional studies were performed revealing peak low-density lipoprotein receptor (LDL-R) expression on days 6–8 ofγδT cell expansion [150]. As LDL-R is the major receptor for VSV-G-pseudotyped lentiviral vectors, this data suggests that greater transduction efficiency can be achieved on these days using lentiviral vectors com-pared to earlier or later in the expansion [151].
To date, numerous preclinical studies have evaluated
CAR-modified γδ T cells targeting neuroblastoma [152,
153], melanoma [154], B cell malignancies [153, 155], and epithelial cell adhesion molecule (epCAM)-positive adeno-carcinomas [156]. GD2-CAR-modifiedγδT cells expressing the RQR8 suicide gene were shown to expand 2.5-fold upon antigen exposure [152]. Furthermore, both GD2-CAR- and
CD19-CAR-modified γδ T cells were demonstrated to
se-crete pro-inflammatory cytokines in the presence of GD2-or CD19-expressing tumGD2-or cells, respectively [153]. While these studies utilized viral vectors to express the CAR, elec-troporation of a Sleeping Beauty transposon has also been
shown to result in CD19-CAR expression in γδ T cells,
resulting in anti-tumor cytotoxicity in both the in vitro and in vivo settings [155]. Additionally, expression of a CAR tar-geting melanoma-associated chondroitin sulfate
proteogly-can (MCSP) was established in γδ T cells using mRNA
transfection. Despite comparable anti-tumor cytotoxicity, lower cytokine secretion was observed in
MCSP-CAR-modified γδ T cells compared to that from conventional
CAR-modifiedαβT cells [154]. Reduced pro-inflammatory cytokine secretion is favorable due to anticipated reduced
severity of CRS. Lastly, epCAM CAR-modified γδ T cells
demonstrated high levels of in vitro cytotoxicity of tumor cell lines whenγδT cells were both fresh and cryopreserved [156]. These studies pave the way for additional trials using CAR-modifiedγδT cells targeting T cell malignancies. They demonstrate that engineering ofγδT cells is feasible and re-sults in enhanced in vitro and in vivo cytotoxicity upon CAR expression.
CAR-modifiedγδT cells may be able to overcome the
obstacle of antigen escape seen in some treatment-resistant cases by relying on their innate ability to
recognize tumor cells through other means. Naïve γδT
cells have been shown to have anti-tumorigenic activity against leukemia, neuroblastoma, and colon cancer cell lines as well as primary cancer cells in vitro [157–160]. They are found in peripheral blood, spleen, and lymph nodes, in addition to almost all mucosal tissues,
functioning as immune-surveillance of epithelial tissues by scanning for inflammatory threats [161,162]. The γδ TCR recognizes self-antigens that serve as endogenous danger signals such as heat shock proteins, which are upregulated in cells with increased metabolism, like can-cer cells. Expression of scavenger receptors like the
NKG2D receptor enables γδ T cell activation through
the interactions with antigens of cellular stress such as MICA/B and ULBPs [147, 163–166]. Additionally, γδT cells express chemokine receptors that can detect che-mokines secreted by cancer cells, likely facilitating their migration toward the tumor site [167]. γδ T cells also express FasL (CD95L) as a means of recognizing Fas ex-pression on tumor cells and initiating apoptosis [168].
Another mechanism by whichγδT cells recognize tumor
cells is through stimulation by phosphoantigens, such as isopentenyl pyrophosphate (IPP), which are recognized by
the γδ TCR. While there are many subsets of γδ T cells,
phosphoantigens specifically expand the Vγ9Vδ2 subset. IPP is used as a substrate in the mevalonate pathway by far-nesyl pyrophosphate synthase (FPPS). Bisphosphonates overproduced in cancer cells block FPPS, resulting in a buildup of IPP, which is subsequently recognized by cyto-toxic Vγ9Vδ2 T cells [169–172]. Bisphosphonate stimula-tion ofγδT cells has been applied to in vitro expansion of
γδ T cells in conjunction with IL-2 in serum-free condi-tions [150]. A preclinical study involving nude mice
receiv-ing repeated dosreceiv-ing of γδ T cells resulted in decreased
tumor growth model; however, tumor growth resumed upon completion of theγδT cell infusions [173]. In phase I clinical trials, adoptive transfer ofγδT cells to patients re-ceiving ex vivo expandedγδT cells with a combination of IL-2 and bisphosphonate stimulation demonstrated the safety of the infused product and suggested that the therapy could be efficacious in slowing the progression of the dis-ease. However, mixed results were seen in terms of efficacy, suggesting that genetic modification with CAR expression is likely to be more beneficial compared toγδT cell therapy alone [169,174–177].
While the autologous transfer of CAR-modified αβ T
Prevention of memory cell formation and T cell aplasia
While current CAR T cell therapies for the treatment of B cell malignancies have been hugely successful in indu-cing and maintaining remission, these therapies have prevented the re-emergence of endogenous B cells in pa-tients in whom the CAR T cells have persisted. The CAR T cells can have a memory phenotype that allows them to remain dormant until restimulation with the cognate antigen, CD19, expressed on all endogenous B cells. While B cell aplasia is an undesirable side effect of these therapies, it has been managed by continued
peri-odic intravenous immunoglobulin injections [36]. The
long-term implications of persistent B cell aplasia remain unknown. In contrast, treatment of T cell malignancies using CAR T cells targeting antigens expressed on the majority of normal T cells is predicted to result in T cell aplasia. While B cell aplasia is tolerable, there is no such treatment for T cell aplasia. Patients who develop T cell aplasia will have profound immunosuppression and can potentially succumb to deadly infections [36]. Therefore, prevention of memory cell formation of CAR T cells and subsequent T cell aplasia remains an essential challenge to translating CAR T cell therapy for the treatment of T cell malignancies. While bridging a patient to an allogen-eic HSCT following CAR T cell therapy may eliminate the risk of life-threatening T cell aplasia by clearing out the CAR T cells, safer and less invasive alternatives must also be explored to downregulate CAR activity after tumor clearance.
mRNA electroporation
There are numerous disadvantages to using retroviral vec-tors for CAR T cell therapy, including risk of clonal
dom-inance [178, 179], high cost of production [180],
maximum cargo size [181,182], and the inability to“turn
off” transgene expression and unpredictable integration
sites potentially resulting in insertional oncogenesis [183,
184]. The indefinite period of CAR expression can result in severe on-target off-tumor toxicities, which is particu-larly challenging to manage in T cell disease. To overcome these unintended side effects, groups are alternatively ex-ploring delivery of CAR mRNA through electroporation as a safer method [185–187]. As with the use of effector cells with limited persistence in vivo, therapies with transi-ent CAR expression require multiple infusions into the patients. Use of mRNA electroporation of T cells for CD19-CAR expression has been reported in a preclinical model, demonstrating reduced tumor burden 1 day post-treatment. This study illustrated prolonged survival of a xenograft mouse model after a single injection of CAR mRNA T cells; however, as predicted, as the mRNA levels decreased the tumor burden increased [185]. Published re-sults from the first non-viral CD19-CAR clinical trial
using mRNA electroporation to deliver the CAR into T cells demonstrated the safety and efficacy of this treatment in four relapsed/refractory classical Hodgkin lymphoma
patients [225]. CAR mRNA was detected 48 h
post-infusion; however, no mRNA could be detected by day 21. While only transient responses were seen, no severe toxic-ities were observed using this approach. Utilizing this non-viral strategy in T cell disease can be particularly ad-vantageous, as it prevents the risk of long-term T cell aplasia. While the transient efficacy precludes this ap-proach from being used as a definitive treatment, it could potentially serve as an effective bridge to transplantation.
Adeno-associated viral vector
AAV is an alternative viral delivery method that can over-come some of the disadvantages of using integrating viral vectors as previously discussed. AAV is a single-stranded, non-enveloped DNA virus with a cargo capacity of ap-proximately 4.7 kilobases [188]. Upon deletion of the Rep protein, the viral transgene forms circular concatamers that exist episomally in the nucleus of the cell. AAV ex-pression is therefore diluted upon each mitotic division, resulting in a transient transgene expression limited to the lifespan of the cell [189, 190]. Thus, AAV delivery can control the duration of CAR expression, which is a desired quality to regulate cytokine production and mediate toxic-ities [191–193]. In particular, transient CAR expression may prove to be advantageous in the setting of T cell ma-lignancies, by preventing unintended T cell aplasia.
Efficient transduction of innate immune cells, such as
NK cells andγδT cells, by an AAV vector would be
par-ticularly invaluable in targeting this group of diseases.
As previously discussed, both NK cells and γδ T cells
are excellent candidates as CAR effector cells against T cell antigens. A common challenge reported in using these cell types is the low transduction efficiency using integrating viral vectors, delaying progress in the devel-opment of these therapies. AAV gene transfer of a CAR into innate immune cells would offer the opportunity to develop an allogeneic off-the-shelf CAR therapeutic that can control CAR expression, thereby mitigating CRS and other adverse events. Additionally, the lack of memory cell formation against T cell antigens in these cell types will completely negate the risk of T cell aplasia. The AAV capsid directs the infectivity of different tissues, and therefore the appropriate capsid serotype must be used to maximize transduction of the desired cell type
[194, 195]. AAV6 has been shown to result in higher
transduction of hematopoietic stem and progenitor cells than have other serotypes [196–198].
Suicide genes and safety switches
mediate the severe adverse events commonly reported following extensive expansion of CAR T cells, they can also serve an alternative purpose. Using pharmacologic agents, the apoptotic pathway in CAR T cells can be ac-tivated, triggering selective cell death of the effector cells, without destroying bystander cells. Therefore, they can be valuable in the setting of T cell malignancies as they can prevent T cell aplasia. There are three main classes of suicide gene technologies, classified by the mechanism of action of the incorporated gene. They (i) convert non-toxic compounds to toxic drugs via meta-bolic pathways [199–202], (ii) induce dimerization of
in-ducible caspase-9 [203, 204], or (iii) mediate ADCC
using monoclonal antibodies [205–207]. Co-expression of the suicide gene with the CAR in a bicistronic vector would result in two populations of cells—those that ex-press both the CAR and the suicide gene, and those that express neither. This strategy negates the risk of generat-ing a CAR-positive population without the safety trans-gene; thus, enabling one to confidently eliminate the entire CAR-positive population and thereby, in the con-text of targeting T cell antigens, controlling T cell aplasia.
The first reported suicide gene utilized the herpes sim-plex virus thymidine kinase (HSV-TK) as a method of GvHD abrogation in the context of an allogeneic HSCT. Expression of HSV-TK in donor lymphocytes prior to their infusion into a HSCT patient allows for selective de-pletion of the donor lymphocytes in patients that devel-oped signs of GvHD upon administration of ganciclovir [199–201]. Metabolism of ganciclovir by the thymidine kinase of HSV-TK results in a toxic substance, ultimately killing the cell [208]. However, there are a couple of limi-tations to this system including the potential for immuno-genicity and the slow T cell depletion, which requires about 3 days [209–211].
More recently, the safety mechanism gaining the most attention has been the inclusion of an inducible caspase-9-based suicide gene (iCas9) into the CAR construct. Pharmacologic activation of the iCas9 results in effective and rapid elimination of CAR T cells. iCas9 inclusion in a CD19-CAR construct has been shown to regulate CAR T cells in a dose-dependent manner, allowing for either control over the CAR T cells to reduce toxicities, or complete elimination of all CAR T cells to facilitate B cell reconstitution [212, 213]. This is especially signifi-cant in cases with severe adverse events, such as GvHD or CRS. iCas9 has recently been included in CAR con-structs containing an IL-15 gene to introduce control over CAR T cell function. The IL-15 gene arms the T cells to produce IL-15, which, while increasing T cell survival and enhancing specific cytotoxicity, can also re-sult in unrestricted proliferation and increased toxicity. Inclusion of an iCas9 gene in these CAR constructs can
provide control to this therapy and increase the safety profile [214]. In addition to CD19-CARs, iCas9 has been included in other CAR constructs including an anti-CD20-CAR, demonstrating enhanced tumor clearance in vivo and a 90% reduction in CAR T cells in the per-ipheral blood of mice following activation of the iCas9 suicide gene, compared to CAR T cells detected in
per-ipheral blood of control mice [215]. Additionally, a
GD2-CAR including the iCas9 gene is being assessed for the treatment of neuroblastoma (NCT01822652), sar-coma (NCT01953900), osteosarsar-coma, and melanoma (NCT02107963) in phase I clinical trials.
In terms of utilizing ADCC for CAR T cell clearance, administration of alemtuzumab, an anti-CD52 antibody commonly used in lymphodepleting regimens, has been tested in several studies. Specifically, alemtuzumab has been assessed for CD4-CAR T cell elimination following tumor cell eradication in NSG mice to prevent T cell apla-sia [87]. Within 6 h following alemtuzumab infusion, > 95% of the CAR T cells had been depleted. This approach was also tested in two other preclinical CAR studies tar-geting AML, both showing excellent results [216, 217]. Multiple groups have also evaluated the retroviral transfer of human CD20 into T cells as a novel suicide gene mech-anism for adoptive T cell therapy. Their data supports that infusion of the anti-CD20 antibody, rituximab, an ap-proved antibody for in vivo therapeutic applications, re-sults in efficient, specific elimination of CD20-positive T lymphocytes through ADCC [205,206,218]. Studies have also demonstrated that rituximab can eliminate CD20-positive cells in vivo through inducing complement-dependent cytotoxicity, a rapid and efficient mode of cell
death [219]. CD20 co-expression with a CD123-CAR
demonstrated strong and rapid anti-leukemia activity in a human AML mouse model. Upon the infusion of rituxi-mab, CAR T cells were cleared and mice were successfully engrafted with human bone marrow cells, mimicking an allogeneic HSCT [217]. Thus, ADCC-based safety systems potentially allow for rapid and efficient elimination of CAR T cells [211].
An epitope-based marker/suicide gene system (RQR8) was recently developed to both track the transduced cells and selectively deplete them by combining epitopes from CD34 and CD20 [220]. Use of Miltenyi Biotec’s clinically
the selection of CAR-positive T cells, tracking of the cells in vivo and selective elimination as a safety mechanism, is the truncated human epidermal growth factor receptor (huEGFRt). Manipulation of this protein was done to re-move intracellular signaling domains, leaving it with an in-tact epitope for binding cetuximab, an anti-EGFR monoclonal antibody. Modification of T cells with the CAR and huEGFRt allows for selection using GMP biotin immunomagnetic beads and biotinylated cetuximab, and tracking using flow cytometry or immunohistochemistry. Upon administration of cetuximab, CAR T cells become the targets for ADCC, resulting in in vivo depletion of CAR T cells. Successful T cell engraftment and ADCC-mediated CAR T cell elimination with cetuximab were demonstrated in a murine model [221]. The huEGFRt sui-cide mechanism is currently being assessed in a phase I clinical trial in an anti-MUC-16ectoCAR construct to treat
patients with recurrent MUC16ecto+ solid tumors
(NCT02498912) [222].
A novel alternative approach to suicide genes is the generation of“ON-switch” CARs [223]. In this strategy, the CAR is a split receptor consisting of two distinct polypeptides: the antigen recognition domain and the intracellular signaling domain. In order to act as a func-tional receptor, the two peptides must first dimerize, achieved through activation by a dimerization-inducing small molecule. However, antigen stimulation is still re-quired to facilitate a response. The small molecule can be titrated for optimal response, controlling the timing and dosage of active CAR T cells. Thus, removal of the small molecule can reversibly regulate CAR T cell activ-ity. These ON-switch CAR T cells demonstrate specific cytotoxicity in vitro and in vivo only when exposed to the small molecule. In a mouse xenograft model, mice treated with ON-switch CAR T cells displayed a reduc-tion in K562 cells engineered to express CD19, only in the presence of the small molecule, similar to mice treated with conventional CD19-CAR T cells. However, no benefit was seen in the absence of the small mol-ecule. Given the tight control over CAR expression using this innovative approach, it has the potential to be adapted for T cell malignancies.
Summary and conclusions
CAR therapies targeting CD19 have resulted in unparal-leled success. However, there are many challenges in translating these therapies beyond the treatment of B cell malignancies. We have highlighted some of these challenges as it pertains to targeting T cell disease. While numerous antigens have been identified for the treatment of T cell malignancies, targeting of many of these antigens results in fratricide and T cell aplasia. Multiple gene editing approaches are being evaluated to prevent fratricide by reducing expression of the targeted
antigen on CAR-modified cells. The identification of tumor-specific antigens would greatly enhance CAR therapy targeting T cell malignancies by avoiding fratri-cide. To date, only a few antigens with limited expres-sion on normal T cells have been assessed as CAR targets to treat T cell malignancies; these include CD30, CD37, and CD1a. However, given their expression on only small subsets of T cell cancers, a focus on these an-tigens is unlikely to have a wide-ranging impact on the overall translation of CAR therapy for patients with T cell disease. In contrast, TRBC1 is expressed on a much larger population of T cells and therefore it is likely to be found on a comparatively higher percentage of T cell malignancies. To the best of our knowledge, only one study has evaluated anti-TRBC1-CAR T cell therapy. The data suggests that TRBC1 is a very promising marker for targeting T cell malignancies and the field would benefit from studies further developing this therapy.
Among other target antigens, CD5 has emerged as a promising candidate given its ability to rapidly downreg-ulate from the cell surface upon interaction with the CD5-CAR. Therefore, only transient and limited fratri-cide is observed, allowing for successful expansion of CD5-CAR T cells. While targeting CD5 or other T cell antigens using gene-edited CAR T cells may overcome the issue of fratricide, the concern regarding T cell apla-sia has not been addressed. The potential for life-threatening T cell aplasia emphasizes the need for a safety mechanism that is completely effective at elimin-ating CAR T cells following tumor eradication. Safer al-ternatives other than bridging to an allogeneic HSCT must be explored to limit CAR T cell persistence. Adjusting the effector cell type to NK cells, NK-92 cells, orγδT cells can limit the risk of a memory cell immune response against a T cell antigen. However, given that NK-92 cells require irradiation prior to infusion in a pa-tient, their therapeutic effect may be limited. mRNA electroporation or AAV delivery systems, which result in transient CAR expression, could be utilized, thereby allowing for restoration of normal T cell immunity once the CAR effect has diminished. Additionally, the use of iCas9 and ADCC-based suicide genes, as well as other CAR safety switches should be explored in the context of T cell malignancies.
TRAC locus is required to prevent GvHD, when using
allogeneicαβ T cells for CAR expression. However, NK
cells and γδ T cells can both be used in an allogeneic
setting given their MHC-independent activation, and are thus unlikely to cause GvHD. Use of allogeneic CAR-modified cells also addresses the challenges of high cost and difficulty of production, since healthy donor cells can be expanded more easily and cryopreserved as an off-the-shelf therapy until they are required for use. Additionally, allogeneic cell delivery allows for titratable dosing as well as multiple infusions, if such is required.
Many avenues are currently being explored to enhance the safety and efficacy of CAR therapy. However, the majority of these strategies do not address all three main challenges to utilizing CAR therapy to treat T cell malig-nancies. Of the approaches evaluated in this review, only those incorporating NK cells or NK-92 cells can poten-tially overcome all of these primary challenges (Fig. 2). NK cells (i) are non-alloreactive and can be obtained from healthy donors, eliminating risk of product con-tamination; (ii) do not form memory responses, prevent-ing T cell aplasia; and (iii) do not express the same antigen repertoire as T cells, avoiding fratricidal
concerns. CD7 is an exception as it is expressed on NK cells and therefore fratricide could occur. While several groups have published studies with CAR NK-92 cells targeting T cell malignancies, more effort needs to be put into using primary NK cells for targeting this dis-ease, especially given the limitations of NK-92 cells. Other, equally promising approaches, such as utilizing
γδT cells as the cellular vehicle for CAR therapy repre-sents an alternative, less studied approach. Similar to NK cells,γδT cells are non-alloreactive and are unlikely
to form a memory response against a T cell antigen.γδ
T cells are likely to succumb to fratricide in certain cir-cumstances; however, targeting an antigen such as CD5 that results in only transient and limited fratricide may
be especially advantageous. Furthermore, γδT cells
ex-hibit innate MHC-independent mechanisms of cytotox-icity by which they can recognize tumor cells. Thus,
CAR therapy using γδT cells represents an
understud-ied avenue with the potential of developing into a super-ior cellular product.
Many advances have been made toward translating CAR therapy for the treatment of T cell malignancies. Both academia and industry are focused on the