Electronic Thesis and Dissertation Repository
4-20-2018 10:30 AM
Development and applications of polyglyoxylate self-immolative
Development and applications of polyglyoxylate self-immolative
polymers
polymers
Bo Fan
The University of Western Ontario
Supervisor
Gillies, Elizabeth R.
The University of Western Ontario
Graduate Program in Chemical and Biochemical Engineering
A thesis submitted in partial fulfillment of the requirements for the degree in Doctor of Philosophy
© Bo Fan 2018
Follow this and additional works at: https://ir.lib.uwo.ca/etd
Part of the Polymer and Organic Materials Commons
Recommended Citation Recommended Citation
Fan, Bo, "Development and applications of polyglyoxylate self-immolative polymers" (2018). Electronic Thesis and Dissertation Repository. 5293.
https://ir.lib.uwo.ca/etd/5293
This Dissertation/Thesis is brought to you for free and open access by Scholarship@Western. It has been accepted for inclusion in Electronic Thesis and Dissertation Repository by an authorized administrator of
i
Self-immolative polymers (SIPs) are relatively recent class of stimuli-responsive and
degradable polymers that have attracted significant attention in the past several years. SIPs
consist of polymer backbones and stimuli-responsive end-caps at one or both polymer termini.
Upon detection of a stimulus, the decomposition of the end-cap leads to complete end-to-end
depolymerization. Polyglyoxylates were introduced as a new class of polyacetal based SIPs by
our group in 2014. Compared with other SIPs, polyglyoxylates have two advantages including:
1) readily available monomers and 2) low toxicity depolymerization products. These
advantages may allow polyglyoxylates to be used in a wide range of applications. This thesis
explored the design, synthesis, and study of a series of responsive end-caps for different
potential applications of polyglyoxylates. First, using the previously developed 6-nitroveratryl
carbonate end-capped poly(ethyl glyoxylate) (PEtG) that responded to UV light, it was
demonstrated that PEtG could depolymerize back to volatile monomer at ambient temperature
and pressure. This unusual feature was used to perform a facile polymer
reprogramming/recycling sequence as well as polymer patterning by a simple
irradiation-evaporation sequence. Moreover, end-caps that allowed polyglyoxylates to respond to
oxidizing and reducing conditions, acid, heat, multiple stimuli, and one that enabled
cross-linking and UV-triggered depolymerization, were developed. Furthermore, linker end-caps
were developed to conjugate PEtG with poly(ethylene glycol) to form amphiphilic block
copolymers. These copolymers were self-assembled to form nanoparticles that could load and
release payload molecules in response to stimuli. In addition, the hydrophobicity of PEtG was
tuned by copolymerization with hydrophobic monomers to improve the nanoparticle drug
loading capabilities. Lastly, triphenylmethyl end-capped PEtGs were demonstrated to undergo
temperature-dependent depolymerization. Proof-of-concept studies were performed to
demonstrate the potential of these polymers for smart packaging applications. Overall, the
work presented in this thesis serves to expand the utility of polyglyoxylate-based SIPs for
various applications through the design and synthesis of responsive end-caps and new polymer
ii
Keywords
Self-immolative polymer, stimuli-responsive, degradation, depolymerization, polyglyoxylate,
poly(ethyl glyoxylate), traceless, photo-lithography, amphiphilic block copolymer, micelle,
iii
The research described in this thesis is a result of contributions from the author as well as
coworkers and supervisor Dr. Elizabeth R. Gillies. The detailed contributions for each chapter
are as follows:
Chapter 1 and chapter 2 were written by the author and edited by Dr. Gillies.
Chapter 3 describes a project jointly conceived by the author, Dr. Gillies and postdoctoral
fellow Dr. John F. Trant. The author conducted most of the depolymerization studies with
assistance from Rebecca E. Yardley and Andrew J. Pickering. The photo-patterning study was
conducted with assistance from Dr. Franç ois Lagugné-Labarthet and the Western
Nanofabrication Facility. The manuscript was prepared by the author and was revised with the
assistance of Dr. Gillies and Dr. Trant.
Chapter 4 describes work jointly conceived by the author and Dr. Gillies, with additional
acid-sensitive end-caps contributed by Dr. Trant. The author conducted most of synthesis (except
for the acid-sensitive end-caps), all characterization, and all depolymerization studies. The
manuscript was prepared by the author and was revised with the assistance of Dr. Gillies.
Chapter 5 describes work jointly conceived by the author and Dr. Gillies. The author conducted
all the experiments. The manuscript was prepared by the author and was revised with the
assistance of Dr. Gillies.
Chapter 6 describes work jointly conceived by the author, Dr. Gillies and Dr. Trant. The author
conducted most of the synthesis, characterization, and all depolymerization studies. Dr. Trant
contributed the synthesis of menthyl glyoxylate, Rebecca Yardley conducted the synthesis of
butyl glyoxylate, and Aneta Borecki conducted the cell studies. The manuscript was prepared
by the author and was revised with the assistance of Dr. Gillies.
Chapter 7 describes work jointly conceived of by the author, Dr. Trant, Dr. Gillies, and
collaborator Dr. Olivier Sandre. Dr. Trant conducted the synthesis of end-caps while the author
conducted the synthesis of polymers and all depolymerization studies. The synthesis of iron
iv
prepared by the author and Gauvin Hemery and was edited by Dr. Gillies and Dr. Sandre.
Chapter 8 describes work jointly conceived of by the author, Dr. Gillies and Dr. Romulo
Salazar. The author conducted all synthesis, characterization, and polymer film colour change
studies. The polymer film depolymerization studies were conducted by Dr. Salazar. The
manuscript was prepared by the author and was revised with the assistance of Dr. Gillies.
v
First of all, I would like to give my sincere appreciation to my supervisor Dr. Gillies. I thank
her for accepting me as a member of this caring, mutually helping and creative research group.
More importantly, I want to acknowledge Dr. Gillies for her guidance and encouragement
along the way, without whom this thesis would not be achievable.
My thanks would also go to all the previous and present members of the Gillies group. Thank
you all for creating a caring and mutually helping research environment. You leave me with
lots of unforgettable memories during the past several years.
I also would like to thank my thesis examiners, Dr. Xu, Dr. Zhang, Dr. Gilroy and Dr. Gu for
taking time to read through my thesis. Many thanks to all the faculty and staff in the
Department of Chemical and Biochemical Engineering and the Department of Chemistry for
their support in my study and research.
Furthermore, I would like to express heartfelt appreciation to my girlfriend-Jing Wan who has
been supportive for my all decisions. I also want to thank her for reading through this thesis.
Lastly but not the least, I want to give the deepest thanks to my parents, without the constant
love, supporting and encouragement from both of you, nothing will be possible for me.
vi
Abstract ... i
Co-Authorship Statement... iii
Acknowledgments... v
Table of Contents ... vi
List of Tables ... x
List of Figures ... xi
List of Schemes ... xix
List of Abbreviations ... xxi
Chapter 1 ... 1
1 Introduction ... 1
1.1 Overview ... 1
1.2 Research objectives ... 3
1.3 Thesis outline ... 4
1.4 References ... 6
Chapter 2 ... 9
2 Stimuli-responsive polymers (SRPs) ... 9
2.1 General non-degradable SRPs ... 10
2.1.1 Light-responsive polymers... 10
2.1.2 Thermo-responsive polymers ... 14
2.1.3 pH-responsive polymers ... 15
2.1.4 Gas-responsive polymers ... 17
2.2 Stimuli-responsive and degradable polymers ... 19
2.2.1 Acid-degradable polymers ... 20
vii
2.2.4 Limitations of conventional stimuli-responsive and degradable polymers
... 24
2.3 Self-immolative polymers ... 25
2.3.1 SIPs depolymerizing via elimination reactions ... 26
2.3.2 SIPs depolymerizing via cyclization reactions ... 30
2.3.3 SIPs depolymerizing due to low-ceiling temperature ... 34
2.3.4 Applications of SIPs in drug delivery ... 41
2.4 References ... 45
Chapter 3 ... 55
3 Photo-controlled depolymerization of stimuli-responsive poly(ethyl glyoxylate): Differentiating features and traceless ambient depolymerization ... 55
3.1 Introduction ... 55
3.2 Experimental section ... 56
3.3 Results and discussion ... 61
3.4 Conclusions ... 70
3.5 References ... 71
Chapter 4 ... 76
4 End-capping strategies for triggering end-to-end depolymerization of polyglyoxylates ... 76
4.1 Introduction ... 76
4.2 Experimental section ... 78
4.3 Results and discussion ... 87
4.3.1 Development of redox-responsive PEtG ... 87
4.3.2 Development of acid-responsive PEtG ... 94
4.3.3 Development of a multi-responsive end-cap ... 96
viii
4.4 Conclusions ... 105
4.5 References ... 106
Chapter 5 ... 114
5 Poly(ethyl glyoxylate)-poly(ethylene glycol) nanoparticles: Stimuli-responsive drug release via end-to-end polyglyoxylate depolymerization ... 114
5.1 Introduction ... 114
5.2 Experimental section ... 116
5.3 Results and discussion ... 125
5.3.1 Synthesis of stimuli-responsive PEtG-PEG triblock copolymers ... 125
5.3.2 Self-assembly of PEtG-PEG triblock copolymers in aqueous solution .. 131
5.3.3 Stimuli-responsive properties of the nanoparticles ... 132
5.3.4 Encapsulation and triggered release of drugs and model drugs ... 139
5.4 Conclusions ... 144
5.5 References ... 145
Chapter 6 ... 151
6 Tuning the hydrophobic core of self-immolative polyglyoxylate assemblies ... 151
6.1 Introduction ... 151
6.2 Experimental section ... 152
6.3 Results and discussion ... 161
6.3.1 Polymer synthesis ... 161
6.3.2 Celecoxib loading and release ... 169
6.3.3 In vitro toxicity studies ... 173
6.4 Conclusions ... 174
6.5 References ... 176
ix
7.1 Introduction ... 182
7.2 Experimental section ... 183
7.3 Results and discussion ... 192
7.4 Conclusions ... 200
7.5 References ... 201
Chapter 8 ... 204
8 Temperature-dependent depolymerization of trityl end-capped poly(ethyl glyoxylate): potential applications in smart packaging ... 204
8.1 Introduction ... 204
8.2 Experimental section. ... 206
8.3 Results and discussion. ... 211
8.4 Conclusions. ... 219
8.5 References. ... 220
Chapter 9 ... 224
9 Conclusions and future perspectives ... 224
Appendix 1: Permission to reuse copyrighted material in Chapter 1 ... 228
Appendix 2: Permission to reuse copyrighted material in Chapter 2 and published chapters ... 229
Appendix 3: Supporting information for Chapter 3 ... 235
Appendix 4: Supporting information for Chapter 4 ... 245
Appendix 5: Supporting information for Chapter 5 ... 283
Appendix 6: Supporting information for Chapter 6 ... 307
Appendix 7: Supporting information for Chapter 7 ... 331
Appendix 8: Supporting information for Chapter 8 ... 356
x
Table 4.1 Molar mass and thermal properties of end-capped PEtGs and measured by SEC,
TGA, and DSC. ... 90
Table 5.1 Properties of PEtG with different linker end-caps and PEG-PEtG-PEG copolymers.
These polymers have been previously reported. ... 129
Table 5.2 Hydrophilic mass fractions (f) of the block copolymers and corresponding
characterization of self-assembled nanoparticles by DLS and CAC measurement. Errors on
the measurements correspond to the standard deviations. ... 131
Table 6.1 SEC and thermal analysis results for the polymers. From SEC; From TGA;From
DSC; From previously published work. ... 163
Table 6.2 Characterization data for the amphiphilic block copolymers and their resulting
assemblies. Previously reported; From SEC; Measured at 1 mg/mL of copolymer. ... 166
Table 6.3 Characterization data for the block copolymer assemblies. Previously reported. 168
Table 6.4 Size characterization and celecoxib loading data for the block copolymer
xi
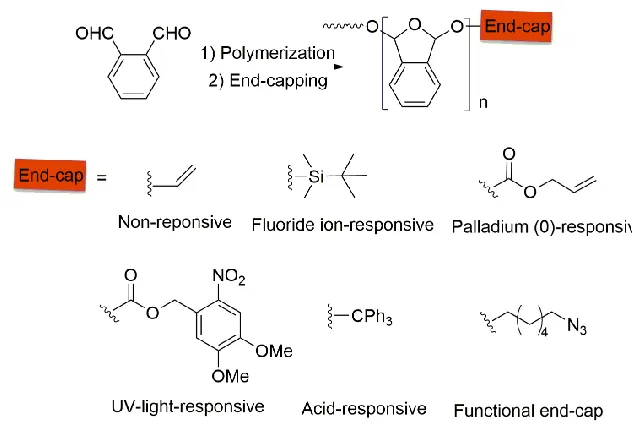
Figure 1.1 Structure of polyglyoxylates and the depolymerization mechanism after end-cap
removal. (Reproduced with permission from reference (29). Copyright 2014 American
Chemical Society.) ... 3
Figure 2.1 Schematic representations of SRPs: a) General non-degradable SRPs that change
in physical properties in response to a stimulus; b) SRPs that can entirely degrade in response
to a stimulus stoichiometrically; c) Self-immolative polymers (SIPs) as a sub-class of SRPs
that can depolymerize in response to a stimulus in an amplified manner. ... 9
Figure 2.2 Representative light-responsive molecules applied in stimuli-responsive polymers:
a) Cis-trans transformation of aromatic azo compounds, b) hydrophobic spiropyran to
hydrophilic merocyanine transformations under different wavelengths of light. ... 11
Figure 2.3 a) Synthesis procedure for the azobenzene liquid crystalline polymer network; b)
Schematic and c) photographs showing light-driven forward moving of a “vehicle” equipped
with a spring-like “motor”. (Reproduced with permission from reference (12). Copyright
2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.) ... 12
Figure 2.4 Photochromic polymersomes exhibiting photo-switchable and reversible bilayer
permeability. (Reproduced with permission from reference (14). Copyright 2015 American
Chemical Society.) ... 13
Figure 2.5 Chemical structures of some typical thermo-responsive polymers. ... 14
Figure 2.6 a) Temperature dependence of water and oil contact angles for a
PMMA-b-PNIPAM film; b) Reversible water and oil contact angle transition of block copolymer film
at different temperatures; c) Diagram of reversible formation of intermolecular hydrogen
bonding between PNIPAM chains and water below and above the LCST. (Reproduced with
permission from reference (23). Copyright 2013 WILEY-VCH Verlag GmbH & Co. KGaA,
Weinheim.)... 15
xii
conformation and behavior of QD-GO at a given pH value. (Reproduced with permission
from reference (24). Copyright 2014 American Chemical Society.)... 17
Figure 2.9 Schematic illustration of: a) Preparation of P(DMAEMA-co-CMA) single-chain
nanoparticles through intrachain photo-cross-linking and the gas-switchable size change of
nanoparticle in aqueous solution; b) Preparation of tadpole-like PS-b-P(DMAEMA-co-CMA)
nanoparticles and their gas-responsive self-assembled micellar assemblies. (Reproduced with
permission from reference (35). Copyright 2017 American Chemical Society.) ... 19
Figure 2.10 Common acid-degradable functional groups and their degradation products. .... 20
Figure 2.11 Polyurethane containing acid-sensitive ketal groups in polymer backbone. ... 21
Figure 2.12 Chemical structure of poly(disulfide) with incorporation of paclitaxel via a PEG
linker. (Reproduced with permission from reference (48). Copyright 2012 Elsevier Ltd.) ... 22
Figure 2.13 Chemical structure of oxidation-responsive polymer and the particle degradation
mechanism in response to H2O2. (Reproduced with permission from reference (52).
Copyright 2012 American Chemical Society.) ... 23
Figure 2.14 a) Synthesis of a photo-degradable amphiphilic block copolymer containing
ortho-nitrobenzyl photolabile groups; b) Schematic illustration of a photo-degradable
micelle. (Reproduced with permission from reference (54). Copyright 2011 American
Chemical Society.) ... 24
Figure 2.15 Synthesis of a poly(benzyl carbamate)-based SIP and its depolymerization
mechanism following end-cap removal. ... 27
Figure 2.16 Modification of poly(benzyl carbamate) SIP via backbone structures and end-cap
design. ... 28
Figure 2.17 Chemical structure and depolymerization mechanism of poly(benzyl ether)s, and
xiii
elimination and decarboxylation reactions, and stimuli-responsive end-caps installed on this
category of SIPs. ... 31
Figure 2.19 SIPs that depolymerize via cyclization and elimination reactions: a) Replacement
of the carbamate from Figure 2.18 with a carbonate leads to faster depolymerization; b)
Replacement of the amine nucleophile in Figure 2.18 with a thiol leads to even faster
depolymerization; c) An SIP that depolymerizes entirely through a series of cyclization
reactions. ... 32
Figure 2.20 Depolymerization profile for linear SIPs involving an initial pseudo zero-order
domain followed by a gradual transition toward first-order behavior. (Reproduced with
permission from reference (60). Copyright 2013 American Chemical Society.) ... 34
Figure 2.21 General synthesis and depolymerization of polyacetals. ... 34
Figure 2.22 Synthesis and end-capping of PPA... 35
Figure 2.23 A patterned film that reveals a cylindrical hole when exposed to the
corresponding stimulus: a) Patterned plastic film design strategy; b) Photograph of the film
before stimulus; c) Photograph of the film after 15 min of exposure to stimulus. (Reproduced
with permission from reference (69). Copyright 2010 American Chemical Society.) ... 36
Figure 2.24 a) Schematic illustration for the preparation of PPA microcapsules by a flow
focusing microfluidic technique; b) SEM images of microcapsules before and after exposure
to stimulus. (Reproduced with permission from reference (75). Copyright 2013 American
Chemical Society.) ... 38
Figure 2.25 Synthesis and end-capping of PEtG-based SIPs. ... 41
Figure 2.26 a) Chemical structure of an amphiphilic polycarbamate-b-PEG; b) TEM image of
particles formed from this polymer; c) Nile red release from the particles over time as
measured by fluorescence spectroscopy. (Reproduced with permission from reference (72).
xiv
and reduction-responsive linkers and their self-assembly into vesicles. (Reproduced with
permission from reference (70). Copyright 2014 American Chemical Society.) ... 43
Figure 2.28 Chemical structure of an amphiphilic PEG-PEtG-PEG triblock copolymer; b)
TEM image of micelles formed from this polymer; c) Micelle disintegration study as
measured by 1H NMR in response to UV irradiation. (Reproduced with permission from
reference (62). Copyright 2014 American Chemical Society.) ... 44
Figure 3.1 Mass loss profiles for UV light irradiated (I) and non-irradiated (N-I)
PEtG-NVOC coatings under different conditions: a) 150 m thickness immersed at 20 oC in buffers
of pH 3-8; b) 150 m thickness immersed at varying temperatures in pH 7.0 buffer; c)
Varying film thicknesses from 25-150 m immersed in pH 7.0 buffer at 20 oC; d) 150 m
thickness immersed in soil with either 10, 20 or 30 mass % of pH 7.0 buffer at 20 oC; e) Film
thicknesses of 50 or 150 m in air (no aqueous immersion) at either 20 or 30 oC; f) Film
thicknesses of 50 or 150 m in air either exposed or not exposed to sunlight in a greenhouse.
In each experiment, the error bars represent the standard deviation of the measurements for
three samples. ... 63
Figure 3.2 a) Number average molar mass (Mn) of polymer remaining on the coating during
the mass loss study, as measured by SEC.; b,c) SEM images of the polymer coating after 5
days of immersion in 0.1 M, pH 7.0 phosphate buffer at 20 °C b) control without UV
irradiation and c) with UV irradiation. ... 65
Figure 3.3 Collection of depolymerized ethyl glyoxylate monomer over time after irradiation
of PEtG-NVOC and the corresponding experimental set-up. ... 68
Figure 3.4 Digital optical microscopy of ambient self-developed patterns: The metal masks
are shown in a) and b) while the corresponding 500 m and 20 m reservoirs fabricated from
these masks are shown in c) and d) respectively. ... 70
Figure 4.1 Schematic illustrating the end-cap cleavage and depolymerization process for
PEtG. ... 78
xv
and at various time points after H2O2 addition (spectra are offset to allow the progression
over time to be clearly observed); b-d) Percent depolymerization versus time in the presence
and absence of stimuli for b) PEtG-boronate with H2O2, c) PEtG-disulfide-a with DTT, and
d) PEtG-azobenzene with DTT. In each case, PEtG-control was also exposed to the stimulus
to confirm that the cleavage was specific to the end-cap. ... 92
Figure 4.4 Percent depolymerization versus time for a) PEtG-MMT and b) PEtG-DMT in the
absence and presence of varying concentrations of acetic acid. ... 96
Figure 4.5 Depolymerization versus time for PEtG-multi following exposure to UV light,
H2O2, Zn/acetic acid, and combinations of these stimuli. ... 99
Figure 4.6 1H NMR spectrum (400 MHz, 9:1 CD3CN:D2O) of PEtG-cross-linked a) before
and b) after UV irradiation. Note that peaks corresponding to derivatized cross-linker 16 are
not visible because assuming a 1:1 ratio of PEtG:16 in the network, there would be >100
EtGH molecules per derivatized 16. ... 101
Figure 4.7 a-b) SEC traces (RI detection) for a) PEtG-disulfide-a and b) PEtG-disulfide-b
following different sonication times; c) Changes in Mn for disulfide-a and
PEtG-disulfide-b following different sonication times. ... 104
Figure 5.1 Depolymerization of PEtG to ethyl glyoxylate (EtG), hydration to form ethyl
glyoxylate hydrate (EtGH), and hydrolysis to glyoxylic acid (GA) and ethanol. ... 116
Figure 5.2 Chemical structures of linker end-caps each containing a chloroformate, one or
more stimuli-responsive moieties, and a site for conjugation of PEG. ... 126
Figure 5.3 TEM images of nanoparticles formed from a) disulfide-PEG, b)
PEtG-nitrobenzyl-PEG, c) PEtG-boronate-PEG, and d) PEtG-multi-PEG. ... 132
Figure 5.4 Stimuli-responsive properties of PEtG-disulfide-PEG nanoparticles: a) % Initial
count rate versus time (measured by DLS) for nanoparticles exposed to varying
concentrations of DTT, b) % Depolymerization versus time for PEtG-disulfide-PEG
xvi
disulfide-PEG nanoparticles in the same solvent. Peaks corresponding to the
depolymerization products EtGH and the DTT adduct appear after the addition of 10 mM
DTT. ... 135
Figure 5.5 %Initial count rate, measured by DLS, versus time for nanoparticles and their
corresponding controls with or without stimuli: a) PEtG-nitrobenzyl-PEG nanoparticles with
UV light, b) PEtG-boronate-PEG with H2O2, c) PEtG-multi-PEG with H2O2, UV light, or
both. The study was carried out at pH 7.4, except for b) which was also performed at pH 5.0.
The temperature was 37 °C. Error bars represent the standard deviation on 3 samples. ... 137
Figure 5.6 Change in Nile red fluorescence intensity as an indicator of its release from
nanoparticles composed of a) disulfide-PEG, b) boronate-PEG, and c)
PEtG-nitrobenzyl-PEG in the presence and absence of their corresponding stimuli. Error bars
correspond to the standard deviation on three samples. ... 141
Figure 5.7 Release of Dox from PEtG-nitrobenzyl-PEG nanoparticles with and without UV
irradiation at a) pH 7.4 and b) pH 5.0. ... 142
Figure 5.8 Curcumin retention in a) PEtG-disulfide-PEG and b) PEtG-boronate-PEG
nanoparticles in the presence and absence of stimuli as well as their corresponding controls.
... 143
Figure 6.1 Chemical structure of the monomers used in this chapter ... 161
Figure 6.2 TEM images of particles formed from a) PEG2000; b)
PEtBuG-PEG5000; c) PEtMenG-PEG2000; d) PMenG-PEG750; e) PEG2000; f)
PEtGC-PEG5000. ... 168
Figure 6.3 Depolymerization of particles following UV light irradiation (or no light for
controls), monitored by DLS based on count rate. ... 169
Figure 6.4 Release of celecoxib over time for assemblies irradiated with UV light and for the
xvii
irradiation and b) after UV irradiation. ... 173
Figure 7.11H NMR spectra of PEtG-DA-Bn incubated in 9:1 CD3CN:D2O at 75 C. Spectra
are offset to allow the progression over time to be clearly observed. ... 195
Figure 7.2 a) Depolymerization of polymers in 9:1 CD3CN:D2O monitored by NMR
spectroscopy; Assembly degradation in pH 7.4 phosphate buffer monitored by b) DLS count
rate changes, c) Nile red fluorescence changes, and d) NMR spectroscopy. ... 196
Figure 7.3 TEM images of a) PEtG-DA-PEG750 vesicles, b) PEtG-DA-PEG5000 micelles,
c) unloaded IONPs, d) IONP-loaded PEtG-DA-PEG5000 micelles. ... 197
Figure 7.4 Bulk temperature, particle diameter, and count rate measured before, during, and
after magnetic hyperthermia using an in situ DLS for 35 mass% IONP-loaded a)
PEtG-DA-PEG5000 micelles and b) Micelle-control. ... 198
Figure 8.1 Structures of trityl end-capped PEtG. ... 205
Figure 8.2 Chemical structure of PEtG-control. ... 213
Figure 8.3 Mass loss profiles for end-capped PEtGs at different temperatures: a) PEtG-DMT;
b) PEtG-MMT; c) PEtG-AMT; d) PEtG-AT; d) PEtG-control. In each experiment, the error
bars represent the standard deviation of the measurements for three samples. ... 214
Figure 8.4 Variable temperature 1H NMR spectra of a) PEtG-AMT and b) PEtG-control at
different temperatures (15 min per temperature increment unless otherwise indicated).
Spectra are offset to allow the progression over time to be clearly observed. ... 215
Figure 8.5 Mechanism for temperature dependent depolymerization of trityl-capped PEtG.
... 216
Figure 8.6 a) and b) Color changes of different PEtG coatings containing either a) 0.1 wt%
Nile red or b) 0.5 wt% IR-780 incubated at 22 °C for different time periods. The rectangular
color patches are computer generated color samples that were sub-sampled from digital
xviii
for 24 h, the inserted pictures in c) are polymer coatings before and after incubation. ... 218
xix
Scheme 3.1 Chemical structure of PEtG-NVOC and its UV light-initiated depolymerization.
This is followed by hydrate formation and hydrolysis of the resulting ethyl glyoxylate in the
presence of water. ... 62
Scheme 4.1 Synthesis of chloroformate end-caps 4 - 6. ... 89
Scheme 4.2 Synthesis and end-capping of PEtG. ... 89
Scheme 4.3 Synthesis of multi-responsive end-cap 11. ... 97
Scheme 4.4 Synthesis of cross-linkable end-cap 15. ... 100
Scheme 4.5 Cross-linking of PEtG-trialkene and UV light triggered depolymerization of the resulting PEtG-cross-linked. ... 101
Scheme 5.1 Synthesis of end-cap 3. ... 127
Scheme 5.2 Synthesis of end-cap 4. ... 127
Scheme 5.3 Synthesis of end-capped PEtGs... 128
Scheme 5.4 Synthesis of PEG-PEtG-PEG copolymers a) PEtG-disulfide-PEG, b) PEtG-nitro-PEG, c) PEtG-boronate-PEtG-nitro-PEG, and d) PEtG-multi-PEG... 130
Scheme 6.1 Synthesis of polymers: a) PEtBuG; b) PMenG; c) PEtMenG; d) PEtGC. ... 163
Scheme 6.2 Synthesis of block copolymers via coupling of PEG-N3 with a) PEtBuG; b) PMenG; c) PEtMenG; d) PEtGC. ... 165
Scheme 7.1 Proposed cap cleavage and depolymerization mechanism of PEtG end-capped with a DA adduct (PEtG-DA). ... 183
Scheme 7.2 Synthesis of end-caps 4a and 4b. ... 193
xx
methoxy groups. ... 212
xxi
AIBN 2,2’azobis(2-methylpropionitrile)
ATR Attenuated total reflectance
Bu Butyl
BuG Butyl glyoxylate
Boc tert-butyloxylcarbonyl
BSA Bovine serum albumin
C Chloral
CuAAC Copper-assisted azide-alkyne cycloaddition
CR Counter rate
CAC Critical aggregation concentration
Cur Curcumin
DBTL Dibutyltin dilaurate
DA Diels-Alder
DLS Dynamic light scattering
DSC Differential scanning calorimetry
DEG Di(ethylene glycol)
DMEM Dulbecco’s modified eagle medium
Dox Doxorubicin
DTT Dithiothreitol
DMAP 4-Dimethylaminopyridine
DMF N, N’-Dimethylformamide
DMSO Dimethyl sulfoxide
Đ Dispersity
EDC 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide
EI Electron impact
ESI Electrospray mass spectrometer
Et Ethyl
EtG Ethyl glyoxylate
EtGH Ethyl glyoxylate hydrate
FBS Fetal bovine serum
xxii
GA Glyoxylic acid
GO Graphene oxide
HRMS High-resolution mass spectrometry
IONPs Iron oxide nanoparticles
LCST Lower critical solution temperature
Me Methyl
MenG L-menthyl glyoxylate
MFH Magnetic field hyperthermia
Mn Number average molecular weight
Mw Weight average molecular weight
MWCO Molecular weight cut-off
MSDS Material safety data sheets
NMDEA N-methyldiethanolamine
NEt3 Triethyl amine
NMR Nuclear magnetic resonance
NIR Near-infrared
NVOC 6-Nitroveratryloxycarbonyl
NVOC-Cl 6-Nitroveratryloxycarbonyl chloride
OPA o-phthalaldehyde
PAA Poly(acrylic acid)
PBuG Poly(butyl glyoxylate)
PDI Polydispersity index
PDMAEMA Poly(dimethylaminoethyl methacrylate)
PDMA Poly(N,N’-dimethylacrylamide)
PE Polyethylene
PEtG Poly(ethyl glyoxylate)
PEtBuG Poly[(ethyl glyoxylate)-co-(butyl glyoxylate)]
PEtGC Poly[(ethyl glyoxylate)-co-chloral]
PEtMenG Poly[(ethyl glyoxylate)-co-(menthyl glyoxylate)]
PEG Poly(ethylene glycol)
xxiii
PMeG Poly(methyl glyoxylate)
PMenG Poly(menthyl glyoxylate)
PMA Poly(methyl acrylate)
PMMA Poly(methyl methacrylate)
PNIPAM Poly(N-isopropylacrylamide)
PPA Poly(phthalaldehyde)
PS Polystyrene
PTEGMA Poly[tri(ethylene glyocol) monoethyl ether methacrylate]
P2VP Poly(2-vinylpyridine)
PVS Poly(vinyl sulfone)
QDs Quantum dots
RAFT Reversible addition-fragmentation chain transfer
ROS Reactive oxygen species
SANS Small angle neutron scattering
SEC Size exclusion chromatography
SEM Scanning electron microscopy
SIPs Self-immolative polymers
SRPs Stimuli-responsive polymers
TBDMS tert-butyldimethylsilyl
TGA Thermogravimetric analysis
THF Tetrahydrofuran
TEM Transmission electron microscopy
To On-set degradation temperature
Tc Ceiling temperatures
Tg Glass transition temperature
Tm Melting point temperature
Chapter 1
1
Introduction
1.1
Overview
Polymers are large molecules composed of many repeating units linked together via
covalent bonds. They can be natural, such as polysaccharides, proteins, and nucleic acids,
or synthetic, such as polyethylene (PE), polystyrene (PS), and nylon.1 Their high molar
mass provides polymers with properties that are significantly different from small
molecules.2 In addition, the physical properties of polymers can be tuned significantly to
achieve specific functions by changing the chemical structures of the polymer backbones
and side groups, as well as their molecular weight, branching, or tacticity.2 For example,
PE is one of the most commonly encountered plastics in our daily lives. Depending on the
molecular weight or chain branching, its physical properties change dramatically.
Ultra-molecular-weight polyethylene has a yield strength that is comparable with
high-strength steels, therefore it can be used in bulletproof vests.3 However, low-density
polyethylene, which contains a high degree of branching, is more frequently used in
packaging due to its low tensile strength.4
Since the first successful commercialization of the thermoplastic polymer-Nylon,5 by
Wallace Carothers at Dupont’s research facility in 1935, polymeric materials have offered
great possibilities for the development of human society in the past century. A series of
polymeric materials have been successfully commercialized in 1940s-1980s.6 However,
since the 1990s, a significant amount of research effort has shifted to polymeric materials
that can respond to their environmental conditions or external stimuli. These polymers are
usually referred to as stimuli-responsive polymers (SRPs) or “smart” polymers.7 SRPs can
receive external signals and exhibit responses by changing their physical properties, such
as shape, color, solubility, and even chemical structure (e.g., cleavage of side groups or
polymer backbone). The chemical or physical property changes associated with SRPs
endow them with a series of novel applications that cannot be achieved by traditional
including biosensors,8 smart coatings,9 drug delivery vehicles,10-12 self-healing,13 and
shape-memory materials.14 However, conventional SRPs usually need significant amounts
of stimuli for a clear response, and one specific polymer backbone can usually respond to
only one stimulus.15 These limitations create a bottleneck for the applications of many
current SRPs.
Self-immolative polymers (SIPs), are a new class of SRPs. They consist of a polymer
backbone and a stimuli-responsive end-cap at one (or both) polymer termini.16-17 Once the
end-cap detects an external signal, the decomposition of the end-cap leads to complete
collapse of the polymer backbone via end-to-end depolymerization. Therefore, compared
with traditional stimuli-responsive polymers, SIPs can easily achieve responsiveness to a
series of different stimuli via changes in the end-cap rather than completely re-engineering
a new polymer backbone. Via this simple strategy, SIPs that respond to light,18-19 heat,20
pH,21 ultrasound,22 oxidizing and reducing conditions,23 and chemicals24 have been
developed. In addition, the response structure of SIPs allows signal amplification, as one
equivalent of stimulus can lead to complete polymer depolymerization and the generation
of hundreds and even thousands of monomers or depolymerization products. In the past l0
years, SIPs including polycarbamates, polycarbonates, poly(benzyl ether)s, and polyacetals
have been reported.15, 25-26However, many of these SIPs produce toxic depolymerization
products such as quinone methides27 or o-phthalaldehyde,28 which may hinder their use in
some applications.
Polyglyoxylates (PGs) were introduced by our group as a new class of polyacetal-based
SIPs in 2014 (Figure 1.1).29 PGs can be readily prepared from inexpensive commercially
available glyoxylates such as ethyl glyoxylate or from other simple glyoxylates prepared
from readily available starting materials such as maleic or fumaric acid via ozonolysis of
the corresponding diesters. Following depolymerization, PGs ultimately degrade to the
alcohol and glyoxylic acid, a metabolic intermediate in the important glyoxylate cycle.30
This is an anabolic variant of the tricarboxylic acid cycle that occurs in plants, bacteria,
protists, and fungi. Glyoxylic acid is also a metabolic byproduct of mammalian
suggesting that the depolymerization products of poly(ethyl glyoxylate) (PEtG) are well
tolerated in the environment by plants and in mammalian models.32 The innocuousness of
these depolymerization products position PGs as ideal materials for applications in vivo or
in the environment.
Figure 1.1 Structure of polyglyoxylates and the depolymerization mechanism after
end-cap removal. (Reproduced with permission from reference (29). Copyright 2014
American Chemical Society.)
1.2
Research objectives
Our group has previously reported the synthesis of a small number of different alkyl and
benzyl polyglyoxylates end-capped with a UV-responsive trigger.29 We also prepared
amphiphilic block copolymers of triggerable hydrophobic poly(ethyl glyoxylate) PEtG
with hydrophilic poly(ethylene glycol) (PEG) and showed that these materials
self-assembled into micelles. Furthermore, when the micelles were exposed to UV light, the
hydrophobic block depolymerized, decomposing the micelle.26 This PG-based
UV-responsive SIP had a fast depolymerization rate in solution. However, its depolymerization
profiles in the solid state under different conditions, such as different pHs, temperatures,
coating thicknesses, and even different environmental media, should be explored for
practical applications. In addition, the UV-responsive PGs may find application in fields
where UV light can be used as a trigger, such as agricultural materials and industrial
photolithography. However, for applications such as drug delivery, UV-responsive SIPs
Therefore, this thesis aims to expand the applications of polyglyoxylate-based SIPs by
increasing the flexibility of the triggering stimuli initiating decomposition by incorporating
different end-caps capable of responding to additional environmental cues besides UV
light. As an extension of this, the self-assembly of PG-based amphiphilic block copolymers
into nano-carriers (such as micelles and vesicles) for on-demand drug release has also been
explored.
1.3
Thesis outline
The thesis is divided into 9 chapters. A broad literature review of all classes of SRPs is
presented in Chapter 2. Following the review, Chapter 3-8 will describe six projects
towards the synthesis, modifications and applications of PG based self-immolative
polymers. The projects details are as follows.
Chapter 3 describes the solid-state depolymerization of UV-sensitive PEtG under different
conditions. This thesis chapter will explore the effects of different environmental
conditions such as pH, temperature, and coating thickness on the depolymerization of PEtG
coatings with the aim to fully understand the depolymerization process in the solid state
and thus understand potential applications of these materials as coatings.
Chapter 4 includes work aimed at increasing the flexibility of the triggering stimuli
initiating decomposition by incorporating different end-caps. New stimuli include
biologically relevant signals such as changes in the concentrations of oxidizing or reducing
agents. An oxidation-sensitive end-cap incorporating a pinacol borane that cleaves in the
presence of hydrogen peroxide and a reduction-sensitive disulfide end-cap that cleaves in
the presence of biologically relevant thiols that would be appropriate for targeting the
reductive environments of cancer tumors were prepared and studied. Novel
multi-responsive end-caps that respond to very different stimuli including combinations of UV
light, hydrogen peroxide, and reducing conditions simultaneously will be introduced.
Furthermore, a cross-linker end-cap that allows SIPs to form cross-linked networks while
at the same time depolymerizing in response to external stimuli, such as UV light will also
Chapter 5 explores the development and application of PG-based nano-assemblies. New
linker end-caps allowed different stimuli-responsive PEtGs to form amphiphilic block
copolymers that were capable of self-assembling to form nano-sized particles in aqueous
solution. Specifically, based on the multi-responsive PEtGs in Chapter 4, the design,
synthesis and self-assembly of H2O2, reduction, and dual-responsive (both UV light and
H2O2) micelles will be described. The signal amplification properties of PEtGs was also
explored in this context. Moreover, loading of anti-cancer drugs into PG-based
nano-carriers and their release in response to stimuli were examined. This work will expand the
applications of PEtG-baseds SIPs into the biomedical field for the release of drugs and
other molecules on demand.
Chapter 6 explores different monomer combinations to increase the hydrophobicity of
PG-based particles and thereby improve their hydrophobic drug loading capabilities.
Specifically, ethyl glyoxylate was copolymerized with n-butyl glyoxylate, choral, and
menthol glyoxylate, respectively, and these monomers were also polymerized to form
hydrophobic homopolymers. These PGs were further coupled with hydrophilic PEG to
form amphiphilic block copolymers. The loading capabilities of the nanoparticles formed
from these copolymers were examined by incorporation of the hydrophobic
drug-celecoxib.
Chapter 7 introduces the design and synthesis of a new generation of thermo-responsive
end-caps based on Diels-Alder adducts that allow SIPs to depolymerize in response to
environmental temperature changes. Furthermore, self-assembly and disassembly of
thermo-responsive micelles and vesicles formed from these polymers in response to direct
and indirect heat were also explored.
Chapter 8: This Chapter explores the temperature dependent depolymerization properties
of triphenylmethyl end-capped PEtG films, and their potential applications in smart
packaging.
Finally, a general discussion with conclusions outlining the significance, limitations and
1.4
References
(1) Gao, Y.; Wei, M.; Li, X.; Xu, W.; Ahiabu, A.; Perdiz, J.; Liu, Z.; Serpe, M. J.
Stimuli-responsive polymers: Fundamental considerations and applications. Macromol.
Res. 2017, 25, 513-527.
(2) Stein, R. S.; Tobolsky, A. V. An investigation of the relationship between polymer
structure and mechanical properties: Part I: relationship between structure, mechanical
properties, and birefringence. Text. Res. J. 1948, 18, 201-223.
(3) Kelly, J. M. Ultra-high molecular weight polyethylene. J. Macromol. Sci., Polym. Rev.
2002, 42, 355-371.
(4) Raj, B. Low density polyethylene/starch blend films for food packaging applications.
Adv. Polym. Technol. 2004, 23, 32-45.
(5) Kauffman, G. B. Wallace Hume Carothers and nylon, the first completely synthetic
fiber. J. Chem. Educ. 1988, 65, 803-808.
(6) Brandrup, J.; Immergut, E. H.; Grulke, E. A.; Abe, A.; Bloch, D. R., Polymer
Handbook. Wiley New York etc: 1989; Vol. 7.
(7) Stuart, M. A. C.; Huck, W. T.; Genzer, J.; Müller, M.; Ober, C.; Stamm, M.;
Sukhorukov, G. B.; Szleifer, I.; Tsukruk, V. V.; Urban, M. Emerging applications of
stimuli-responsive polymer materials. Nat. Mater. 2010, 9, 101-113.
(8) Hu, J.; Liu, S. Responsive polymers for detection and sensing applications: current
status and future developments. Macromolecules 2010, 43, 8315-8330.
(9) Nath, N.; Chilkoti, A. Creating “smart” surfaces using stimuli responsive polymers.
Adv. Mater. 2002, 14, 1243-1247.
(10) Mura, S.; Nicolas, J.; Couvreur, P. Stimuli-responsive nanocarriers for drug delivery.
Nat. Mater. 2013, 12, 991-1003.
(11) Ganta, S.; Devalapally, H.; Shahiwala, A.; Amiji, M. A review of stimuli-responsive
nanocarriers for drug and gene delivery. J. Controlled Release 2008, 126, 187-204.
(12) Cabane, E.; Zhang, X.; Langowska, K.; Palivan, C. G.; Meier, W. Stimuli-responsive
polymers and their applications in nanomedicine. Biointerphases 2012, 7:9.
(13) Habault, D.; Zhang, H.; Zhao, Y. Light-triggered self-healing and shape-memory
(14) Meng, H.; Li, G. A review of stimuli-responsive shape memory polymer composites.
Polymer 2013, 54, 2199-2221.
(15) Fan, B.; Gillies, E. R. Self-Immolative Polymers. Encycl. Polym. Sci. Technol. 2015,
1-35.
(16) Sagi, A.; Weinstain, R.; Karton, N.; Shabat, D. Self-immolative polymers. J. Am.
Chem. Soc. 2008, 130, 5434-5435.
(17) Roth, M. E.; Green, O.; Gnaim, S.; Shabat, D. Dendritic, oligomeric, and polymeric
self-immolative molecular amplification. Chem. Rev. 2015, 116, 1309-1352.
(18) de Gracia Lux, C.; McFearin, C. L.; Joshi-Barr, S.; Sankaranarayanan, J.; Fomina, N.;
Almutairi, A. Single UV or Near IR triggering event leads to polymer degradation into
small molecules. ACS Macro Lett. 2012, 1, 922-926.
(19) Liu, G.; Wang, X.; Hu, J.; Zhang, G.; Liu, S. Self-immolative polymersomes for
high-efficiency triggered release and programmed enzymatic reactions. J. Am. Chem. Soc. 2014,
136, 7492-7497.
(20) Peterson, G. I.; Church, D. C.; Yakelis, N. A.; Boydston, A. J. 1, 2-oxazine linker as
a thermal trigger for self-immolative polymers. Polymer 2014, 55, 5980-5985.
(21) Esser-Kahn, A. P.; Sottos, N. R.; White, S. R.; Moore, J. S. Programmable
microcapsules from self-immolative polymers. J. Am. Chem. Soc. 2010, 132, 10266-10268.
(22) Diesendruck, C. E.; Peterson, G. I.; Kulik, H. J.; Kaitz, J. A.; Mar, B. D.; May, P. A.;
White, S. R.; Martínez, T. J.; Boydston, A. J.; Moore, J. S. Mechanically triggered
heterolytic unzipping of a low-ceiling-temperature polymer. Nat. Chem. 2014, 6, 623-628.
(23) DeWit, M. A.; Beaton, A.; Gillies, E. R. A reduction sensitive cascade biodegradable
linear polymer. J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 3977-3985.
(24) Zhang, H.; Yeung, K.; Robbins, J. S.; Pavlick, R. A.; Wu, M.; Liu, R.; Sen, A.;
Phillips, S. T. Self-powered microscale pumps based on analyte-initiated depolymerization
reactions. Angew. Chem., Int. Ed. 2012, 51, 2400-2404.
(25) Phillips, S. T.; DiLauro, A. M. Continuous head-to-tail depolymerization: an emerging
concept for imparting amplified responses to stimuli-responsive materials. ACS Macro
(26) Fan, B. Polyglyoxylates: a new class of triggerable self-immolative polymers. 2014.
Electronic Thesis and Dissertation Repository. 2568. https://ir.lib.uwo.ca/etd/2568.
(27) Monks, T. J.; Jones, D. C. The metabolism and toxicity of quinones, quinonimines,
quinone methides, and quinone-thioethers. Curr. Drug Metab. 2002, 3, 425-438.
(28) Peterson, G. I.; Boydston, A. J. Kinetic analysis of mechanochemical chain scission
of linear poly (phthalaldehyde). Macromol. Rapid Commun. 2014, 35, 1611-1614.
(29) Fan, B.; Trant, J. F.; Wong, A. D.; Gillies, E. R. Polyglyoxylates: a versatile class of
triggerable self-immolative polymers from readily accessible monomers. J. Am. Chem.
Soc. 2014, 136, 10116-10123.
(30) Springsteen, G.; Yerabolu, J. R.; Nelson, J.; Rhea, C. J.; Krishnamurthy, R. Linked
cycles of oxidative decarboxylation of glyoxylate as protometabolic analogs of the citric
acid cycle. Nat. Commun. 2018, 9, 91.
(31) Lorenz, M. C.; Fink, G. R. The glyoxylate cycle is required for fungal virulence.
Nature 2001, 412, 83-86.
(32) Belloncle, B.; Bunel, C.; Menu-Bouaouiche, L.; Lesouhaitier, O.; Burel, F. Study of
the degradation of poly (ethyl glyoxylate): biodegradation, toxicity and ecotoxicity assays.
Chapter 2
2
Stimuli-responsive polymers (SRPs)
SRPs are polymers that can detect external signals and respond with changes in physical
properties (e.g., shape, color, solubility) or chemical structures (e.g., cleavage of side
groups or polymer backbone).1 The chemical or physical property changes associated with
SRPs endow them with a series of novel applications that cannot be achieved by traditional
materials.2-3 Therefore, SRPs have attracted significant attention from both academia and
industry. SRPs can be classified into two main categories according to their physical and
chemical property changes. However, depending on their degradability, SRPs can also be
separated into 1) general non-degradable SRPs (Figure 2.1a), and 2) stimuli-responsive and
degradable polymers (Figure 2.1b, 2.1c).
Figure 2.1 Schematic representations of SRPs: a) General non-degradable SRPs that
change in physical properties in response to a stimulus; b) SRPs that can entirely
degrade in response to a stimulus stoichiometrically; c) Self-immolative polymers
(SIPs) as a sub-class of SRPs that can depolymerize in response to a stimulus in an
amplified manner.
Over the past decade, stimuli-responsive and degradable polymers have evolved into two
different classes. The first class of SRPs can entirely degrade stoichiometrically in response
to stimuli (Figure 2.1b). However, a key limitation of these stimuli-responsive and
complete degradation.4 In addition, for each different stimulus, a completely new polymer
backbone is usually required, which significantly increases the barrier for practical
applications. To address the limitations associated with conventional stimuli-responsive
and degradable polymers, new efforts have been directed to the amplified degradation of
polymeric materials in response to external stimuli in the past 10 years. This new
generation of stimuli-responsive and degradable polymers is usually referred as
“self-immolative polymers” (SIPs) (Figure 2.1c).5
This chapter systematically reviews the development and applications of
stimuli-responsive polymers (SRPs) in three main sections: 1) General non-degradable SRPs that
undergo changes in physical properties in response to external stimuli; 2) SRPs that can
entirely degrade stoichiometrically in response to stimuli; 3) Self-immolative polymers
(SIPs) as a sub-class of SRPs that can depolymerize in response to stimuli in an amplified
manner.
2.1
General non-degradable SRPs
The general non-degradable SRPs are polymers that can detect external stimuli and respond
in form of changes in physical properties such as shape, solubility, or color. The triggering
stimuli can be light, heat, specific chemicals or gases, or changes in pH, depending on the
responsive sites on polymer structures. Each stimulus has its own advantages and can
usually meet the demand for a specific application. For example, light can be applied in a
situation that does not require external additives and can be controlled spatiotemporally.
Heat can be readily applied from outside of polymer environment. Gases are easy to add
and remove, especially, in large volume operations and are of great interest in industrial
applications. This section reviews four representative non-degradable SRPs that can
respond to light, heat, pH changes, and gases.
2.1.1
Light-responsive polymers
Light has been one of the most extensively investigated stimuli for controlling polymer
properties and functions.6-8 It can be easily applied and regulated remotely and possesses
external additives. Reversible light-controllable polymers have been heavily investigated
since the 1980s, and the potential applications of these polymers in optical-to-mechanical
conversion actuators and polymer nanoparticles for drug delivery have been explored.
In 1980, by incorporating aromatic azo chromophores as cross-linkers for rubbery
poly(ethyl acrylate), Eisenbach and coworkers demonstrated that the rubbery network
could contract and extend when it was irradiated with different wavelengths of light.9 This
photomechanical effect was believed to be caused by the conformational change resulting
from the trans-cis isomerization of the aromatic azo cross-linker. At the same time,
Riordan’s group observed the same effects when they incorporated the aromatic azo
functional group into polyamides, specifically,
poly(3,3’-zaodibenzoyl-trans-2,5-dimethylpiperazene) and poly(4,4’-azodibenzoyl-trans-poly(3,3’-zaodibenzoyl-trans-2,5-dimethylpiperazene).10 The
photomechanical effect of aromatic azo compounds is an interesting phenomenon as it
allows the direct conversion of light into mechanical energy. Now, it has been well
established that aromatic azo compounds can undergo trans-cis isomerization when
irradiated by light with a wavelength of ~330-380 nm, and that this process is completely
reversible.11 The cis isomer can convert back to the trans isomer when the compound is
exposed to light with wavelength of ~420 nm, depending on the specific structure (Figure
2.2a)
Figure 2.2 Representative light-responsive molecules applied in stimuli-responsive
polymers: a) Cis-trans transformation of aromatic azo compounds, b) hydrophobic
spiropyran to hydrophilic merocyanine transformations under different wavelengths
Following Eisenbach and Riordan’s pioneering work, much research was devoted to
aromatic azo-containing polymers in order to develop them as light-triggerable actuators.
For example, Zhao and coworkers recently reported a light-driven actuator based on an
azobenzene-containing liquid crystalline polymer network (Figure 2.3a).12 In this work,
they demonstrated that they could use UV light to generate mechanical force using light to
mechanical energy conversion and also light-triggered release of prestored strain energy in
a azobenzene-containing liquid crystalline polymer network that was synthesized from
4,4’-diglycidyloxyazobenzene and dodecanedioic acid. They achieved large and tunable
photoinduced contractile stress up to 7 MPa, which was much higher than previously
reported. Moreover, the azobenzene-containing polymer enabled continuous motions of
large rolling objects under irradiation of UV light (Figure 2.3b, c).
Figure 2.3 a) Synthesis procedure for the azobenzene liquid crystalline polymer
network; b) Schematic and c) photographs showing light-driven forward moving of a
“vehicle” equipped with a spring-like “motor”. (Reproduced with permission from
reference (12). Copyright 2017 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.)
In addition to aromatic azo compounds, spiropyran (Figure 2.2b) is another well-known
photochromic molecule that has been widely studied in photo-responsive dynamic
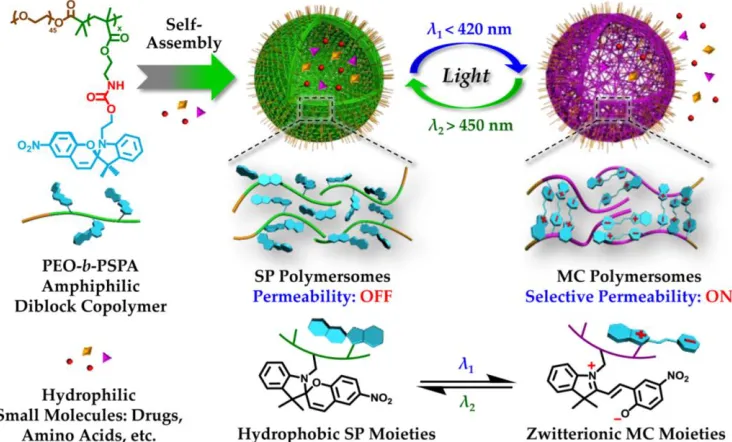
materials. Spiropyran can respond to light irradiation (wavelength < 420 nm) and undergo
reversible isomerization from a colorless hydrophobic spiropyran to a colored hydrophilic
by light with wavelengths above 450 nm. Recognizing the possible application of this
molecule for drug delivery, in 2015 Liu’s group reported the fabrication of photochromic
vesicles that exhibited reversible changes in bilayer permeability upon triggering by
UV-Vis light irradiation (Figure 2.4).14 The photochromic amphiphilic block copolymer was
poly(ethylene glycol)-b-PSPA (PEG-b-PSPA) diblock copolymer, where SPA was a
spiropyran-based monomer containing a carbamate linkage. Upon self-assembly into
vesicles, the spiropyran moieties within the vesicle bilayers underwent reversible
photo-triggered isomerization between spiropyran and zwitterionic merocyanine states. The
microstructures of both vesicles were stabilized by multiple cooperative noncovalent
interactions including hydrogen bonding, π-π stacking, and zwitterionic interactions.
Interestingly, they found that the UV-actuated merocyanine vesicle possessed both
sustained release upon short UV irradiation and on-demand switchable release under
alternating UV-Vis light irradiation.
Figure 2.4 Photochromic polymersomes exhibiting photo-switchable and reversible
bilayer permeability. (Reproduced with permission from reference (14). Copyright
2.1.2
Thermo-responsive polymers
Temperature change as a stimulus for polymers has also played a key role among all
available stimuli, as it can be readily applied from outside of the polymer environment.15
In particular, poly(N-isopropylacrylamide) (PNIPAM) is one of the most studied
thermo-responsive polymers that undergoes a phase transition in solution with temperature
changes.16 Once the temperature is above a certain limit, the polymer chain transforms
from a coil state to a globular state to minimize the free energy of the system. This leads to
the macroscopic phenomenon of an increase in solution turbidity. This temperature
associated phase transition is usually called the lower critical solution temperature (LCST).
Besides PNIPAM, there are several other polymers such as, poly[tri(ethylene glyocol)
monoethyl ether methacrylate] (PTEGMA),17 poly(dimethylaminoethyl methacrylate)
(PDMAEMA),18 and poly(2-isopropyl-2-oxazoline) (PIPOZ)19 that have the property of an
LCST as well (Figure 2.5). Because of this unique property, thermo-responsive polymers
have been widely studied for numerous applications such as drug delivery,20 catalysis,21
tissue engineering,22 and surface engineering.23
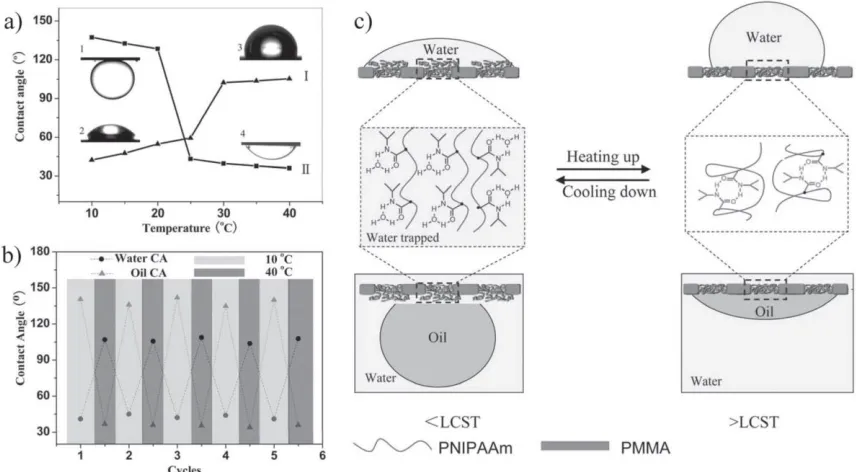
Figure 2.5 Chemical structures of some typical thermo-responsive polymers.
In 2013, Liu and coworkers reported a temperature-controllable dual water/oil on-off
switch mesh allowing the separation of water and oil by controlling the temperature.23 The
steel mesh was coated with block copolymer poly(methyl methacrylate)
(PMMA)-b-PNIPAM. Due to the hydrophilic-hydrophobic transition around the LCST of PNIPAM
and the resultant surface roughness change, the mesh could be open to water and closed to
2.6). This work provided a smart solution to the controllable separation of water and oil
mixtures.
Figure 2.6 a) Temperature dependence of water and oil contact angles for a
PMMA-b-PNIPAM film; b) Reversible water and oil contact angle transition of block
copolymer film at different temperatures; c) Diagram of reversible formation of
intermolecular hydrogen bonding between PNIPAM chains and water below and
above the LCST. (Reproduced with permission from reference (23). Copyright 2013
WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.)
2.1.3
pH-responsive polymers
Change in pH is another stimulus that can trigger the change of polymer physical properties.
For some organic functional groups, such as organic acids, pyridines, and amines,
environmental pH changes lead to the protonation or deprotonation of these functional
groups and consequently result in solubility changes of these molecules. Therefore,
polymers containing these functional groups also exhibit pH-dependent solubility.
poly(2-vinylpyridine) (P2VP) (pKa = 3.0), and poly(2-N,N’-dimethylaminoethyl methacrylate)
(PDMAEMA) (pKa= 7.5) (Figure 2.7).
Figure 2.7 Chemical structures of some representative pH-responsive polymers.
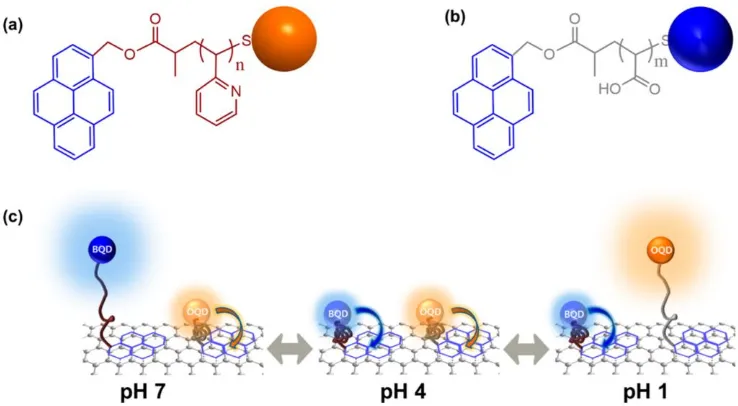
Based on the properties of pH-responsive polymers, in 2014 Kim and coworkers reported
a versatile platform for a highly stable and wide-range pH sensor that could respond to pH
variations from 1 to 7.24 This sensor platform consisted of the pH-responsive polymers
PAA and P2VP, and quantum dots (QDs). The PAA chain was grafted onto the surface of
blue-colored cadmium sulfide/zinc sulfide QDs, and the P2VP chain was grafted onto the
surface of orange-colored cadmium selenide/zinc sulfide QDs. The hybrids were deposited
on the surface of single graphene oxide (GO) sheets via π-π staking interactions between
the pyrene functional groups on the polymer termini and the basal plane of the GO surfaces.
The distances between the two color-emitting QDs and the GO were controlled by the
linker polymers PAA and P2VP. The sensing scheme is shown in Figure 2.8. Specifically,
when the pH was lower than 3, the protonation of P2VP led to the swelling and extension
of polymer chains, while the PAA chains were protonated and less soluble in water,
resulting in their collapse and attachment to the GO. This led to the quenching of blue
colored QDs, and consequently the orange colored QDs on P2VP dominated the emission.
When the pH was higher than 4.5, the deprotonation of PAA led to the swelling and
extension of PAA and the collapse of P2VP, so emission from the blue QDs dominated.
When the pH was between these two values, both QDs emitted light and therefore white
Figure 2.8 Structures of a) P2VP-QD and b) PAA-QD; c) Schematic illustration of the
conformation and behavior of QD-GO at a given pH value. (Reproduced with
permission from reference (24). Copyright 2014 American Chemical Society.)
In addition to pH sensors, pH-responsive polymers have also been exploited to deliver
drugs or genes to specific tissues and trigger the release of payloads at target sites. The
basic theory behind this application is that healthy tissues have a pH of 7.4, whereas in
inflamed tissues or tumors the environment is slightly acidic. These pH variations can lead
to solubility and morphology changes of pH-responsive polymers, enabling the delivery of
the payload.25-26
2.1.4
Gas-responsive polymers
Gases can be easily added and removed from a system, especially, in large volume
operations. Therefore, gaseous triggers are of great interest in industrial applications.
Several gaseous triggers have been reported so far including CO2,27 CO,28 NO,29 and H2S.30
Numerous applications have been explored for these gas-responsive polymers, such as drug
delivery vehicles, cell signaling systems, microgels, and nanoreactors.31 As a non-toxic and
abundant gas, CO2 is the most studied gas trigger for gas-responsive polymers. A number
of CO2-reactive functional groups have been explored including tertiary amines, amidines,
as those in PDMAEMA (Figure 2.7) are the most frequently explored because the pKa
values of polymeric tertiary amines typically range from 6.5 to 8.1.34 The protonation and
deprotonation of this class of polymers can readily occur upon the addition or removal of
CO2 through bubbling of CO2 or an inert gas (nitrogen or argon) respectively, resulting in
solubility changes of the polymer.
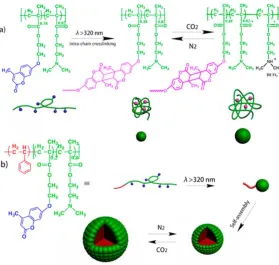
For example, in 2017 Zhao and coworkers reported CO2-responsive polymer nanoparticles
and micellar aggregates as gas-controlled nanoreactors for gold nanoparticle synthesis with
the capability to tune the gold nanoparticle size and formation rate.35 The nanoparticles
were prepared from the random copolymer poly{(N,N’-dimethylaminoethyl
methacrylate)-co-4-methyl-[7-(methacryloyl)oxyl-ethyl-oxyl]coumarin} (P(DMAEMA-co-CMA).
Tadpole-like single-chain nanoparticles were prepared from an amphiphilic block
copolymer of PS-b-P(DMAEMA-co-CMA). (PS represents polystyrene) (Figure 2.9).
Both particles underwent reversible swelling/shrinking with CO2/N2 stimulation. In
addition, they found the rate of gold nanoparticle formation using these particles as
nanoreactors increased under CO2 stimulation and slowed down by bubbling with N2. CO2
-induced swelling likely provided easier access of AuCl4 − counterions into the nanoreactors
for association with protonated amine groups, and residual non-protonated tertiary amine
groups led to AuCl4 − in-situ reduction to zerovalent gold. They also demonstrated that the
size of the gold nanoparticles could be controlled by the amount of CO2 in solution via the
Figure 2.9 Schematic illustration of: a) Preparation of P(DMAEMA-co-CMA)
single-chain nanoparticles through intrasingle-chain photo-cross-linking and the gas-switchable
size change of nanoparticle in aqueous solution; b) Preparation of tadpole-like
PS-b-P(DMAEMA-co-CMA) nanoparticles and their gas-responsive self-assembled
micellar assemblies. (Reproduced with permission from reference (35). Copyright
2017 American Chemical Society.)
2.2
Stimuli-responsive and degradable polymers
In addition to the physical property changes that occur in non-degradable SRPs as
described above, there is another class of SRPs that can undergo complete backbone
degradation in response to specific stimuli. In general, there are three major class of
stimuli-responsive and degradable polymers including 1) acid-degradable polymers; 2)
redox-degradable polymers; 3) photo-degradable polymers. In this section, examples and
applications for these three classes of stimuli-responsive and degradable polymers will be
2.2.1
Acid-degradable polymers
Acid-responsive and degradable polymers are usually designed with the incorporation of
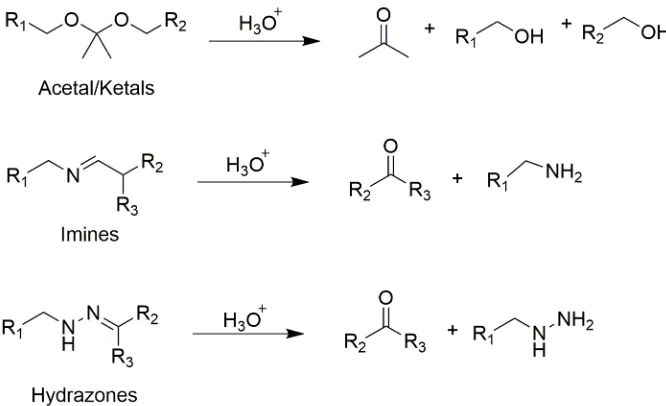
acid-sensitive functional groups such as acetals, ketals, imines, and hydrazones (Figure
2.10).36 Despite the structural diversity of these groups, they all tend to be stable or have
slow degradation at neutral pH and are prone to degrade much faster at acidic pH. These
acid-responsive and degradable polymers are commonly used to form drug delivery
vehicles for the targeted and triggered release of therapeutics, as healthy tissues have a pH
around 7.4, whereas inflamed tissues or tumors have mildly acidic pHs ranging from
5.7-7.8. The endosomal and lysosomal compartments of cells also have acidic pHs ranging
from 4.5-5.5.3,36-37
Figure 2.10 Common acid-degradable functional groups and their degradation
products.
The degradation of acetals and ketals has been a subject of interest since 1960s,38-39 and
has attracted significant attention in the past decades, as these moieties yield charge-neutral
and potentially non-toxic products upon cleavage.40 In addition, the hydrolysis rate is
linearly proportional to the H+ concentration, ensuring a predictable response to pH
changes. Therefore, acetal- and ketal-based polymers have been the most widely studied