THE ROLE OF MACROPHAGE GLUT1-MEDIATED GLUCOSE METABOLISM IN ATHEROSCLEROSIS
Liyang Zhao
A thesis submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Master of Science in the Department of
Nutrition in the Gillings School of Global Public Health.
Chapel Hill 2015
iii ABSTRACT
Liyang Zhao: The Role of Macrophage GLUT1-Mediated Glucose Metabolism in Atherosclerosis
(Under the direction of Liza Makowski)
Macrophages play a key role in the pathogenesis of atherosclerosis. We recently created a novel murine model with GLUT1 specifically deleted from monocyte/macrophages. Preliminary results from our lab suggest that macrophages lacking GLUT1 reduce the pro-inflammatory response. We therefore hypothesized that macrophages with GLUT1 deletion will have reduced pro-inflammatory activation during atherogenesis, which will reduce formation of atherosclerosis in a mouse model lacking the LDL receptor (Ldlr-/-). We transplanted bone marrow from Glut1fl/fl
or Glut1MΦ-/- mice into Ldlr-/- mice and fed mice a Western Diet (WD) for 12 weeks. Glut1 MΦ-/-Ldlr-/- mice exhibited significantly less plasma total cholesterol and LDL cholesterol compared to Glut1fl/flLdlr-/- mice. Additionally, our results demonstrated that mice with macrophages lacking GLUT1 displayed more and larger necrotic cores in atherosclerotic lesions compared to floxed transplanted controls. These results suggest that macrophage glucose metabolism
iv
ACKNOWLEDGEMENTS
v
TABLE OF CONTENTS
LIST OF FIGURES ... vi
LIST OF ABBREVIATIONS ... vii
CHAPTER I - BACKGROUND ...1
Introduction ... 1
Risk factors of Atherosclerosis ... 4
Mouse Models of Atherosclerosis ... 7
Roles of Macrophages in Atherosclerosis ... 10
Glucose Transporter 1 and its function on Macrophage ... 21
Rationale and Hypotheses ... 22
CHAPTER II - MANUSCRIPT...24
Introduction ... 25
Experimental Methods ... 27
Results ... 33
Discussion ... 36
CHAPTER III- DISCUSSION ...55
vi
LIST OF FIGURES
Figure 1 - Illustration of the different stages of atherosclerosis………...3
Figure 2 - Aorta anatomy……… 9
Figure 3 - The role of efferocytosis and apoptosis on atherosclerosis ………..17
Figure 4 - Diagram of Study Design………..….…41
Figure 5 - Deletion of macrophage GLUT1did not increase Ldlr-/- mice’s susceptibility to weight gain……….…...42
Figure 6 - Deletion of macrophage GLUT1 did not alter body composition in recipient Ldl-/-mice………...43
Figure 7 - Lack of macrophage GLUT1increased plasma total cholesterol and LDL cholesterol……….……….…..44
Figure 8 - Lack of macrophage GLUT1did not alter plasma HDL cholesterol and triacylglycerol………...45
Figure 9 - Deletion of macrophage GLUT1 did not change blood pressure after 6 weeks on WD……….…..46
Figure 10 - Lack of macrophage GLUT1 did not change fasting plasma glucose……….…47
Figure 11 - Deletion of macrophage GLUT1 did not affect atherosclerotic lesion size in the aorta sinus……….…..48
Figure 12 - Increased necrotic core formation in Ldlr-/- mice reconstituted with Glut1MΦ-/-bone marrow……….….50
Figure 13 - Lack of macrophage GLUT1decreased collagen content in aorta sinus……….…..51
Figure 14 - Lack of macrophage GLUT1decreased phagocytosis in thioglycollate-elicited peritoneal macrophages………...53
vii
LIST OF ABBREVIATIONS
AAM Alternatively activated macrophage
ABCA1 ATP binding cassette transporter ABCA1
ABCG1 ATP binding cassette transporter ABCG1
ACC Acetyl CoA carboxylase
BMDMs bone marrow derived macrophages
BMT Bone marrow transplant
CAM Classically activated macrophage
CD36 Cluster of differentiation 36
CETP Cholesterol ester transfer protein
CLS Crown like structure
ER Endoplasmic reticulum
FAT/CD36 Long-chain fatty acyl coenzyme A
FFPE Formalin fixed paraffin embedded
GLUT1 Glucose transporter 1
GLUT1-EV GLUT1 empty vector
Glut1MΦ-/- Macrophage specific GLUT1 knock out
Glut1fl/fl Floxed Glut1 gene
GLUT1-OE GLUT1 over expresser
GSH Reduced glutathione
GSSG Oxidized glutathione
HFD High fat diet
viii
ICAM Intercellular adhesion Molecule
IFN Interferon gamma
IL-1β Interleukin-1β
IL-6 Interleukin-6
iNOS Inducible nitric oxide synthase
JNK C-Jun NH2 terminal kinase
LFA1 Lymphocyte function-associated antigen 1
LPS Lipopolysaccharide
LXR Liver X receptor
MCP-1 Macrophage chemotactic protein-1
M-CSF Monocytes colony stimulating factor
MMP Matrix metalloproteinase
NO Nitric oxide
OCT Optimal Cutting Temperature
ORO Oil Red O
OxLDL Oxidized low density lipoprotein
PAI-1 Plasminogen activator inhibitor-1
PPP Pentose Phosphate Pathway
ROS
Reactive oxygen species SMC
Smooth muscle cells
SR-A Scavenger receptor class A type I/II
SREBP1 Sterol regulatory element binding protein 1
ix
TNF-α Tumor necrosis factor-α
VCAM Vascular cells adhesion molecule
1
CHAPTER I - BACKGROUND Introduction
Atherosclerosis, affecting large and medium sized arteries, is the underlying cause of about 50% of all deaths in Westernized societies (Lusis, 2000). Factors including obesity, smoking, hypertension, elevated serum cholesterol, triglycerides, and free fatty acids, as well as decreased insulin sensitivity, are associated with increased risk of atherosclerosis. To date, there are limited relevant options to treat atherosclerosis besides prevention; therefore, further research into biological mechanisms is necessary.
Atherosclerosis is a chronic inflammatory disease characterized by four major steps including 1) initiation of endothelial activation and inflammation; 2) promotion of intimal lipoprotein deposition, retention, modification, and foam cell formation; 3) progression of complex plaques by plaque growth and enlargement of the necrotic core, fibrosis, thrombosis, and remodeling; and 4) precipitation of acute events (Hopkins, 2013).
Lesion development is initiated by deposition of cholesterol-rich and apolipoprotein B-containing lipoproteins, particularly low-density lipoprotein (LDL) in the subendothelial space of blood vessels. Physical interaction between LDL and proteoglycans causes accumulated LDL to remain in the subendothelial space. Mouse experiments show that mice expressing the
2
oxidation, aggregation, enzymatic or non-enzymatic cleavage, and hydrolysis all of which trigger local maladaptive responses to modified LDL (Moore, Sheedy, & Fisher, 2013). Modified LDL is pro-inflammatory, which enables it to activate overlying endothelial cells to express cell adhesion molecules such as intercellular adhesion molecule (ICAM) and vascular cell adhesion protein (VCAM) and secrete chemokines. Monocytes, attracted by activated endothelial cells are slowed down through rolling and stopped by attachment to the endothelial cells followed by transmigration to the subendothelial space. Upon stimulation by macrophage colony-stimulating factor (M-CSF) and other differentiation factors, monocytes differentiate into macrophages (Moore & Tabas, 2011). Scavenger receptor-mediated engulfment of oxidized LDL (oxLDL) transforms infiltrated macrophages to cholesterol-laden foam cells. The combination of macrophages taking up oxLDL through scavenger receptors and a lack of negative feedback keeps this engulfing process going non-stop. Eventually, accumulated cholesterol esters confer the macrophage “foam cell” characteristics. When the intracellular cholesterol content exceeds the foam cell’s processing capacity, foam cells undergo apoptosis. High concentrations of cytokines such as TNFa, unesterified cholesterol or oxysterols , oxLDL, activation of the Fas death pathway by Fas ligand (32), and ER stress all promote apoptosis processes (Seimon & Tabas, 2009). Apoptotic cells are readily cleared by macrophages through phagocytosis in the early lesion, however with the progression of lesion; phagocytosis of dead cells by lesion
3
2002). Stages of atherosclerosis are shown in Figure 1. Pathological studies suggest that the development of thrombus-mediated acute coronary events depend principally on the composition and vulnerability of a plaque rather than the severity of stenosis (Lusis, 2000). Macrophages play critical roles in promoting lesion progression through secreting pro-inflammatory cytokines, matrix degrading enzymes, and reactive oxygen species which all contribute to lesion structural disorganization. Eventually, under the stress of various detrimental factors, lesions become vulnerable, and hematoma-hemorrhage and thrombus ensue, often leading to lesion rupture, myocardial infarction, and death.
4
Through investigating the underlying causative mechanisms promoting atherogenesis, we can find more effective prevention and treatment methods for atherosclerosis and clinical events. Atherosclerosis is a multi-factorial disease and macrophages play an unparalleled role in almost every stage of its pathogenesis. Therefore, studying macrophage biology has become a primary interest in the field. A body of evidence has demonstrated that macrophages, a critical
component of the innate immune system, contribute to chronic inflammation-related pathology including obesity, insulin resistance, and atherosclerosis. Moreover, substrate metabolism is one of the mechanisms that affects the macrophage inflammatory response. Hence, the research presented herein examined the mechanistic links between macrophage substrate metabolism and the inflammatory response in atherogenesis.
Risk factors of Atherosclerosis
A large body of evidence has shown that dyslipidemia, hypertension, smoking, metabolic syndrome, diabetes, and other underling disease are atherosclerosis risk factors (Altman, 2003). 1) Dyslipidemia
5
intracellular cholesterol activates sterol regulatory element binding protein (SREBP) which then enters the nucleus to up-regulate LDL receptor and HMG-CoA reductase, and other enzymes in cholesterol regulationand de novo cholesterol synthesis pathway. Conversely, high intracellular cholesterol down-regulates LDL receptor and HMG-CoA reductase and subsequently reduces endogenous cholesterol production (Horton, Goldstein, & Brown, 2002). Uptake of plasma cholesterol through LDL receptor-mediated endocytosis not only reduces plasma cholesterol concentrations, but also signals cellular lipid synthesis machinery to stop synthesizing endogenous cholesterol. Therefore, any genetic variation or mutation affecting the process of receptor-mediated LDL endocytosis leads to elevated plasma LDL and hypercholesterolemia. Evidence indicates that high LDL promotes atherosclerosis at every stage of its pathological development. Discoveries of LDL receptor defects in familial hypercholesterolemia patients, along with clinical trials of statin drugs that decrease plasma LDL and reduce coronary morbidity and mortality, proved that LDL cholesterol is an independent risk factor of coronary heart disease (Grundy, 2002).
2) Hypertension
Hypertension is associated with increased risk of atherosclerosis and, importantly, it shares similar cellular and molecular mechanisms with atherosclerosis, particularly from the
endothelium perspective. Endothelial dysfunction appears to be one of the initiators of pathogenesis for both diseases (Alexander, 1995). Removing endothelial cells facilitates
6
endothelial cell morphology changes. Under chronic hypertensive conditions, smooth muscle cells (SMC) of blood vessels experience adaptive hypertrophy and proliferation, salient features of hypertension, which confer blood vessels greater contractility. Thickening of blood vessels by SMC hypertrophy increases the distance required for diffusion of oxygen from the lumen. This in turn results in incomplete oxidation and eventually increases free radical production, which oxidizes subendothelial-retained LDL to oxLDL. Importantly, hypertension is one of the leading causes of atherosclerotic renal disease in the US. Prehypertensive state is associated with
increased presence of inflammatory markers linked to atherosclerosis and even mild
hypertension serves as an important risk factor for progression of chronic kidney disease toward irreversible renal failure (Chade, Lerman, & Lerman, 2005). The pathogenesis of hypertension, like atherosclerosis, is extremely complicated. More importantly, it can enhance atherosclerosis through various other processes and remains one of the most relevant independent risk factors of atherosclerosis (Alexander, 1995).
3) Diabetes
Patients with type 2 diabetes (T2D) have an increased risk of cardiovascular morbidity and mortality. T2D and atherosclerotic vascular disease have shared pathogenic pathways, in which insulin resistance and abdominal obesity play central roles (Wassink et al., 2008). Macrophages, as an important component of the innate immune system, have been discovered to be the
7
TNF-α, disrupt insulin signaling pathways by phosphorylating insulin receptor substrate 1 (IRS1) 307 serine and result in insulin resistance and eventually T2D. Furthermore, insulin resistance and T2D lead to hypercholesterolemia through increased lipolysis and decreased lipid clearance. Through sharing the same pathogenesis and directly causing dyslipidemia, T2D accelerates atherosclerosis.
4) Other risk factors
Atherosclerosis is a multifactorial disease: both genetic and environmental factors promote its development. Recent studies have characterized monocytosis, which is defined as elevated monocyte numbers in the circulation, as another independent risk factor for coronary artery disease in humans (Randolph, 2014). Studies have shown that hypercholesterolemia is associated with monocytosis through promotion of hematopoietic cell proliferation, signaled by alteration of cholesterol levels on hematopoietic stem cells, in mice, pigs, and rabbits (Moore et al., 2013; Randolph, 2014). Moreover, recent compelling evidence has shown that ApoE is able to regulate monocyte pool size (Randolph, 2014), demonstrated by the fact that ApoE-/- mice show a 50% increase in monocyte numbers relative to wild type mice (Moore et al., 2013). Interestingly, in hypercholesterolemic mice, monocytosis is mainly derived from an increase in the more inflammatory LY6Chi subset (Moore et al., 2013).
Mouse Models of Atherosclerosis
8
major lipoprotein in mice, and subsets of HDL in mice differ from human. Both of these characteristics render mice resistant to atherogenesis (Getz & Reardon, 2012). Murine models including mutations of LDL receptors or ApoE that increase LDL cholesterol, have distinct features: 1) most mouse models do not develop unstable atherosclerotic plaques, whereas in humans, a vulnerable plaque is the leading cause for a clinical acute cardiovascular event; 2) mouse models do not form thick fibrous caps which are frequently seen in human chronic atherosclerotic plaques; 3) mouse models develop the majority of plaques in the aortic root, but rarely in coronary artery, unlike humans; 4) in the mouse peripheral blood, there are
approximately 50% pro-inflammatory monocytes (LY6Chi) and 50% anti-inflammatory monocytes (LY6Clow) circulating; whereas in humans the ratio of pro-inflammatory to anti-inflammatory monocytes is approximately 9:1 (Moore et al., 2013; Randolph, 2014). Although differences in the characteristics of atherosclerosis exist between men and mice, murine models undeniably have contributed tremendously to our understanding of the mechanisms of
atherogenesis (Getz & Reardon, 2012). Due to LDL receptor and ApoE’s critical functions in plasma lipoprotein levels, Apoe-/- and Ldlr-/- mouse models are two of the most frequently used mouse models for atherosclerosis research (Getz & Reardon, 2012).
9
of age and aortic sinus plaques at 8 months of age. When exposed to an atherogenic diet, Apoe
-/-mice develop abundant and large aortic plaques after 14 weeks of diet exposure. ApoE is expressed in bone marrow-derived cells, mostly in the monocyte/macrophage lineage. Transfer of bone marrow cells from ApoE-/- mice into wild type mice increases aortic root atherogenesis independent of effects on plasma lipid concentration; therefore, macrophage-derived ApoE plays a large role in this model of atherosclerosis (Getz & Reardon, 2012).
The Ldlr-/-mouse model was developed by Dr. Joachim Herz one year after the
successful development of ApoE-/- mice. Previous studies have demonstrated that Ldlr−/− mice display moderate increases plasma lipids and develop no or only mild atherosclerosis when fed a regular chow diet. The Paigen diet (1.25% cholesterol, 7.5% cocoa butter, 7.5% casein, and 0.5% sodium cholate) and Western Diet (21% fat and 0.15% to 0.25% cholesterol) are the most widely used high-fat regimens to accelerate atherosclerosis in the Ldlr-/-mouse. When exposed to
Western Diet (WD), Ldlr-/- mice show the presence of advanced lesions in the aortic sinus after 3 months and in the innominate artery at 6 to 9 months on diet (Ma et al., 2012). Anatomy of aorta is shown in Figure 2.
10
Bone marrow transplant (BMT) serves as an extremely powerful tool for atherosclerosis studies. Transplanting bone marrow cells into atherogenic mouse models is useful in studying the roles of hematopoietic cells on atherosclerosis (Getz & Reardon, 2012). To accomplish BMT, atherogenic mice receive a lethal dose of irradiation prior to receiving bone marrow from either wild type mice or genetically modified mice. After reconstitution, chimeric atherogenic mice carry transplanted hematopoietic stem cells, allowing for the study of specific roles from newly transplanted blood cells. While BMT is a powerful tool to study the contribution of
hematopoietic cells, some disadvantages include that irradiation in the recipient may not ablate 100% of the hematopoietic system, irradiated mice do not gain as much weight on high fat diets (HFD) after irradiation, and, finally, irradiation can cause permanent damage to various tissues.
Roles of Macrophages in Atherosclerosis
Macrophages, central effectors of innate immunity, are recognized as key
pathophysiologic agents in wide-spread disease processes associated with chronic inflammation and aging, including cancer, obesity-induced insulin resistance, and atherosclerosis (Moore & Tabas, 2011). Macrophages play a central role in the pathogenesis of atherosclerosis and investigating macrophage function has become a fundamental interest in understanding initiation, progression, and regression of atherosclerosis (Dickhout, Basseri, & Austin, 2008).
The roles of macrophages in atherosclerosis are diverse, including: 1) being the
precursors of foam cells; 2) secreting pro-inflammatory cytokines, chemokines, reactive oxygen species (ROS), reactive nitrogen species (RNS), matrix degrading enzyme, and
11
rupture (Randolph, 2014). During the initiation stage of atherosclerosis, through continual uptake of oxidized apolipoprotein B containing lipoproteins by scavenger receptor type A (SR-A) and CD36, macrophages clear sequestered lipoproteins in the subendothelial space. This is at first beneficial in restricting lesion expansion. However, there is little negative feedback following lipid uptake, and macrophages continually engulf lipoproteins and ultimately become
cholesterol-laden foam cells (Moore et al., 2013). Unregulated lipid accumulation in
macrophages results in increased secretion of pro-inflammatory cytokines, accelerated apoptosis and necrosis, defective phagocytic capacities, and reduced cell migration. All dysregulated macrophage functions lead to progression of atherosclerosis and finally cause “complex” atherosclerotic lesion formation. During the progression of atherosclerosis, macrophage inflammatory responses accelerate changes in lesion morphology, notably necrotic core formation and thinning of the fibrous cap, which are characteristic of the “vulnerable” lesions that most often lead to severe cardiac events and death (Moore & Tabas, 2011). Combined effects of increased macrophage apoptosis and defective macrophage clearance result in necrotic core formation. Besides pro-inflammatory cytokines, foam cells also secrete matrix
metalloproteinases which degrade collagen, elastin, and proteoglycans in the extracellular matrix, thus promoting thinning of the fibrous cap. Macrophages orchestrate the aforementioned activities, highlighting their essential contributions to the underlying mechanism(s) for the progression and aggravation of atherosclerosis. There are many steps in the process by which macrophages contribute to atherogenesis.
1) Monocyte recruitment: traffic into lesions
12
activation. Turbulent flow, hypertension, or other pro-atherogenic factors, such as oxLDL, activate endothelial cells. Upon activation, endothelial cells express chemokines that attract monocytes expressing cognate chemokine receptors. Activated endothelial cells subsequently express selectins and cell adhesion molecules, specifically ICAM and ACAM, which recognize respectively monocyte cell surface selectin ligands and cell adhesion molecules receptor
integrins. Finally, firm adhesion of monocytes is followed by their entry into the subendothelial space (a process called diapedesis) (Moore & Tabas, 2011).
Since the discovery of distinct subsets of monocytes in peripheral circulation systems by Geissmann in 2003 (Geissmann, Jung, & Littman, 2003), monocyte subsets have become an extensively investigated topic. In Both human and mouse monocytes fall into at least 2
phenotypically distinct subsets: LY6Chi (which are also phenotypically Gr-1+CCR2+CX3CR1lo) and LY6CloW (which are also phenotypically Gr-1–CCR2–CX3CR1hi) mouse monocytes
13 2) Macrophage becomes foam cells:
Modified LDL is able to activate endothelial cells, and endothelial activation in turn attracts more apoB-containing lipoproteins entering atherosclerotic lesion and retains them there. Accumulated LDL is modified by oxidation, acetylation, and aggregation. Upon arrival,
macrophages starts engulfing modified LDL that are recognized by several macrophage pattern recognition receptor including SR-A1, CD36, SR-A2, CD68, SR-B1, and LOX-1. The majority of cholesterol in LDL exists in the form of cholesterol ester which, after internalization by the macrophage, is hydrolyzed to free cholesterol and fatty acids in late endo-lysosome, while free cholesterolundergoes re-esterification by ACAT to cholesterol ester. Cholesterol ester is stored in membrane-bound lipid droplets, therefore remains as a relatively inert form which confers foam cells “foam” characteristics (Moore et al., 2013). Unesterified or “free” cholesterol traffic from lysosomes to the plasma membrane and thus may become available for efflux out of the cell. However, free cholesterol has cytotoxic effects on cells, accumulation of which causes cell apoptosis or even necrosis (Moore & Tabas, 2011). SR-A1 and CD36 are among the most studied macrophage scavenger receptors. In vivo experiments demonstrate that deleting either SR-A1 or CD36 significantly reduces atherosclerosis in ApoE-/- mice, clearly demonstrating that uptake of oxLDL by macrophages is critical in promoting atherogenesis (Boullier et al., 2001). Phospholipid constitutes the largest majority of lipid on the surface of LDL while neutral lipids are mainly located in lipid core. Evidence suggests that oxidized phospholipid is the most relevant component in mediating binding between scavenger receptors and oxLDL.
14
oxidized phospholipids, with apoptotic and bacterial cell membranes and are therefore taken up as foreign bodies. Although the uptake of oxLDL by scavenger receptors could cause detrimental effects for cardiovascular systems, the role of scavenger receptors in innate immunity have persisted due to natural selection (Boullier et al., 2001).
3) Foam cell inflammation
Cholesterol uptake or accumulation drives macrophage activation through direct or indirect mechanisms. Directly, oxLDL acts as a ligand for pattern recognition receptors such as
scavenger receptors and toll like receptors (TLRs). Depending on the extent of oxidation, oxLDL serves as agonist of either CD14-TLR4-MD2 or CD36-TLR4-TLR6 immune receptor complexes to activate a pro-inflammatory signal cascade which involves IL-1
15
Besides activating a pro-inflammation pathway, cholesterol loading in macrophages affects cells in various other aspects. Proteins involved with cytoskeletal regulation, vesicle-mediated transport, lipid binding, and apolipoprotein E are among the proteins most upregulated in peritoneal macrophages obtained from chow- vs. WD fed Ldlr−/− mice (Becker et al., 2010; Moore & Tabas, 2011). Boyle et al. demonstrated that TNFα secreted by macrophages together with Fas ligand (FasL) on macrophage cell membrane contributes to the vascular SMC apoptosis (Boyle, Weissberg, & Bennett, 2003). SMC is the only cell type that synthesizes extracellular collagens in an atherosclerotic lesion. Decreased collagen synthesis through impairing SMC function or reducing SMC numbers, and increased collagen degradation mediated by
macrophage derived matrix metalloproteinases (MMP) result in thinning of the fibrous cap, which leads to “vulnerable lesion” formation. Alternatively activated macrophages (AAM) secrete TGFβ which is an important inducer for SMC collagen biosynthesis; however, TGFβ secretion is reduced in complex lesions, which substantially decreases SMC collagen synthesis and increases the vulnerability of these lesions (Moore & Tabas, 2011).
4) Foam cell apoptosis and secondary necrosis
Mechanisms promoting foam cell apoptosis include existence of extracellular oxLDL,
accumulation of high concentrations of intracellular free cholesterol, production of pro-apoptotic cytokines such as TNF-α, excessive ROS production, growth factor withdrawal, hypoxia and direct cell–cell interactions such as binding of Fas-ligand to Fas (Schrijvers, De Meyer, Herman, & Martinet, 2007). Death of macrophages elicits complicated consequences regarding
16
absence of clearance, apoptotic cells rapidly expose phosphatidylserine on the surface, which is a potent activator in the generation of thrombin and activation of the coagulation cascade;
simultaneously, apoptotic cells progress to secondary necrosis and autolyse, releasing potentially dangerous cellular components (Schrijvers et al., 2007). With the progression of atherosclerosis, a majority of subendothelial macrophages become cholesterol-laden foam cells and gradually demonstrate less phagocytic capacity; hence, macrophage apoptotic cells and extracellular lipid accumulate in lesion plaques, form the necrotic core which is the hallmark of an unstable plaque and ultimately may lead to cardiovascular events through rupture of the lesion.
5) Efferocytosis
Macrophage apoptosis by itself will not trigger plaque necrosis. Rather, plaque necrosis results when apoptotic macrophages are not sufficiently cleared by phagocytes (efferocytosis) (Moore & Tabas, 2011). If apoptotic foam cells are not cleared efficiently by phagocytes, they eventually undergo a secondary necrosis process and subsequently release toxic intracellular contents including large amounts of tissue factors which trigger the coagulation cascade and thrombosis formation. Therefore, effective removal of apoptotic cell debris prevents unstable plaque development, whereas, reduced phagocytosis of apoptotic cells increases vulnerability of the atherosclerotic plaque. Furthermore, efferocytosis activates phagocytes to express IL-10 and TGFβ which mount an anti-inflammatory response, and it promotes survival of the efferocytes themselves so that they do not succumb to potentially toxic factors in apoptotic cells (Moore & Tabas, 2011). Professional phagocytes, such as macrophages and dendritic cells, rapidly
17
known that efferocytosis in atherosclerotic lesions is defective, however the underlying
mechanisms are poorly studied. Venter et al. recently demonstrated that the capacity to undergo LPS-induced cell shape changes and to phagocytize particles does not depend on oxidative phosphorylation activity but is fueled by glycolysis. They postulated that in macrophages, glucose critically participates in non-metabolic regulation of actin cytoskeletal remodeling via posttranslational modification of receptors or signaling molecules (Venter et al., 2014). Other evidence suggests that effective phagocytosis requires macrophages to have intact lipid metabolism to maintain effective cholesterol esterification and efflux (Moore et al., 2013). Hence, while the role of metabolism in macrophage-mediated phagocytosis has been
demonstrated, the exact substrates and metabolic pathways remain unclear. Illustration of the role of apoptosis and efferocytosis on atherosclerosis is shown in Figure 3.
18 6) Macrophage egress and lipid efflux
It has been shown that macrophages can emigrate out of atherosclerotic lesions, and this phenomena happens more in the early stage of atherosclerosis. Moore et al. demonstrated that both cholesterol loading and hypoxia increase the expression of the neuro-immune guidance cues Netrin-1 and Semaphorin-3E which are encoded by gene Ntn-1 and Sema3e in mice , both of which function to induce macrophage chemostasis in vitro. Chimeric Ldlr-/- mice carrying deficient macrophage netrin1 display reduced atherosclerosis progression and increased macrophage emigration from lesions, suggesting that Netrin-1 may function to retain macrophages in plaques (Moore et al., 2013). Cells sense increased intracellular cholesterol content and decrease endogenous cholesterol synthesis and LDL receptor expression regulated by SREPB1, and increase expression of ATP binding cassette transporter A1 (ABCA1) and ATP binding cassette transporter G1 (ABCG1) through activation of LXR by cholesterol derivatives oxysterols and desmosterol. ABCA1 and ABCG1 are members of ATP-binding cassette
superfamily participating in cholesterol efflux. Specifically, ABCA1 transfers cholesterol to lipid-poor APOA1 of HDL, and ABCG1 pumps cholesterol to mature HDL particles (Moore et al., 2013). This is a process called reverse cholesterol transport and understanding the role of macrophages in removal of cholesterol from plaques is vital.
7) Macrophage polarization and inflammatory response
19
2015), notably classically activated macrophages (CAM), alternatively activated macrophages (AAM), and a spectrum of intermediate classes. Macrophages are activated by infection, cytokines such as IFNγ, or saturated fatty acids through classical activation pathway. Upon activation, CAMs secrete high concentrations of TNFα, IL-1β, IL-12, and IL-23, but low levels of IL-10 and TGFβ. INFγ-/- /apoE-/- double knock-out mice display significantly smaller atherosclerotic lesion than ApoE-/- mice, and exhibit a substantial increase in their collagen content, which indicate that loss of Th1 signaling polarizes macrophages to the
less-inflammatory state (Vats et al., 2006). From a functional point of view, CAMs participate in the removal of pathogens during infection through activation of the NADPH oxidase system and production of pro-inflammatory cytokines. In contrast, AAMs stimulated by type 2 T-helper cytokines IL-4 or IL-13 express IL10, IL-1 receptor antagonist, arginase-1, transforming growth factor; therefore, they are implicated in tissue remodeling and wound repair and categorized as anti-inflammatory. Functionally, AAMs can scavenge debris and apoptotic cells and promote plaque fibrosis (Chinetti-Gbaguidi et al., 2015; Johnson, Milner, & Makowski, 2012). In addition to the CAM and AAM subpopulations, other specific and distinct macrophage phenotypes exist in the heterogeneous and complex environment of the atherosclerotic plaque. Macrophages are extraordinarily plastic and can switch from one phenotype to another depending on the
(Chinetti-20
Gbaguidi et al., 2015). Thus, the composition of CAM to AAM ratios in the microenvironment of the vessel and plaque are very important.
8) The Role of macrophage substrate metabolism in their inflammatory response Metabolism and immunity are two fundamental systems of metazoans. The presence of immune cells such as macrophage in metabolic tissues suggests a dynamic and on-going crosstalk between these two regulatory systems (Chawla et al., 2011). In 2003, Xu et al. and Weisberg et al. demonstrated that macrophage infiltration caused elevated expression of pro-inflammatory cytokines in adipose tissue of obese mice. TNFα, a cytokine largely produced by CAMs, promotes insulin resistance through phosphorylating serine 307 of insulin receptor substrate 1 (IRS1) - further demonstrating that macrophages play a critical role in metabolic disease (Chawla et al., 2011). Vats et al. demonstrated that in response to IL-4, signal transducer and activator of transcription 6 (STAT6) and PPARγ coactivator-1β (PGC-1β) induce
macrophage metabolic programs for fatty acid oxidation and mitochondrial biogenesis. Increased glucose uptake and suppressed fatty acid uptake in CAMs, increased fatty acid uptake and
elevated β-oxidation in AAMs indicated for the first time that there is a switch in nutrient utilization when macrophage shift from CAMs to AAMs (Vats et al., 2006). Etomoxir, mitochondrial inhibitor of fatty acid metabolism completely abolished macrophages’ anti-inflammatory effects stimulated by IL-4. In contrast, inhibition of mitochondrial respiration did not have effect on inflammatory activation of macrophages stimulated by INFγ and
21
From a functional point of view, macrophage metabolic pathways that are either dependent upon glycolysis or oxidative phosphorylation, are critical for maintaining proper host defense.
Glycolysis rapidly provides ATPs which are required by the respiratory burst to produce reactive oxygen species (ROS) and reactive nitrogen species (RNS) by CAMs to combat acute bacterial infection. In contrast, fatty acid oxidation provides a long-term energy supply which is needed by AAMs to secrete or express TGFβ, IL-10, and arginase I to fight intracellular parasite infections (Vats et al., 2006).
Glucose Transporter 1 and its function on Macrophage
Glucose transporter (GLUT) family and the sodium-dependent glucose transporter (SGLT) family are two classes of hexose transporters moving glucose into the cells. The GLUT family includes fourteen hexose transporters (twelve in mouse) which are facilitative transporters that transport sugars along the concentration gradient. The most widely expressed hexose
transporter is GLUT1 which is thought to maintain basal glucose transport in most types of cells (Young et al., 2011). Glucose is a critical component in the pro-inflammatory response of macrophages. Previous work from our group demonstrated that GLUT1 is the primary rate limiting glucose transporter on pro-inflammatory CAMs. Furthermore, in HFD-fed rodents, macrophages in crown-like structures and inflammatory loci in adipose and liver, respectively, stain positively for GLUT1. Freemerman et al. stably over-expressed the GLUT1 transporter in RAW264.7 macrophages (GLUT1-OE). Gene expression and proteome profiling analysis revealed that GLUT1-OE macrophages developed a hyper-inflammatory state characterized by elevated secretion of inflammatory mediators. Finally, ROS production and evidence of
22
inflammatory mediators such as PAI-1, suggesting that glucose-mediated metabolism and oxidative stress were driving the pro-inflammatory response in a GLUT1-dependent manner. These results indicated that increased utilization of glucose, induced a ROS-driven pro-inflammatory phenotype in macrophages. Based on the in vitro overexpression data, the Makowski lab created a novel macrophage GLUT1-deficient mouse model to determine the contribution of GLUT1-mediated glucose metabolism to in vivo metabolic dysfunction. Recent work suggests that lack of GLUT1 reduced the pro-inflammatory response and the ability for GLUT1-/- macrophages to polarize to the classically activated state in vitro, which may play an integral role in the promotion of obesity-associated insulin resistance or atherogenesis.
Rationale and Hypotheses
Our previous work indicates that when macrophage fuel substrates are modulated in vitro
23
white adipose tissue from Glut1MΦ-/- mice compared to Glut1fl/fl controls. However, the
macrophages in Glut1MΦ-/- were shifted towards AAM-polarized, not CAM-polarized as would be expected in obesity. Therefore, it appears that the lack of GLUT1-mediated glucose
metabolism in macrophages blocked CAM-polarization and release of pro-inflammatory cytokines, even in an obese state.
The objective of this proposal is to ask if reduced macrophage glucose metabolism will also halt the formation of atherogenesis using the Ldlr-/- atherosclerosis mouse model. We hypothesize that macrophages with blunted glucose metabolism due to lack of GLUT1 will have limited inflammatory activation during atherogenesis and reduced plaque burden. In order to investigate the effect of deletion of macrophage GLUT1 on the pathogenesis of
24
CHAPTER II - MANUSCRIPT
Macrophages play a key role in the pathogenesis of atherosclerosis. Metabolic programs powered by glucose or lipids enable macrophages to elicit pro- or anti-inflammatory responses, respectively. Although the chronic inflammatory feature of atherosclerosis has been well-established, the role of macrophage substrate metabolism in atherogenesis remains unclear. We previously demonstrated that macrophages with elevated GLUT1-mediated glucose metabolism have an increased inflammatory response. Therefore, we created a novel macrophage GLUT1-deficient murine model by crossing floxed GLUT1 mice to LysM-Cre mice. Pilot studies suggest that lack of GLUT1 reduces the pro-inflammatory response and the ability for Glut1
-/-macrophages to polarize to the classically activated state in vivo. Therefore, the objective of this study was to examine how macrophage metabolic reprogramming affects the development of atherosclerosis. We hypothesized that macrophages with restricted glucose metabolism due to lack of glucose transporter GLUT1 will have reduced pro-inflammatory activation during atherogenesis. We transplanted bone marrow from Glut1fl/flor Glut1MΦ-/- mice into Ldlr-/- mice and fed mice a WD for 12 weeks. Glut1fl/fl Ldlr-/- or Glut1MΦ-/- Ldlr-/- chimeric mice did not exhibit significant differences in body weight, body composition, blood pressure, fasting blood glucose, or triacylglycerol and HDL cholesterol concentrations. However, Glut1MΦ-/- Ldlr-/- mice had significantly less plasma total cholesterol and LDL cholesterol compared to Glut1fl/fl Ldlr
25
mice lacking macrophage GLUT1 had higher plasma cholesterol and developed increased necrotic cores in lesions of the aorta root relative to mice with wild type macrophage GLUT1, suggesting that macrophage glucose metabolism mediates systemic cholesterol metabolism; and, importantly, maintenance of atherosclerotic lesion stability may be regulated by macrophage-specific glucose-dependent mechanisms.
Introduction
Atherosclerosis, a major cause of cardiovascular disease (CVD), is chronic inflammation that can proceed to acute clinical events by thrombosis and plaque rupture (Frostegard, 2013; Lusis, 2000). Atherosclerosis is tied to many risk factors including obesity, smoking,
hypertension, elevated serum cholesterol, triglycerides, and free fatty acids, as well as decreased insulin sensitivity. Similar to obesity, one important driving force of atherogenesis is a
microenvironment of chronic inflammation. Macrophage infiltration, partially due to
overexpression of either MCP1 or its receptor CCL2, is implicated in local inflammation and insulin resistance (Odegaard et al., 2007a). There has been substantial progress in understanding the role of macrophages contributing to inflammation of vessels and adipose; however, the role of metabolism in regulating inflammation in atherosclerosis is poorly understood. Macrophages are involved in almost every stage of atherosclerosis, including but not limited to: 1) in the early stages: macrophages become foam cells through unabated engulfment of lipids, clearing
26
proliferator activated receptor gamma (PPARγ), modulation of reverse cholesterol transport, the inflammatory Iκk-NFκB kinase pathway, and endoplasmic reticulum (ER) stress, as well as manipulation of macrophage glucose metabolism through deleting GLUT1 to alter the inflammatory response (Erbay et al., 2009; Makowski, Brittingham, Reynolds, Suttles, & Hotamisligil, 2005).
27 Experimental Methods
Reagents
All reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. Carboxylate microspheres were obtained from Polysciences, Inc (Warrington, PA). Antibodies were purchased from the following sources: F4/80 (AbD Serotec/BioRad, CA); Annexin V PE Apoptosis Detection Kit PE (eBioscience, CA); Mouse IgG (Life technologies, CA); Goat anti-Mouse IgG, Rhodamine Red conjugate (Life technologies, CA); Goat Anti-anti-Mouse IgG Cy5 (Life technologies, CA); Alexa Fluor oxL488 Phalloidin (Life technologies, CA). DL and acetyl LDL were purchased from Alfa Aesar Company (Ward Hill, MA). Fluoro-Gel II with DAPI was purchased from Electron Microscopy Sciences (Hatfield, PA). Glucometer and Glucose strips were purchased from Abbott Diabetes Care (Abbott Park, IL).
Animals and Maintenance
All animal experiments were performed in compliance with protocol approved by the University of North Carolina at Chapel Hill Institutional Animal Care and Use Committee.
Glut1fl/flmice were generously provided by Dr. Dale Abel (University of Iowa). LysM-Cre mice
(stock number. 004781) and Ldlr-/-mice (stock number. 002207) were obtained from Jackson Laboratories (Bar Harbor, ME). Glut1fl/fl mice were backcrossed with C57BL/6J mice for 2 (Erbay et al., 2009) generations in the University of North Carolina at Chapel Hill animal facility and then crossed with LysM-Cre mice. All mice were housed in a12-hour light-dark cycle with
ad libitium access to food and water. To generate littermate Glut1fl/fl and Glut1MΦ-/- mice, male
LysM-Cre+/+ and female Glut1fl/fl mice were bred and kept Glut1fl/fl/LysM-Cre+/- as breeders for experiments to generate littermates. Upon conducting BMT, chimeric Ldlr-/- mice were
28
were maintained on chow diet for four weeks before challenge with WD (TD88137, 42% of Kcal from milk fat with 0.15% added cholesterol) from Harlan Teklad (Indianapolis, IN) for 12
weeks.
DNA isolation and GLUT1 genotyping
DNA was isolated from mouse-tail tissues or BMDMs using a DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA) and DNA concentration was measured by NanoDrop 2000 (Thermo scientific, DE). Genotyping was performed using the following primers: Flanked loxP forward – CTGTGAGTTCCTGAGACCCTG; Flanked loxP reverse – CCCAGGCAAGGAAGTAGTTC; FRT forward: TCCATTCTCCAAACTAGGAAC; FRT-R2:
GAAGGCACATATGAAACAATG. iProof High-Fidelity PCR super mix (Bio-Rad, Hercules, CA)-based genotyping was performed on a C1000 thermocycler (Bio-Rad, Hercules, CA).
Bone Marrow Transplantation
29
5x106 bone marrow cells through retro-orbital injection under anesthesia. Control animals were transplanted with the HBSS buffer only and died within 10 days of lethal irradiation.
Body weight and composition
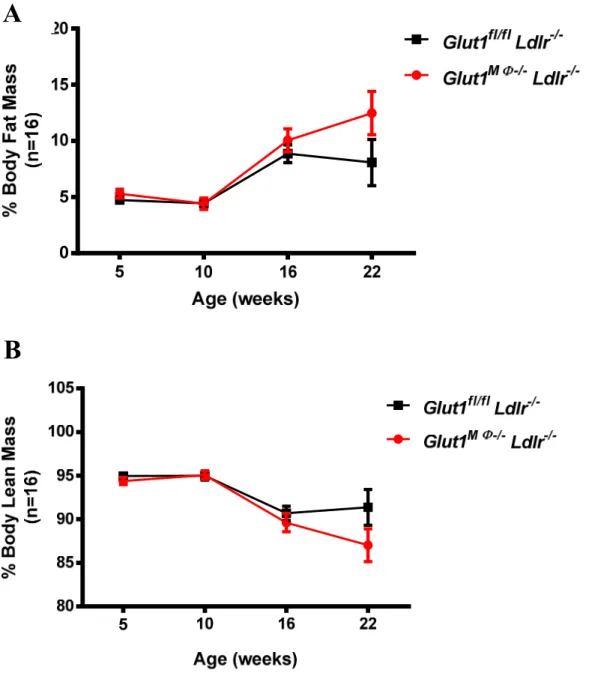
Body weight was measured prior to starting mice on diet and weekly until sacrifice. Body composition including lean mass, fat mass, free water content, and total water content of non-anesthetized mice was measured at age of 5, 10, 16, and 22 weeks old using the EchoMRI-100 quantitative magnetic resonance whole body composition analyzer (Echo Medical Systems, Houston, TX). Fat mass or lean mass is presented as percent fat mass or lean mass over total body weight (Sundaram et al., 2013).
Histology and immunohistochemistry
30 Histology quantification
Quantification of atherosclerotic lesions was done as described with modification (Bjorkbacka et al., 2004; Rotllan et al., 2015). Serial interrupted sections were cut through the aorta at the origins of the aortic valve leaflets, and every other section (10 µm) throughout the aortic sinus (1600 µm) was stained with ORO and hematoxylin. Aortic lesion size of each animal was obtained by averaging 20 sections from the same mouse. 5µm serial interrupted sections were stained with Masson’s trichrome and used for quantification of collagen content and necrotic cores. A color deconvolution algorithm, developed by Bentley Midkiff from UNC Translational Pathology Laboratory, was used to separate collagen for digital quantification. Scores of collagen amount were given based on the product of algorithm calculated as optical density (OD) x percent of total positive area stained with collagen. Necrotic cores were quantified by Aperio ePathology software after manually circling necrotic area.
Fasting Plasma Lipids
After 6 hours of fasting, 200µl blood was collected into an ETDA coated tube by submandibular bleeding. Blood was centrifuged at 10,000g for 2 minutes at 4°C; plasma was then transferred to another tube. Plasma was diluted to 1:10 with PBS. From this diluted plasma, total cholesterol, HDL-cholesterol, and triglycerides were measured in the UNC Animal Clinical Chemistry and Gene Expression Core Facility by Vt350 automated chemical analyzer from Ortho-Clinical Diagnostics Company (Rochester, NY).
Blood Pressure
31
Mice (n=8) received 5 consecutive days of training on the tail-cuff blood pressure system (CODA, Kent Scientific, CA) prior to data collection. Starting on day 6, systolic and diastolic blood pressure was measured in conscious mice daily for 3 days. Mice were acclimated to the warm (32°C) restrainer for 15 minutes and blood pressure was recorded during 10 acclimation followed by 25 measurement cycles.
Cell culture
32
were counted. Finally, the cells were diluted into different concentrations for plating 6-well and 24-well plates.
Phagocytosis assay
Coverslips were coated in a 24-well plate with 1ml of 10µg/ml rat tail collagen I (Corning, NY) overnight in a sterile cell culture hood. Collagen-coated coverslips were then washed with sterile PBS twice and air dried. Mouse peritoneal macrophages were seeded at a concentration of 8x105 cells per well and left to attach to coverslips overnight. The carboxylated microspheres were opsonized with normal mouse IgG by mixing 3 ml of sterile PBS, 60µl of microspheres, and 60µl of mouse IgG and rotating this suspension at 37°C for one hour.
33
from wells and mounted to slides with Fluoro-Gel II with DAPI (Electron Microscopy Sciences, PA) mounting media for future analysis. ZEISS LSM 700 confocal microscope (UNC
Microscopy service lab) was used to capture the images. Phagocytosis Index (PI) was calculated by numbers of microspheres took up by cells divided by total microspheres in the field. Results from 5 fields were averaged.
Statistics
For all in vivo and ex vivo data, statistical differences between experimental groups were determined by unpaired Student’s t-tests using statistical software within GraphPad Prism (GraphPad Software, Inc., La Jolla, CA). All data are shown as mean ± standard error of the mean (SEM). P values less than 0.05 were considered statistically significant.
Results
Deletion of macrophage GLUT1 did not alter metabolic parameters in Ldlr-/- mice Weekly body weight excluding 2 weeks after BMT and body composition including before BMT, before diet, and 6 weeks on diet and 12 weeks on diet were measured to determine if abolishing glucose metabolism in macrophages affects systemic metabolic parameters of Ldlr -/- mice. In addition, fasting blood glucose was measured every 3 weeks and fasting total
cholesterol, HDL cholesterol, and triglyceride in plasma was measured at 12 weeks on WD.
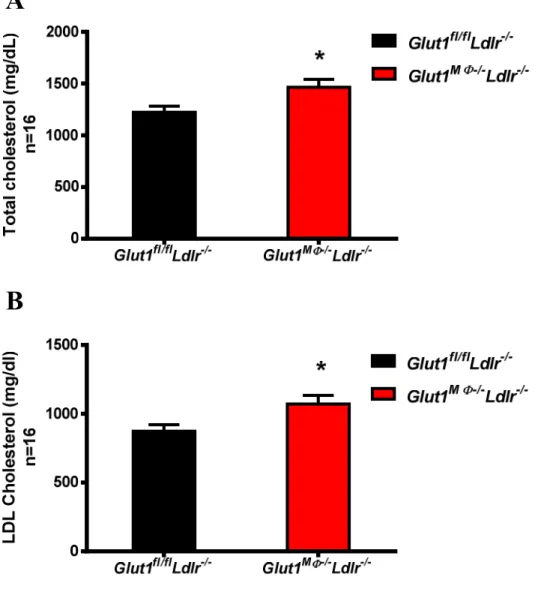
Glut1MΦ+/+ Ldlr-/-and Glut1MΦ-/- Ldlr-/- mice body weights did not differ after a 12 weeks WD exposure (Figure 5). Likewise, fat and lean muscle percentages were not significantly different between Glut1fl/fl Ldlr-/- and Glut1MΦ-/- Ldlr-/-mice (Figure 6 A-B). Interestingly, circulating total cholesterol and LDL cholesterol concentrations in plasma were significantly higher in Glut1
34
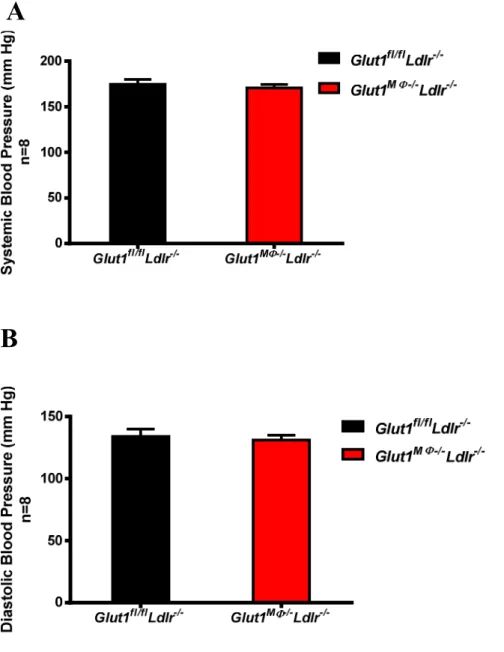
were no differences in plasma HDL cholesterol or triglycerides (Figure 8 A-B). Additionally, systolic and diastolic blood pressure was recorded at the age of 16 weeks of age, but there was no genotype effect (Figure 9 A-B). In addition, 6-hour fasting glucose was recorded at 5, 10, 13, 16, 19, and 22 weeks of age with no GLUT1- mediated differences detected (Figure 10).
Lack of macrophage GLUT1 does not affect atherosclerosis lesion size
Preliminary data from the Makowski Lab indicated that Glut1MΦ-/-BMDMs failed to elicit a pro-inflammatory response. Glut1MΦ-/-expressed 80% less IL-1β and 60% less iNOS messenger RNA compared to Glut1fl/fl BMDMs. We sought to understand whether GLUT1-mediated modulation of the macrophage inflammatory phenotype could reduce atherosclerotic plaque size since atherosclerosis is dependent on inflammation. To achieve our purpose, we conducted BMT by transplanting bone marrow collected from Glut1MΦ-/- or Glut1fl/fl mice or vehicle control HBSS buffer into Ldlr-/-mice through retro-orbital injection. After 12 weeks of WD exposure, we assessed aortic root lesion formation by histology. All mice that received vehicle transplantation died at 10 or 11 days post-irradiation, while all of the mice that received BMT survived. At the end point of the experiments, when mice were 22 weeks of age, we assessed lesion size by quantifying the average of lesion areas/section in both groups. After analyzing ORO stained aortic sinus lesions, we observed no difference in lesion size between
Glut1fl/fl Ldlr-/- and Glut1MΦ-/- Ldlr-/- mice (Figure 11 A-C).
Glut1MΦ-/- Ldlr-/- chimeric mice develop fewer and smaller necrotic cores than Glut1fl/fl Ldlr -/-mice
35
death and necrotic core formation. We next sought to investigate whether abolishing glucose metabolism in macrophages affected the necrotic area. We analyzed Masson’s trichrome stained FFPE sections (Figure12 A-B) and observed more than a 50% increase in the necrotic area in the
Glut1MΦ-/- Ldlr-/- mice compared to Glut1fl/fl Ldlr-/- mice (Figure 12 C). 87.5% (7 out of 8) of
Glut1MΦ-/- Ldlr-/-mice developed necrosis but only 37.5% ( 3 out of 8) Glut1fl/fl Ldlr-/- mice developed a necrotic core (Figure 12 D).
Deletion of macrophage GLUT1 decreased collagen amount in aorta sinus
Collagen is majorly secreted by lesion SMCs. MMP, secreted by pro-inflammatory macrophages and necrotic foam cells, degrades collagen and increases lesion vulnerability. Surprisingly, we observed a 20% decrease in collagen content in the aorta sinus of Glut1MΦ-/-
Ldlr-/- mice compared to Glut1fl/fl Ldlr-/- mice (Figure 13 A-B, p=0.0161 and p=0.0065).
Lack of GLUT1 decreased macrophage phagocytic capacity
36 Discussion
A body of evidence has demonstrated that macrophages are implicated in chronic adipose tissue inflammation, obesity, insulin resistance, and T2D. Macrophage metabolism and the immune response are closely intertwined with each other (Odegaard & Chawla, 2011). The link between fatty acid oxidation and macrophage alternative activation has been elucidated
(Odegaard et al., 2007b; Vats et al., 2006). However, the role of glucose metabolism in regulating macrophage polarization is still poorly understood. GLUT1 is the predominant glucose transporter on macrophages (Freemerman et al., 2014). In order to understand how glucose metabolism affects macrophage polarization, our lab created a mouse macrophage RAW 246.7 cell line which stably expressed GLUT. Freemerman et al. demonstrated that over-expression of GLUT1 significantly increased glucose uptake, lactate production, and
simultaneously TNF-α, IL-1β, IL-6, and PAI-1 gene expression, in a glycolysis and
ROS-dependent manner, indicating that glucose metabolism plays a key role in driving macrophage to mount a robust, pro-inflammatory response (Freemerman et al., 2014). We generated a novel
Glut1MΦ-/- mouse model wherein Glut1 was specifically deleted from hematopoietic myeloid
37
play a central role in the pathogenesis of atherosclerosis, and because obesity and atherosclerosis share certain common etiologies such as chronic inflammation, we intended to expand our knowledge garnered from the in vitro GLUT1 gain of function model and the in vivo GLUT1 loss of function obesity mouse model to investigate how macrophage glucose metabolism affects atherosclerosis. We chose to use atherogenic Ldlr-/- mice as bone marrow recipient mice because transplanting ApoE+/+ macrophages into ApoE-/- mice decelerates atherogenesis in ApoE-/- mice (Getz & Reardon, 2012). Our initial hypothesis was that mice with mutated macrophage glucose metabolism would develop reduced atherosclerosis because of decreased macrophage pro-inflammatory response. However, after 12 weeks of WD exposure, deletion of macrophage GLUT1 surprisingly did not affect atherosclerotic lesion size per se but strikingly altered the composition of lesions compared to floxed controls. Lack of macrophage GLUT1 increased necrotic core formation and decreased aorta sinus collagen content.
We showed that deletion of macrophage GLUT1 did not affect Ldlr-/-mouse body weight or fasting glucose. We also did not observe any difference on other systemic measurements including body composition, blood pressure, plasma HDL cholesterol, and plasma
triacylglycerol. Of note, Glut1MΦ-/- Ldlr-/- mice fed 12 weeks of WD had significantly higher plasma total cholesterol and LDL cholesterol than Glut1fl/fl Ldlr-/-mice. Plasma cholesterol concentration is determined by endogenous cholesterol synthesis, dietary intake, and clearance. All mice received WD during the 12-week diet exposure; therefore it was likely that deletion of macrophage GLUT1 affected liver cholesterol synthesis or secretion. Our preliminary data indicated that Glut1MΦ-/-mice developed more crown like structure (CLS) than Glut1fl/fl mice in epididymal white adipose tissue when mice were made obese on a HFD. The elevated
38
expression of MCP-1 in Glut1MΦ-/-adipose tissue; MCP-1 expression was 70-fold higher in
Glut1MΦ-/- epididymal white adipose compared to Glut1fl/fl adipose tissue. One explanation for increased plasma total cholesterol and LDL cholesterol is that deletion of macrophage GLUT1 could have increased macrophage liver infiltration, and consequently increased total activity of pro-inflammatory cytokines such as IL-1β. It is well established that pro-inflammatory
cytokines, including TNF-α, IL-1β, and IL-6, increase hepatic de novo cholesterol and lipogenesis (Popa, Netea, van Riel, van der Meer, & Stalenhoef, 2007; Zhao et al., 2011).
Phagocytosis is the process of engulfing particles larger than 0.5µm and requires receptor participation. Macrophages play critical roles in defending against infection, maintaining tissue homeostasis, and remodeling through phagocytizing foreign bodies such as bacteria, fungi, and apoptotic bodies. Cells undergoing apoptosis release various molecules such as ATP,
lysophosphatidylcholine, and sphingosine 1-phosphate which function as chemokines, recruiting phagocytes to the vicinity of dying cells (Flannagan, Jaumouille, & Grinstein, 2012). Venter et al. demonstrated that glycolysis fuels LPS-induced macrophage cell shape changes and
phagocytosis of zymosan particles in the RAW macrophage 246.7 cell line, which highlights the role of glucose in maintaining functional macrophage phagocytosis (Venter et al., 2014).
39
Therefore we speculated that lack of GLUT1 decreased PFKFB3 activity which further reduced actin cytoskeleton remodeling in macrophages. Macrophages with reduced ability to rearrange actin cannot form proper pseudopodia, and therefore phagocytic capacity is impaired (Coppolino et al., 2002). In the subendothelium space of blood vessels, decreased macrophage phagocytic capacity eventually leads to necrotic core formation in atherosclerotic lesion.
To characterize the effect of GLUT1 on phagocytic capacity, we performed an ex vivo
phagocytosis assay to test the capacity of thioglycollate-elicited peritoneal macrophages to phagocytose normal mouse IgG opsonized 2µm microspheres as a model of engulfing
Lipoproteins. Results of the pilot study suggested that Glut1MΦ-/-macrophagesdisplayed reduced phagocytic capacity relative to Glut1fl/fl macrophages. These findings corroborated our findings on histologic quantification of necrotic cores. Furthermore, preliminary data in diet-induced obese mice suggested that although macrophages infiltrated in greater numbers in Glut1
MΦ-/-mice, they failed to polarize to CAMs. One hypothesis is that the macrophages infiltrate but fail to activate because of a lack of phagocytic capacity of macrophages lacking GLUT1. We also observed decreased collagen content in Glut1MΦ-/-Ldlr-/-mice compared to Glut1fl/fl Ldlr-/-mice which may have resulted from increased MMP released by necrotic Glut1MΦ-/- foam cells. Taken together, we have provided preliminary data that macrophage GLUT1 is required to maintain the functional phagocytic capacity and therefore sustain atherosclerotic lesion stability.
40
necrotic areas between GLUT1 overexpressed macrophage and wild type macrophage
transplanted Ldlr-/-mice (Nishizawa et al., 2014). This was the first article to document the role of macrophage glucose metabolism in atherosclerosis. Our results were consistent with their finding that altering macrophage glucose metabolism did not affect atherosclerotic lesion size, but by utilizing a loss of function model we observed that mice with deleted macrophage GLUT1 developed more and larger necrotic core areas in atherosclerotic lesions. We speculate that overexpression of macrophage GLUT1 either maintains or increases PFKFB3 activity and as a result, decreased macrophage phagocytic capacity was not seen in Nishizawa’s study.
In summary, deletion of macrophage GLUT1 increased plasma total cholesterol and LDL cholesterol but did not affect body weight, body composition, or fasting glucose. In the aorta sinus, more and larger necrotic cores but less collagen content were observed in Glut1
MΦ-/-mice. We hypothesize that deletion of macrophage GLUT1 may have increased liver lipogenesis and furthermore that maintenance of atherosclerotic lesion stability may be regulated by
macrophage specific glucose-dependent mechanisms. In order to test our hypothesis we will measure total lipid content in liver from frozen liver tissues; also stain liver FFPE sections with macrophage surface marker F4/80 to examine liver macrophage infiltration. In future studies, we aim to repeat ex vivo phagocytosis assays and further measure whether deletion of GLUT1 increases macrophage apoptosis. Last, we will stain aorta sinus sections with Mouse
Macrophages/Monocytes antibody (Moma-2) to quantify aorta root macrophage infiltration. Taken together, this study provides evidence for the first time that macrophage
4
1
Figure 4. Diagram of Study Design. Littermate male Glut1fl/fl and Glut1MΦ-/- donor mice were maintained at the UNC animal facility.
Recipient male Ldlr-/- mice were purchased from Jackson laboratory at 4 weeks of age. At 6 weeks of age Ldlr-/- mice were lethally
irradiated and transplanted with 5x106 bone marrow cells which were collected from sex- and age-matched donor mice. Chimeric Ldlr
42
Figure 5. Deletion of macrophage GLUT1 did not increase Ldlr-/- mice’s susceptibility to weight gain. Body weights of Ldlr-/- mice were measured weekly starting at 5 weeks of age. Irradiation and BMT occurred at 6 weeks of age (black arrow). WD started at 10 weeks of age (purple arrow). Blood pressure was measured at 16 weeks of age (red arrow) (n=16 per Glut1
43
Figure 6. Deletion of macrophage GLUT1 did not alter body composition in recipient Ldl -/-mice. (A) Body fat mass and (B) lean mass percentage were measured by Echo MRI at 5, 10, 16, and 22 weeks of age corresponding to before BMT, before diet, 6 weeks on diet, and 12 weeks on diet (at sacrifice). Data are presented as means ± SEM, n = 16 per Glut1 genotype.
A
44
Figure 7. Lack of macrophage GLUT1 increased plasma total cholesterol and LDL cholesterol. Six hour fasted plasma lipids at 22 weeks of age were measured in the UNC Animal Clinical Chemistry and Gene Expression Core Facility. (A) Total cholesterol was 1464±77 mg/dl for Glut1MΦ-/-Ldlr-/- mice, and 1223 ± 59.07 mg/dl for the Glut1fl/fl Ldlr-/- mice. (*P<0.05). (B) LDL cholesterol was calculated by subtracting HDL cholesterol from total cholesterol. LDL cholesterol were 1071 ± 62.62 mg/dl for the Glut1MΦ-/-Ldlr-/-mice, and 873.1 ± 46.93 mg/dl for the Glut1fl/fl Ldlr-/- mice. (*P<0.05). Data are presented as means ± SEM, n = 16 per Glut1
45
Figure 8. Lack of macrophage GLUT1 did not alter plasma HDL cholesterol and triacylglycerol. (A) 6 hours fasted plasma HDL and (B) triacylglycerol were measured in UNC Animal Clinical Chemistry and Gene Expression Core Facility as above. Data are presented as means ± SEM, n = 16 per Glut1 genotype.
A
46
Figure 9. Deletion of macrophage GLUT1 did not change blood pressure after 6 weeks on WD. Blood pressure was measured at 16 weeks of age using a tail-cuff system. (A) Systolic and (B) diastolic blood pressure were measured in Glut1MΦ-/-Ldlr-/-and Glut1fl/fl Ldlr-/- mice. Data are presented as means ± SEM, n = 8 per Glut1 genotype.
47
G
lu
c
o
s
e
(
m
g
/d
l)
n
=
1
6
48
Figure 11. Deletion of macrophage GLUT1 did not affect atherosclerotic lesion size in the aorta sinus. Serial interrupted 10µm sections were stained with ORO and counter stained with Hematoxylin. Slides were scanned by Aperio Digital Slides Scanner. (A-B) 20X representative photomicrographs of ORO staining. Black arrow indicates representative lesions quantified. (C) Quantitative analysis of atherosclerotic lesion size in Glut1fl/fl Ldlr−/−or Glut1MΦ-/-Ldlr−/− mice. Data are presented as means ± SEM, n = 8 per Glut1 genotype.
A
Glut1fl/fl Ldlr-/-B
Glut1MΦ-/- Ldlr49
A
Glut1fl/fl Ldlr-/--/-50
Figure 12. Increased necrotic core formation in Ldlr-/- mice reconstituted with Glut1MΦ-/-bone marrow. (A-B) 20X representative photomicrographs of Masson’s trichrome staining. Black arrow indicates representative necrotic core quantified. (C) Average necrotic area per section. Data are presented as means ± SEM, n=8. (D) Percent of Glut1fl/fl Ldlr−/− versus Glut1MΦ-/- Ldlr−/− mice that developed necrotic core at sacrifice. n=8 per Glut1 genotype.
C
51
Figure 13. Lack of macrophage GLUT1decreased collagen content in aorta sinus. Histologic analysis of collagen was performed using a color deconvolution algorithm in Aperio ScanScope software. Scores were assigned based on collagen content in regions of atherosclerotic lesions. (A) Collagen content score. (*P<0.05). (B) Calculated intensities of positive collagen staining (**P<0.01). Data are presented as means ± SEM, n = 6 per Glut1 genotype.
52
A
Glut1fl/fl peritoneal macrophagesB
Glut1MΦ-/- peritoneal macrophages RhodamineExtracellular beads
DAPI nucleus Phalloidin Actin DIC
Cy5
Intracellular beads MERGE
DAPI nucleus Phalloidin Actin DIC
Rhodamine
Extracellular beads MERGE
Cy5
53
Glut1fl /fl Ldlr-/- Glut1M -/- Ldlr-/- 0.0
0.2 0.4 0.6
0.8
Glut1
fl/flLdlr
-/-Glut1
M -/-Ldlr
-/-*
Figure 14. Lack of macrophage GLUT1decreased phagocytosis in thioglycollate-elicited peritoneal macrophages.
(A-B) Thioglycollate-elicited peritoneal macrophages were isolated from Glutfl/fl and Glut1MΦ -/-mice. Macrophages were incubated with carboxylated conjugated microspheres for 20 minutes. Macrophages were stained with DAPI for nuclei (blue), phalloidin for actin (green), rhodamine for extracellular beads (red), and after permeabilization Cy5 for intracellular beads (magenta). A ZEISS LSM 700 confocal microscope was used to capture representative images (20X). (C) Phagocytosis index was calculated by numbers of microspheres taken up by macrophages (magenta)/ total microspheres in field (red and magenta). (n=1 experiment).
54
Figure 15. Working model for how decreased GLUT1-mediated glucose metabolism impairs phagocytosis. PFKFB3 maintains and supports regular cellular actin remodeling
55
CHAPTER III- DISCUSSION
Atherosclerosis is a chronic inflammatory disease and macrophages play an unparalleled role in its pathology but to date, the role of macrophage glucose metabolism on atherosclerosis remains unclear; therefore the intent of this study was to address the gap in the knowledge regarding the mechanistic link between macrophage glucose metabolism and the inflammatory response in atherogenesis by examining nutrient-responsive pathways. In 2006, Ajay Chawla demonstrated that fatty acid β-oxidation is required for alternative activation of macrophages which is involved with inflammation resolution and wound healing (Vats et al., 2006). To study the role of glucose metabolism on macrophage inflammatory response, the Makowski lab has previously published in vitro GLUT1 gain of function (Freemerman et al., 2014) and has
obtained preliminary data using ex vivo GLUT1 loss of function BMDM models. These studies have demonstrated that glucose is a substrate that is required for activation of CAMs which launch the pro-inflammatory response in response to products derived from or associated with bacterial infections, such as lipopolysaccharide (LPS) and interferon gamma (IFNγ). Briefly, Freemerman et al. demonstrated that glucose transporter 1 (GLUT1) was the predominant glucose transporter on mouse BMDMs and mouse macrophage cell line RAW 264.7. Over-expression of GLUT1 on RAW cells increased their glucose uptake, glycolytic rate, and lactate production, and, more importantly, increased TNFα, IL-6, and MCP-1 gene expression
(Freemerman et al., 2014). Furthermore, preliminary data showed that lack of GLUT1