PHARMACOLOGICAL AND BIOPHYSICAL DISSECTIONS OF THE TRAFFICKING AND INFECTION OF ADENO-ASSOCIATED VIRUS (AAV) IN VITRO AND IN VIVO
Ping-Jie Xiao
A dissertation submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Cell and Developmental Biology.
Chapel Hill 2013
Approved by:
©2013 Ping-Jie Xiao
ABSTRACT
PING-JIE XIAO: Pharmacological and Biophysical Dissections of the Trafficking and Infection of Adeno-Associated Virus In Vitro and In Vivo.
(Under the direction of Dr. R. Jude Samulski)
Adeno-associated virus (AAV) is a defective and non-pathogenic human parvovirus that is dependent on the co-infection of a helper virus, such as Adenovirus or Herpes Virus, to fulfill its productive life cycle. While many traits of this virus have made it an attractive vector for gene therapy as well as the wide application in over 100 clinical trials, there remains a great need to further improve the performance of AAV in gene delivery. As a most promising solution to fit such need, development of novel AAV vectors with improved efficiency on cellular trafficking and processing was partially achieved by several strategies in the last decade, including isolation of natural serotypes, directed evolution, and rational design. Being a most effective approach for the development of new AAV vectors in the future, rational design requires extensive knowledge on AAV-host interaction, which is mainly a multi-step trafficking and intracellular processing from cell surface binding to nuclear entry. However, much is unknown about the details of interactions between AAV and its host, which has limited one’s ability to improve the delivery efficacy of AAV vectors and therefore restricted their medical applications.
To my parents and wife,
ACKNOWLEDGEMENTS
With the completion of this dissertation, I’d like to show my gratitude to the nurturing environment surrounding me in Chapel Hill and back home in Shanghai that has fostered my intellectual growth. I would like to specifically thank Dr. Jude Samulski for his guidance, his inspiration, and for setting an excellent scientific example for me to follow. Thank Dr. Samulski for providing a great environment to further foster my research independence that was initially established during my Master’s study in Dr. Xu’s lab at Fudan University in Shanghai. As an international student, I also received valuable help from him to improve my language. In the Gene Therapy Center and Department of Cell Biology there are many faculty members that go above and beyond their profession, as teachers and as mentors. In this regard, the Drs. Ken Jacobson, Keith Burridge, Tal Kafri, Ellen Weiss, Aravind Asokan have been outstanding. I would also like to thank Drs. Robert Bagnell, Ken Jacobson, Michael Chua, Aaron Neumann, and Vladimir Ghukasyan for the productive discussions on the principles and rationales of fluorescence microscopy.
TABLE OF CONTENTS
LIST OF TABLES ...x
LIST OF FIGURES ... xi
ABBREVIATIONS ...xv
I. INTRODUCTION TO GENE THERAPY AND AAV BIOLOGY ··· 1
VIRAL VECTORS FOR GENE THERAPY ··· 1
AAV BIOLOGY AND VECTOROLOGY ··· 9
APPLICATIONS AND CHALLENGES OF AAV IN CLINIC ··· 15
SUBCELLULAR EVENTS DURING AAV INFECTION ··· 35
MICROSCOPY AS A METHOD TO STUDY VIRAL TRAFFICKING ··· 39
MICROTUBULES IN VIRAL TRAFFICKING ··· 43
OVERCOME CELLULAR BARRIERS: MOMENTUM OF VECTOR DEVELOPMENT ··· 45
II. QUANTITATIVE 3D TRACING OF GENE-DELIVERY VIRAL VECTORS IN HUMAN CELLS AND ANIMAL TISSUES ··· 50
SUMMARY ··· 50
INTRODUCTION ··· 52
MATERIALS AND METHODS ··· 55
RESULTS··· 60
DISCUSSION ··· 83
III. CYTOPLASMIC TRAFFICKING, ENDOSOMAL ESCAPE, AND PERI-NUCLEAR ACCUMULATION OF AAV2 PARTICLES ARE FACILITATED BY MICROTUBULE NETWORK ··· 92
MATERIALS AND METHODS ··· 97
RESULTS··· 103
DISCUSSION ··· 124
IV. PERINUCLEAR RETENTION LIMITS THE INFECTION OF BOTH ENVELOPED AND NON-ENVELOPED VIRUSES ··· 134
SUMMARY ··· 134
INTRODUCTION ··· 136
MATERIALS AND METHODS ··· 140
RESULTS··· 147
DISCUSSION ··· 167
V. CONCLUSIONS AND FUTHER EXPLORATIONS ··· 173
SUMMARY OF RESULTS ··· 173
UNPUBLISHED DATA AND FUTURE DIRECTIONS ··· 180
APPENDICES: PLASMIDS ...197
LIST OF TABLES
TABLE 1. COMPARISON OF METHODS OF GENE DELIVERY ... 4
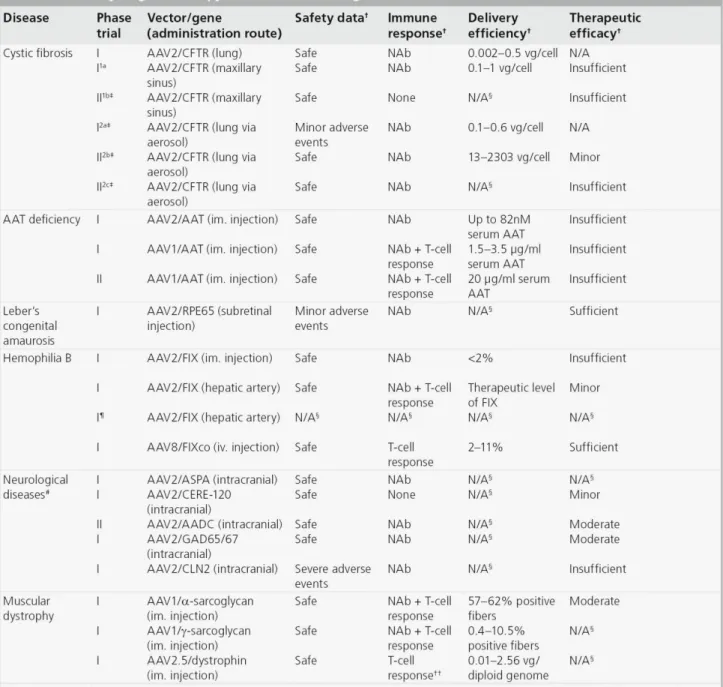
TABLE 2. SUMMARY OF AAV MEDIATED CLINICAL TRIALS ... 17
LIST OF FIGURES
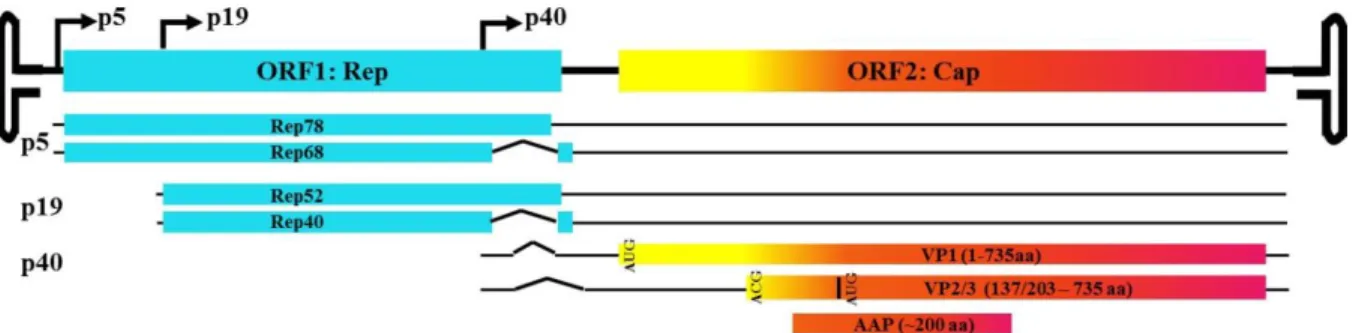
FIGURE I-1. AAV GENOME. ... 11
FIGURE I-2. STRUCTURAL DISSECTION OF THE AAV CAPSID. ... 13
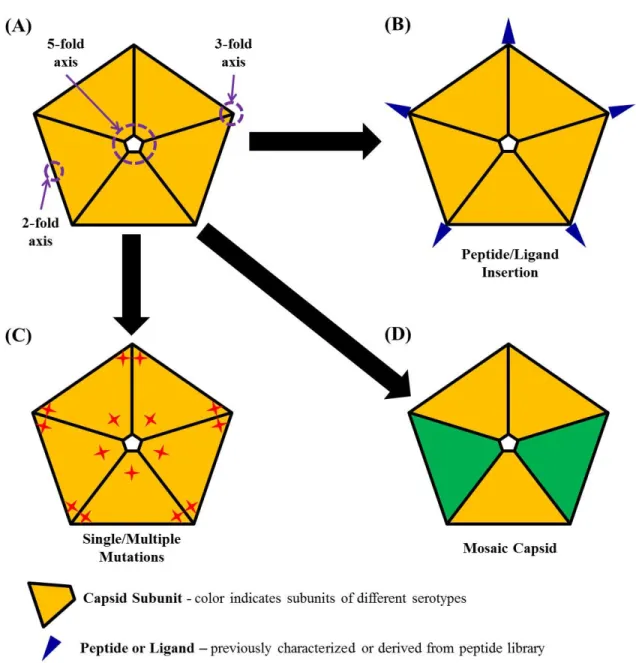
FIGURE I-3. ILLUSTRATION FOR CAPSID RATIONAL DESIGN. ... 33
FIGURE I-4. EXPOSURE OF VP1/2 N-TERMINI THROUGH THE
5-FOLD PORE. ... 37
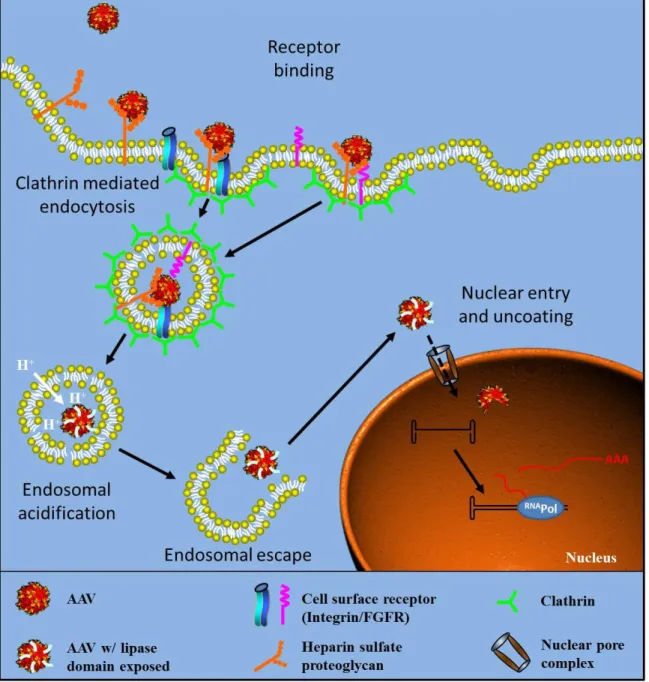
FIGURE I-5. AAV TRAFFICKING SCHEME. ... 40
FIGURE I-6. MT MEDIATED TRAFFICKING OF CELLULAR
FACTORS AND VIRUSES. ... 46
FIGURE II-1. OBJECT-BASED QUANTITATIVE 3D DISTRIBUTION
MICROSCOPY. ... 61
FIGURE II-2. THEORETICAL POINT SPREAD FUNCTION (PSF) OF CONFOCAL IMAGES AND OPTIMIZATION OF 3D
DECONVOLUTION. ... 62
FIGURE II-3. TOTAL FLUORESCENCE INTENSITY (TFI) IS PROPORTIONAL TO THE AMOUNT OF DYES AND
DYE-LABELED BEADS. ... 64
FIGURE II-4. EVALUATION OF AAV2 MORPHOLOGY AND INFECTIVITY AFTER CHEMICAL CONJUGATION
WITH CY5. ... 66
FIGURE II-5. CHARACTERIZATION OF FLUORESCENCE SIGNAL
FOR SINGLE CY5-AAV2 PARTICLES. ... 68
FIGURE II-6. VISUALIZATION OF CY5-AAV2 PARTICLES WITHIN
FIGURE II-7. FLUORESCENCE PROPERTIES OF SINGLE
CY5-AAV2 PARTICLES WITHIN CELLS. ... 71
FIGURE II-8. MEAN FLUORESCENCE INTENSITY OF SINGLE CY5-AAV2 ON COVERSLIPS AND IN CELLULAR
ENVIRONMENT. ... 72
FIGURE II-9. ASSOCIATION OF CY5-AAV2 WITH THE
LYSOSOMES. ... 75
FIGURE II-10. KINETICS OF CY5-AAV2 NUCLEAR TARGETING. ... 76
FIGURE II-11. THE LEVEL OF TRANSGENE EGFPSC EXPRESSION
AT TWO DIFFERENT VIRION DOSAGES ... 78
FIGURE II-12. TRAFFICKING OF AAV2 IN MOUSE MUSCLE. ... 80
FIGURE II-13. SCHEMATIC AND QUANTITATIVE
DOCUMENTATION OF AAV2 TRAFFICKING IN VITRO
AND IN VIVO. ... 81
FIGURE III-1. MICROTUBULE DISRUPTION BEFORE VIRAL INOCULATION REDUCES EARLY AAV2
TRANSDUCTION. ... 105
FIGURE III-2. NORMALIZED RATIO OF THE PERCENTAGE OF
GFP POSITIVE CELLS AT VARIOUS VIRAL DOSAGES ... 106
FIGURE III-3. MT DISRUPTION BY NOCODAZOLE IMPAIRED VIRAL TRANSDUCTION AFTER THE CELLS WERE
PULSE INFECTED WITH AAV2 AT 37 DEGREE. ... 107
FIGURE III-4. MT DISRUPTION BY NOCODAZOLE DOES NOT INTERFERE WITH THE ACTIVITY OF PROMOTER IN
VIRAL TRANSGENE CASSETTE. ... 108
FIGURE III-5. VARIOUS ANTI-MICROTUBULE DRUGS CAN
IMPAIR AAV2 TRANSDUCTION. ... 110
FIGURE III-7. EARLY PERI-NUCLEAR ACCUMULATION AND NUCLEAR ENTRY IS REDUCED BY THE DISRUPTION
OF MT NETWORK. ... 113
FIGURE III-8. AAV2 CO-LOCALIZES WITH MTS. ... 115
FIGURE III-9. FAST AND UNI-DIRECTIONAL MOVEMENT OF
AAV2 TOWARDS THE PERI-NUCLEAR REGION. ... 116
FIGURE III-10. TRAJECTORIES OF AAV2 TRAFFICKING. ... 119
FIGURE III-11. FAST AND UNI-DIRECTIONAL MOVEMENT OF AAV2 PARTICLES IS DEPENDENT ON INTACT
MICROTUBULES. ... 120
FIGURE III-12. ASSOCIATION OF AAV2-CONTAINING
ENDOSOME WITH MTS AND DELAYED ENDOSOMAL
ACIDIFICATION UPON MTS DISRUPTION. ... 123
FIGURE III-13. A MODEL FOR THE ROLE OF MICROTUBULES ON THE CYTOPLASMIC TRAFFICKING AND
ENDOSOMAL ACIDIFICATION FOR AAV2 ESCAPE. ... 125
FIGURE IV-1. FLUORESCENT LABELING OF AAV PARTICLES. ... 149
FIGURE IV-2. PERINUCLEAR RETENTION OF AAV BY THE
MT-MTOC. ... 150
FIGURE IV-3. DISRUPTION OF MT-MTOC INCREASES
TRANSDUCTION OF RAAV, RAD, AND LENTIVIRUS. ... 153
FIGURE IV-4. MT-MTOC DISRUPTION DOES NOT AFFECT
PROMOTER ACTIVITY AND VIRAL DEGRADATION. ... 156
FIGURE IV-5. RELEASE OF AAV AND INCREASED NUCLEAR
ENTRY UPON MT-MTOC DISRUPTION. ... 157
FIGURE IV-6. DISRUPTION OF THE MT-MTOC INCREASES VIRAL TRANSDUCTION AND NUCLEAR ENTRY
FIGURE IV-7. PERINUCLEAR ACCUMULATION IN MOUSE
TISSUES. ... 165
FIGURE IV-8. PERINUCLEAR ACCUMULATION OF AAV PARTICLES IS DISRUPTED AND VIRAL
TRANSDUCTION IS INCREASED UPON NOCODAZOLE
TREATMENT IN VARIOUS MOUSE TISSUES. ... 166
FIGURE V-1. UPDATED SCHEME FOR AAV TRAFFICKING... 175
FIGURE V-2. PERI-NUCLEAR LOCALIZATION OF AAV2
REQUIRES INTACT MTS AND ASSOCIATED WITH
MTS THROUGHOUT CELL CYCLE. ... 182
FIGURE V-3. TRANSITION OF AAV PARTICLES FROM EARLY
ENDOSOME TO LATE ENDOSOME. ... 185
FIGURE V-4. AAV2 CO-LOCALIZES WITH NUCLEAR PORE COMPLEX (NPC) AND DISRUPTION OF VIRAL ACCUMULATION AT MTOC RESULTS IN VIRAL
ACCESS TO THE NPC DISTAL FROM THIS REGION. ... 191
FIGURE V-5. AAV TRAFFICKING IN RODENT CEREBRUM. ... 195
FIGURE S-1. MAPS OF TR-CBA-SSDNA AND
DSEMBOL-TR-CMV-EGFPSC. ... 199
FIGURE S-2. THE MAP OF PACKAGING PLASMID ‘PXRS’. ... 200
FIGURE S-3. THE REPRESENTATIVE MAP OF AD HELPER
PLASMID PXX680. ... 201
FIGURE S-4. THE REPRESENTATIVE MAP OF PLASMID
EGFP-RAB5. ... 202
FIGURE S-5. THE REPRESENTATIVE MAP OF PLASMID
ABBREVIATIONS
3D Three-dimensional
AAV Adeno-associated virus
Ad Adenovirus
BR1- Basic region 1 mutation 120QAKKR/QANNR BR2- Basic region 2 mutation 140PGKKR/PGNNR BR3- Basic region 3 mutation 168PARKR/PANNR
CBA Chicken beta actin
CF Cystic fibrosis
CFTR Cystic fibrosis transmembrane conductance regulator
CMV Cytomegalovirus
CPV Canine parvovirus
DAPI 4′,6′-diamidino-2-phenylindole DIC Differential interference contrast DMEM Dulbecco’s modified Eagle’s medium
DMSO Dimethyl sulfoxide
DNA Deoxyribonucleic acid
EGFP Enhanced green fluorescent protein
FIX Factor IX
G551D Glycine at position 551 mutated to aspartic acid
GFP Green fluorescent protein
HEK Human embryonic kidney
HIV Human immunodeficiency virus
HS Heparin sulphate
ITR Inverted terminal repeat
Luc Luciferase
MFI Mean fluorescence intensity
MOI Multiplicity of infection
mRNA Messenger ribonucleic acid
MTOC Microtubule organizing center
MVM Minute virus of mice
NLS Nuclear localization signal
ORF Open reading frame
PBS Phosphate-buffered saline
PCR Polymerase chain reaction
PEI Polyethyleneimine
PLA2 Phospholipase A2
qPCR Quantitative polymerase chain reaction rAAV Recombinant adeno-associated virus
RPE65 Retinal pigment epithelium-specific 65kD protein
SA Sialic acid
siRNA Short interfering ribonucleic acid SV40 Simian vacuolating virus 40
VP Viral protein
CHAPTER 1
INTRODUCTION TO GENE THERAPY AND AAV BIOLOGY
Viral vectors for gene therapy
Developing a method to introduce therapeutic nucleic acids into specific cells or tissues within the body is not straightforward. Many different strategies have been employed. In general, all of these strategies can be classified into two categories based on the mechanism of delivery, non-viral or viral. Non-viral methods are typified by the delivery of naked nucleic acids (DNA or RNA) by physical means (e.g. gene gun or ultrasonic) or with the help of chemical reagents (e.g. cationic polymers) (Table 1). Viral methods rely on packaging nucleic acid encoding the functional protein or RNA inside of a viral particle and subsequent delivery to target cells via viruses’ natural infection pathways.
Non-viral delivery. Delivery of ‘naked’ nucleic acid is probably the most straight forward
nucleic acids from exposure to nuclease degradation (Luo et al. 2000). These molecules coat the nucleic acid with excessive positive charges to improve association with the cell surface (Chen et al. 2007).The effect of these carriers is limited, however, due to promiscuous binding, which leads to low circulation times and the inability to target specific tissues or cell types (e.g. tumors) (Luo et al. 2000). Tissue specific delivery of naked nucleic acids can be achieved, to a certain degree, by conjugating specific ligands, such as transferrin, peptides, or growth factors (Gupta et al. 2005; Watson et al. 2005). The addition of these ligands targets the molecule to certain cell surface receptors, thus improving specificity. Sugars, such as galactose, mannose and saccharides, can also be added to enhance cellular uptake of nucleic acids and provide some level of specificity in tissue targeting (Hashida et al. 2001; Yu et al. 2009). However, most carriers and ligands do not assist crossing of the nuclear membrane and thus, transfer of plasmid to the nucleus only occurs when there is breakdown of the nuclear membrane during cell division (Wells 2010).
Table 1. Comparison of methods of gene delivery
Retroviruses. Retroviruses are characterized by the ability to reverse transcribe a single-stranded RNA (ssRNA) genome into double-stranded DNA (dsDNA) and integrate into the host genome as part of their natural life cycle. The retroviral virion consists of an exterior envelope, averaging 80-100nm in diameter, which surrounds a protein capsid. The capsid contains two copies of the linear ssRNA genome, each 7-12kb in length (Osten et al. 2007). Viral infection begins with binding to the cell via interaction between glycoproteins on the viral envelope and specific receptors on the host cell surface. The viral envelope then fuses with the cell membrane and releases the capsid into the cytoplasm. Subsequently, the genetic material is released and transported to the nucleus with the aid of viral and cellular proteins. Once inside the nucleus, the viral genome integrates into the host chromosomes as a natural feature of its replication cycle.
or are used in 2.3% clinical trials, treating diseases such as adrenoleukodystrophy (ALD), parkinson’s disease, sickle cell anemia, and cancer immunotherapy (Bank et al. 2005; Li et al. 2005; Cartier et al. 2009; Williams 2009). Several advancements in vector engineering have made lentiviruses a viable option for clinical therapy. Among these, pseudotyping of vectors with different surface glycoproteins, such as vesicular stomatitis virus glycoprotein, has expanded targeting to a wider variety of cell/tissue types. Additionally, insights into the mechanism of vector integration have yielded non-integrating lentiviral (NIL) vectors that have nearly eliminated the risk of insertional mutagenesis. Despite these promising improvements, several hurdles remain. One problem is that these vectors are subject to transgene silencing (Bonci et al. 2003; Ellis 2005). The mechanism of this is not well understood, but long-term gene expression is limited. The innate immune response and preexisting immunity to glycoproteins used to pseudotype these vectors have also limited their efficacy (Towers 2007). Finally, a means of scalable production of clinical grade vector is lacking and challenges future advancement of this system.
Development of Ad as a gene therapy vector has undergone several generational advancements. First generation vectors were created by removal of the E1 region of the genome. This deletion prevents replication of the virus and provides space in the genome for insertion of exogenous DNA (Kovesdi et al. 1997). To propagate these vectors, the E1 proteins must be provided in trans. The major concern with this method is that recombination events during vector production between the vector and complementing E1 sequence can result in restoration of wild type (WT) Ad. Second generation vectors remove a greater portion of the genome, including regions E2, E3, and E4. This reduces, but does not eliminate the risk of recombination events resulting in WT virus and expands the capacity for transgenes. Third generation ‘gutless’ vectors take this principle to the maximum by removing all non-essential sequence in the genome (Chen et al. 1997).
Ad-based vectors can infect dividing and non-dividing cells, have high transgene capacity, and a low rate of insertional mutagenesis. Additionally, scalable methods of production allow high titer preparations. Ad vectors have been widely used to treat a variety of diseases and account for ~24% of clinical trials ((Ghosh et al. 2006; Roth 2006; Gray et al. 2008; Mata-Espinosa et al. 2008); www.wiley.co.uk/genmed/clinical). The greatest hurdle to use of Ads is that they are highly immunogenic and inflammatory. Many patients have preexisting immunity to the virus and therapeutic effect of derived vectors can be limited by the host immune response (Douglas 2007). As a result, Ad vectors may be best suited for treating diseases in which only transient gene expression is needed, such as cancer.
containing a proteinaceous layer (i.e. tegument) and an icosahedral protein capsid. The packaged genome is a linear double-stranded DNA of ~152kb in length. Similar to retroviruses, HSV binds to specific receptors on target cells via glycoproteins on the surface of the viral envelope. The envelope then fuses with the cell membrane and the capsid and tegument are released into the cytoplasm. The capsid is trafficked to the nucleus with the aid of viral and cellular factors and the genome released through the nuclear pore. Once in the nucleus, the linear genome circularizes and persists in the cell as an extrachromosomal episome. Vectors derived from HSV are able to transduce dividing and non-dividing cells and can be targeted to various cell types by surface protein pseudotyping, much like retroviruses. Due to its size, HSV can deliver large pieces of exogenous DNA, the largest capacity among commonly used viral vectors (Fink et al. 1997; Geller 1997). HSV vectors account for ~3% of clinical trials. However, also much like retroviruses, a limitation of these vectors has been silencing of gene expression caused by DNA methylation, which has made these vectors less effective for long-term therapy (Kass et al. 1997; Lilley et al. 2001; Sun et al. 2003; Suzuki et al. 2008). Host immune responses to the vector and lack of scalable production have limited the application of these vectors as well (Fraefel et al. 1996).
AAV biology and vectorology
AAV shares many features in common with the viral vectors discussed above, however, it also has unique advantages that make it an exceptional vector for gene delivery. Unlike other viral systems, WT AAV is not associated with disease or pathology and derived vectors have an excellent safety profile in humans (Mueller et al. 2008). Moreover, the natural persistence of this virus in many tissues facilitates long-term transgene expression (Berns et al. 1975). A number of innovations have contributed to the success of AAV as a vector for gene therapy as well. For example, the ability to package AAV serotype-2 genome into capsids from various serotypes (transencapsidation) gives this vector a wide range of potential tissue tropisms (Rabinowitz et al. 2002). The following sections review the biology of AAV, successes and challenges that are already visible from clinical trials, and current strategies to improve AAV vectors further.
genotoxic stimuli, such as hydroxyurea or UV irradiation, can induce AAV replication as well (Grossman et al. 1984; Yakobson et al. 1987; Yalkinoglu et al. 1988).
AAV vectorology. Since generation of the first infectious clone of AAV2, the potential of recombinant AAV (rAAV) as a vector for gene therapy has become increasingly evident (Samulski et al. 1982; Samulski et al. 1983). Among many desirable attributes, AAV can transduce a variety of cell types, both dividing and non-dividing, and it is capable of long-term gene expression. Vectors derived from AAV retain only the ITR sequence, which is required for viral packaging and assembly, from the WT virus and do not express any viral genes (Xiao et al. 1997). This removes the possibility of unwanted consequences from introducing viral genes or genomic sequence. Additionally, scalable systems of production have been developed. Production of vector requires co-expression of Rep/Cap genes. Current methods supply these proteins as well as necessary helper virus proteins in trans and have virtually eliminated the risk of producing WT AAV (Samulski et al. 1989; Grieger et al. 2006). rAAV vectors utilize the same mechanisms and pathways of entry and trafficking as WT AAV, except that they do not integrate into the host chromosome due to the absence of the Rep proteins. Instead, the genomes form predominantly concatamers and persist as circular episomes (Miao et al. 1998; Nakai et al. 2001).
Applications and challenges of AAV in clinic
The first AAV vector to be delivered to human subjects carried the CFTR gene in a clinical trial to treat cystic fibrosis in 1995 (Flotte et al. 2003). Since then the number of AAV-based clinical trials has increased to more than 60. Here, I will briefly summarize these trials and discuss the challenges AAV vectors must overcome. A brief summary of these clinical trials is provided in Table 2.
other methods of delivery, reported a dose-dependent response in gene expression (Aitken et al. 2001). In an attempt to reach therapeutic levels of expression, a multi-dose approach was conducted in which patients received multiple injections of vector. In this trial, repeated doses were well tolerated by the host immune system and an encouraging trend in improvement of pulmonary function was suggested by measurement of interleukin-8 (IL-8) and forced expiratory volume (Moss et al. 2004). However, a follow up study, which expanded treatment to additional patients, did not find the same effect, which brings into question the efficacy of this treatment protocol (Moss et al. 2007). Overall, these results provide data on the safety of rAAV delivery to the airway epithelium, but indicate that more efficient gene transfer is required to achieve therapeutic effect. A more comprehensive review can be found at Griesenbach et al (Griesenbach et al. 2012).
Alpha1-antitrypsin deficiency. Alpha1-antitrypsin (AAT) deficiency is an autosomal recessive disorder caused by reduced circulating levels of AAT protein (normally 1.5-3.5 mg/ml), which is secreted into the serum by hepatocytes and provides protease protection of the lung. Certain mutations of the AAT gene can also result in accumulation of abnormal protein in hepatocytes, causing disease of the liver. The symptoms of AAT-deficiency are emphysema, chronic obstructive lung disease, and liver damage. Current treatments include protein replacement therapy and avoidance of risk factors, such as smoking. Protein replacement therapy is costly and requires weekly infusions. Gene therapy has been explored for this disease primarily because restoration of only low levels of AAT (~800 g/ml) can have therapeutic effect.
Based on successes in preclinical studies, several clinical trials have been initiated to treat AAT
T-cell immune response against the viral capsid were observed, but this did not adversely affect patient health and was dependent on vector dose. Although the level of AAT expression was below that required for therapeutic effect, transgene expression persisted for over 1 year and the safety data revealed no adverse reactions. These results suggest this method of delivery may produce therapeutic effect with increased vector dosage (Brantly et al. 2006; Brantly et al. 2009). This was tested in a subsequent study in which dose-escalation was performed in conjunction with IM delivery of rAAV serotype 1, shown to transduce muscle with high efficiency in animal models (Flotte et al. 2011). At three months of observation, vector administration was well tolerated, but all subjects developed neutralizing antibodies to AAV1 capsid. Antibody response was not observed against the transgene product, WT AAT. In this context, the level of transgene expression corresponded with vector dose and the level of circulating AAT in the serum reached >20 g/ml in all patients (~2.5% of the required level). Thus, significant progress has been made toward developing a therapeutic for AAT deficiency, but further improvement is necessary to achieve rescue of function. Flotte et al provides a more comprehensive review on this disease (Flotte et al. 2011).
et al. 2001). When this strategy was extended into patients, restoration of vision was observed in most patients several weeks after treatment (Bainbridge et al. 2008; Cideciyan et al. 2008; Maguire et al. 2008). Continued observation revealed improvement in vision for up to 1.5 years and no adverse effects or pathology was observed (Simonelli et al. 2010). This is now regarded as the first successful AAV-based gene therapy trial in humans. These results demonstrate the safety and efficacy of delivering rAAV to the eye by sub-retinal injection and introduce a method of therapy that could potentially be used to treat other ocular diseases in addition to LCA. A more comprehensive review can be found at Stein et al (Stein et al. 2011).
used (Kay et al. 2000; Manno et al. 2003). The second trial altered targeting to the liver via infusion through the hepatic artery. This approach did achieve expression of therapeutic levels of FIX, but only transiently (~2-4 weeks). Host immune response mediated by T lymphocytes against the vector capsid has been suggested to clear the transduced hepatocytes and thus limit the efficiency of delivery (Manno et al. 2006; Mingozzi et al. 2007). Based on these results, a third trial has been initiated using the same strategy as the second trial, targeting the liver, with co-administration of immunosuppressive drugs (High 2011). Results of this trial have not yet been reported, but it will be interesting to see if vector transduction is increased as a result. More recently, Nathwani and colleagues have taken a different angle to evade the immune system and increase gene expression, including utilization of serotype 8 capsid, which transduces the liver with higher efficiency and is less immunogenic than serotype 2 in animal models, and codon optimization of the transgene expression cassette (High 2011; Nathwani et al. 2011). With this strategy, therapeutic levels of FIX were observed (2-11% of normal) in all patients and gene expression persisted up to 16 months (Nathwani et al. 2011). Thus, efforts are close to a successful therapy. This evolution of treatment for FIX deficiency offers insight into the planning of future trials with AAV. Foremost, that there may be considerable flexibility in the immunogenicity and efficiency of transduction with capsids of different serotypes. High et al provides a more comprehensive perspective of gene therapy to treat this disease (High 2011).
al. 2010). A third strategy is infusion of the subthalamic nucleus with rAAV expressing two isoforms of the enzyme glutamic acid decarboxylase (GAD65 and GAD67), which are responsible for the production of inhibitory neurotransmitter GABA (Luo et al. 2002). The subthalamic nucleus (STN) region of the brain has a central role in regulating movement and its dis-inhibition is believed to be responsible for the hyperkinetic motor activity symptoms in PD patients. The hypothesis is that introduction of these enzymes will alleviate the motor symptom of PD (Luo et al. 2002). As a result of this treatment most patients experienced significant improvement in motor function through one year post treatment (Kaplitt et al. 2007). A follow-up phase II trial with this approach is now underway. All the above results demonstrate the safety and long-term efficacy of rAAV for delivery into the CNS delivery. A more comprehensive review can be found at Weinberg et al (Weinberg et al. 2012).
one patient developing a T-cell immune response to the capsid. In addition to LGMD-2C, clinical efforts have begun to treat Duchenne muscular dystrophy (DMD), an X-linked disease caused by mutation of the gene that encodes dystrophin (Bowles et al. 2011; Mendell et al. 2011). In trial, six children with DMD were given vectors by intramuscular delivery. This trial was the first instance in which an engineered capsid (AAV2.5), derived from serotypes 1 and 2, has been used. The novel capsid did not elicit a significant host immune response and there were no unanticipated side effects from the treatment. However, the therapeutic dystrophin protein did elicit an auto-reactive T-cell response in one patient (Mendell et al. 2011). Diprimio et al provides a more comprehensive view on this disease (DiPrimio et al. 2010).
Other diseases. A number of clinical trials have been conducted using rAAV to treat diseases other than those described above as well, diseases such as inflammatory arthritis, heart disease, and metabolic diseases. Currently, there is one phase I trial to treat inflammatory arthritis using a vector to express a fusion protein of the human tumor necrosis factor-immunoglobulin Fc domain (TNFR:Fc), an antagonist of TNF- For delivery, vector was introduced into fifteen patients via intra-artery injection with the goal of reaching the joints (Mease et al. 2009). The objective of this trail was to determine safety, and though successful, was dosed too low to see therapeutic effect.
has been linked with heart failure (Lipskaia et al. 2010). Results of this study have not yet been reported.
Lipoprotein lipase (LPL) deficiency, a monogenic metabolic disease leading to high serum triglyceride levels and pancreatitis, is another disease that may benefit from gene therapy. The approach in this trial was to introduce rAAV1 expressing LPL via IM injection. Eight patients were treated and three months after treatment average serum triglyceride levels were reduced by 27% and 41% in the low-dose and high-dose cohorts, respectively. Levels returned to baseline, however, at 18-31 months after treatment (Stroes et al. 2008). Transient expression in this trial may have been caused by a neutralization antibody and T-cell immune response against the capsid (Mingozzi et al. 2009).
Challenges associated with AAV in clinic
retinal ganglion cells (RGC), will require cell-type-specific vectors or promoters (Martin et al. 2002).
Strategies for enhancing rAAV capsid
The limitations of rAAV, learned from pre-clinical and clinical studies alike, have given momentum to efforts to improve this vector. Various strategies have been pursued, including development of novel capsids, transgene cassettes, and employment of pharmacological reagents to enhance gene delivery and expression. This section focuses on perhaps the most promising of these strategies, development of novel vector capsids to improve specific targeting and transduction efficiency (Table 3, Fig.I-3). The immune response associated with AAV vectors and potential strategies to tackle this issue have been extensively summarized in several most recent reviews (Bartel et al. 2011; Mingozzi et al. 2011; Rogers et al. 2011).
has also been shown to have specificity for transducing non-neuronal cells in the retina. Alternatively, serotypes 4 and 5, though not tropic for muscle, have a greater specificity for transduction of the eye, specifically the retinal pigment epithelium (RPE), than 1 or other serotypes (Martin et al. 2002). rAAV5 is currently the most efficient for transduction of photoreceptor cells in the eye (Sun et al. 2010). Serotype 6 capsid has been shown to be efficient at transducing human airway epithelium cultures (Limberis et al. 2008). Based on these results, rAAV6 is currently in phase I clinical trial to deliver the human placental alkaline phosphatase gene to the upper airway in cystic fibrosis patients. Serotype 7 transduces muscle as efficient as AAV1, the most efficient serotype for muscle transduction identified so far (Gao et al. 2002).Serotype 8 displays ~100-fold higher transduction of the liver than serotype 2 and other serotypes (Gao et al. 2002; Cooper et al. 2009). rAAV8 is being used extensively in pre-clinical trials and has gone into patients to treat Hemophilia B (Nathwani et al. 2011). Serotypes 8 and 9 can also transduce myocardium much more efficiently (20-fold and 200-fold, respectively) than AAV1 (Pacak et al. 2006). In fact, AAV9 appears to be superior to other serotypes in a majority of tissues tested (Gao et al. 2004). A brief summary of AAV serotypes 1-9 is provided in Table 3.
property. Recovery is accomplished by subsequently amplifying the capsid variants that were successful. One of the first attempts to use this approach created a mutant capsid library (~10^6-10^7 variants) by error-prone PCR. For a function-based selection, the library was incubated with rabbit or human anti-AAV2 sera and transduced onto 293T or Hela cells, respectively (Maheshri et al. 2006; Perabo et al. 2006). This yielded two distinct groups of capsid variants, both with reduced reactivity with antibodies against serotype 2 (Maheshri et al. 2006; Perabo et al. 2006). A later design shuffled the Cap gene sequence among several natural serotypes in combination with error-prone PCR to create a potentially more diverse library (Grimm et al. 2008; Koerber et al. 2008; Li et al. 2008). Shuffling was performed between human serotypes 2, 4, 5, 8, and 9 along with avian, bovine, and caprine capsids. The resulting library was selected for transduction of human hepatocytes in the presence of human sera. A chimeric capsid of serotypes 2/8/9, AAV-DJ, was recovered (Grimm et al. 2008). This variant was shown to have higher efficiency of transduction than parent serotypes in several different tissues and transduced mouse liver much more efficiently than serotype 2.
(AAVM41) with similar transduction efficiency to AAV9 in cardiac-muscle, but de-targeted from the liver and other tissues (Yang et al. 2009). Gray et al. also employed selection of a chimeric capsid library in the CNS of rats whose blood brain barrier (BBB) was compromised by kanic acid-induced seizure. Following recovery, a chimera (clone83) was obtained that efficiently transduces neurons and oligodendrocytes, while being de-targeted for most other tissues (Gray et al. 2009).
Directed evolution has proven to be a very efficient approach for generating novel vector capsids with enhanced transduction efficiency for specific tissues. This strategy may provide a way to advance clinical approaches otherwise limited by the tissue tropisms of parental rAAV vectors. For example, therapy for cystic fibrosis (CF) has been hindered by the low efficiency with which rAAV transduces the airway epithelium. Using a directed evolution approach, two groups were able to generate several AAV chimeras, which display significantly higher transduction of the apical airway compared with its parental serotypes (Excoffon et al. 2009; Li et al. 2009). Efforts using this method are ongoing and will likely make a significant contribution to AAV-based gene therapy in the future.
al. 2003; Waterkamp et al. 2006; Michelfelder et al. 2007). Similar approaches have also been used to select for vectors that survive circulation in vivo, where selective pressures more closely reflect those in patients (Michelfelder et al. 2007; Grimm et al. 2008; Michelfelder et al. 2009).
Rational design. For AAV vector, rational design is an engineering method to make artificial capsids with enhanced performance in gene delivery based on current understanding of AAV or other related materials’ properties. To date, rational design of vector capsids has been used to enhance tissue-specific targeting, reduce non-specific targeting, and improve vector production (Zhang et al. 2002; Zhong et al. 2008; Petrs-Silva et al. 2009; Asokan et al. 2010; Bowles et al. 2011). Currently used strategies for rational design are illustrated as in figure I-3.
capsids AAV1/5, 2/5, or 3/5. In contrast, inclusion of AAV2 capsid as high as 75% was unable to make AAV2/5 bind to heparin. This phenomenon may be a result of binding affinities of those two capsids for their respective receptors, but this remains to be shown.
Rational design by peptide insertion. Peptides with specific cell binding properties, when inserting onto the surface of the vector, can alter the cell specificity of rAAV. It has been determined that the N-terminus of VP2 tolerates insertion of peptide sequences without disrupting capsid structure or vector production. Initially this was tested by insertion of peptides with known receptor binding properties or identified to bind specific targets in phage-display assays. Based on use of well-characterized peptides, insertion of the 14 amino-acid peptide L14 re-targeted vector to cells expressing integrin receptor, which were otherwise resistant to rAAV transduction (Girod et al. 1999). Similarly, insertion of the ligands for the serpin receptor or the luteinizing hormone receptor directed rAAV to lung epithelia and ovarian cancer cells, respectively (Wu et al. 2000; Shi et al. 2001). Alternatively, insertion of peptides identified by phage display has been used to generate tropism for CD13- and integrin-expressing cells, endothelial cells, brain, lung, and muscle tissues (Grifman et al. 2001; Nicklin et al. 2001; Shi et al. 2003; White et al. 2004; Work et al. 2006; White et al. 2008; Yu et al. 2009).
other tissues, including the heart (Michelfelder et al. 2009). This indicates that additional binding motifs exist on the capsid surface and contribute to the virus’s natural tropism.
rational design can be used to derive new vectors well suited to clinical applications in gene therapy.
The use of AAV-based vectors continues to increase compared to other viral and non-viral gene transfer systems in clinical trials. Due to its unique properties, the ability to transduce a broad spectrum of dividing or non-dividing cells, low immunogenicity, and sustained gene expression, this vector is well suited to most gene therapy applications. The potential to treat human monogenic disorders has been demonstrated with recent successes in clinical trials for LCA and Hemophilia B. However, there is still a great need for improvement in the specificity and efficiency of gene delivery. In the last decade, significant advancement has been achieved with various methods of rAAV engineering to address these challenges. Increasing knowledge of AAV trafficking promises to extend strategies of capsid design to these processes of transduction as well (Johnson et al. 2008; Xiao et al. 2011; Kaminsky et al. 2012). In addition, our increased understanding of the interactions between AAV and the host immune system will allow further reduction of vector immunogenicity (Mingozzi et al. 2007; Bartel et al. 2011; Mingozzi et al. 2011; Rogers et al. 2011).
Subcellular events during AAV infection
Despite many advances in vector developments, administration of high dose viral particles is required to achieve efficient transduction due to rate-limiting steps including pre-existing immune response and non-specific targeting (Rogers et al. 2011). In addition to these limitations, host cells also exploit various cellular components as barriers to AAV productive infection, including the critical steps required for viral trafficking (e.g. cell surface uptake, cytoplasmic trafficking, endosomal escape, nuclear entry, etc.) (Wang et al. 2011). Better understanding of the AAV cellular trafficking will advance our knowledge in AAV biology and facilitate the development of enhanced AAV vectors.
compartments until acidification of the compartment triggers exposure of the amino-terminus of VP1 (Ding et al. 2005). It is suggested that the N-termini of VP1 and VP2 initially reside inside the capsid and are exposed on the surface in the acidic environment of the endosome (Bleker et al. 2005; Kronenberg et al. 2005), which allows virions to escape from these vesicles (Fig.I-4). It is believed that the N-terminus of VP1 carries phospholipase activity and facilitates the escape of the viral particle by breaking down the endosome membrane (Girod et al. 2002). This endosomal processing of AAV is required for successful infection (Vihinen-Ranta et al. 1998; Ding et al. 2005).
The N-termini of VPs are suggested to be very important for virus-host interactions. For instance, two functional elements of the capsid, a phospholipase A2 (PLA2) domain and several putative nuclear localization signals (NLSs), have been identified in that region. Most parvoviruses contain a motif of ~70 amino acids in VP1 highly homologous to the catalytic domain of PLA2, which is comprised of histidine and aspartic acid residues (75HD) (Zadori et al. 2001). Mutation of these residues to alanine and asparagine (75HD/AN) does not affect cell surface attachment or endocytosis, but strongly inhibits PLA2 activity and viral infectivity (Zadori et al. 2001). PLA2 activity is proposed to penetrate the endosomal membrane to permit the escape of AAV into the cytosol.
residues of VP1 in CPV and many other parvoviruses is a canonical NLS (Vihinen-Ranta et al. 1997), several conserved, hydrophilic, basic regions have been identified in AAV2 as potential NLSs, named BR1 (120QAKKR), BR2 (140PGKKR), and BR3 (168PARKR) (Wu et al. 2000). Interestingly, one study shows that MVM is able to disrupt the nuclear envelope during infection, suggesting an alternative mechanism for the nuclear entry of parvovirus that PLA2 domain may also act at the nuclear membrane, in addition to the endosomal membrane (Cohen et al. 2006).
Upon reaching the nucleus, the virion must uncoat, while the details of this event and underlying mechanism are not well understood. It is unclear whether virion uncoating occurs prior to nuclear entry, concomitant with nuclear entry, or inside the nucleus. Intact capsids have been found to enter the nucleus, but whether the capsid disassembles completely or partially to release the genome and if this process is active or passive remain to be determined (Xiao et al. 2002; Grieger et al. 2006). The details of cellular trafficking of AAV are illustrated in figure I-5. More detailed information regarding the AAV trafficking may be found in two most recent reviews (Wang et al. 2011; Nonnenmacher et al. 2012).
Rep proteins (Kotin et al. 1992; Miller et al. 2005; Nakai et al. 2005). The episome scenario of AAV is exploited by gene therapy.
Microscopy as a method to study viral trafficking
Therapeutic gene delivery is becoming one of the major strategies to improve human health. For example, both viral and non-viral vectors (e.g. adenovirus, AAV), liposomes and nanospheres) have recently been engineered to transfer therapeutic agents in an effort to treat human diseases (Warrington et al. 2006; Rissanen et al. 2007). Encapsulation of the intracellularly-acting materials (i.e. nucleotides, proteins) into specialized delivery vehicles (i.e. viral capsids, nanospheres) that can deliver these agents to specific organelle in a controlled manner is critical to achieve efficient and selective pharmacological effects (Torchilin 2006; Breunig et al. 2008). To accomplish such functions, these vectors have to partially or fully pass through a biological jungle, which is mainly a multi-step trafficking process from cell surface binding to nuclear entry (Bartlett et al. 2000; Brandenburg et al. 2007). As a result, development of therapeutic delivery vectors has concentrated on pharmacological reagents and vector variants that affect these pathways (Duan et al. 2000; Maheshri et al. 2006; Asokan et al. 2009; Yang et al. 2009). Effective achievement of such efforts requires quantitatively evaluating the biological effect(s) of those reagents or vector variants on these complex trafficking routes and bio-distribution of delivery vehicles.
the assistance of 2D co-localization assay helped to demonstrate that AAV2 differentially traffics through late and recycling endosomes in a dose dependent manner (Ding et al. 2006), while TIRF microscopy assisted live cell imaging helped to show that actin disruption drugs could block the retrograde flow of Human papillomavirus on cell surface (Schelhaas et al. 2008).
cells because of the fact that particles may aggregate or move into a sub-resolutional region like vesicles to give a single fluorescent spot, which is common for nanoparticle trafficking. As a result, no method to date is available for quantitatively determining the bio-distribution of nano-scaled vectors in three dimensions (such as outside or inside nucleus, or traversing nuclear membrane).
In Chapter-2, based on recent advances with computational image processing (Sedarat et al. 2004; Feng et al. 2007), we developed a sensitive and reliable methodology by integrating single particle imaging and 3D quantification into classical immunofluorescence to quantitate the trafficking kinetics and bio-distribution of nanoparticles in three dimensional animal cells and tissues. Using Cy5-labeled AAV as a working model, we quantitatively investigated the nuclear entry kinetics and bio-distribution of AAV2 in human cells and mouse tissues. This study demonstrates the potential of this methodology in screening pharmacological reagents and vector variants for the development of therapeutic-material delivery strategies, as well as in understanding the intracellular behavior of delivery viral vectors in vitro and in vivo.
Microtubules in viral trafficking
particularly kinesin and dynein (Leopold et al. 2006). MTs forms a radial filamentous network with minus end anchored at a perinuclear site and plus end reaching cell surface area. This structural architecture of MTs allows the shuttling of intracellular molecules and vesicles between peri-nuclear region and other areas of a cell (Giannakakou et al. 2000; Niklinski et al. 2000) (Fig. I-6). For instance, intact MTs are required for the nuclear import of several transcription factors (e.g. p53, NF-kB, pRb) and on other hand, these structures are required to sequester other proteins (e.g. c-myc, MIZ-1, smad3) in the cytoplasm to block their nuclear entry (Giannakakou et al. 2000; Niklinski et al. 2000; Ziegelbauer et al. 2001; Roth et al. 2007; Gong et al. 2011).
influenza virus migrates on the MTs towards the perinuclear region while staying inside of the endocytic vesicles (Lakadamyali, Rust et al. 2003). Acidification of endosomes is essential for escape of influenza virus from vesicles into cytoplasm, which can be blocked by Bafilomycin and Chloroquine.
For AAV, previous studies have reported that MT disruption impairs AAV transduction (Sanlioglu et al. 2000) and AAV can bind the cytoplasmic dynein in an in vitro binding assay (Kelkar et al. 2006). However, these studies were contrasted by another publication suggesting AAV transduction was not affected by the disrupted MT network or dynein motor activity (Hirosue et al. 2007). Additionally, it remains controversial whether AAV escapes from endosomes at early or late stage. As a result, the exact roles of MTs and endosomes on AAV transduction as well as the underling mechanisms are yet unclear.
In chapter 3, we have investigated the exact role(s) of MTs on AAV transduction and corresponding potential mechanisms using multiple techniques including confocal microscopy, live cell imaging, quantitative 3D microscopy, pharmacological reagents, and single particle tracking. Moreover, we also determine for the first time that AAV2 traffics on MTs in endosomal compartment and acidification of such structure is dependent on intact MTs. This study strongly supports an as yet undocumented model in which AAV2 exploits MTs for its rapid-directed cytoplasmic transportation towards the peri-nuclear sites where acidification of endosomes for viral escape takes place.
Overcome cellular barriers: momentum of vector development
envelope sever as additional barriers for viruses to reach their replication sites. As a consequence, viruses have to overcome these cellular barriers in host cells before delivering their genome to the target region for successful infection. Like many other viruses, AAV employs receptor-mediated endocytosis to penetrate the plasma membrane of host cells. Although not clear for AAV yet, many viruses exploit MTs as an effective strategy to traverse the crowded cytoplasm in order to deliver their genetic materials to the replication sites as discuss above. Moreover, like most DNA viruses, it is critical for AAV to deposit its genome in the nucleus. Typically there are three possible mechanisms for viruses to shuttle their genomes into the nucleus. The first mechanism, as exemplified by polyomavirus infection, is importing DNA into the nucleus through an intact virion, followed by intra-nuclear capsid uncoating and genome release (Whittaker et al. 2000). The second approach, as demonstrated by Herpes-simplex-virus (HSV) and Adenovirus (Ad), involves pre-steps of capsid uncoating before the viral genome itself is imported into the nucleus (Greber et al. 1997; Ojala et al. 2000). These two nuclear entry approaches are typically mediated by nuclear pore complex (NPC) on the nuclear envelope. Finally, as exemplified by retrovirus infection, some viruses utilize mitosis to import DNA into the nucleus. This presents a unique situation where the virus takes advantage of the breakdown of the nuclear envelope during the cell cycle.
these barriers or designing capsid with enhanced ability to penetrate these barriers will offer promising opportunities to improve current vectors for gene therapy.
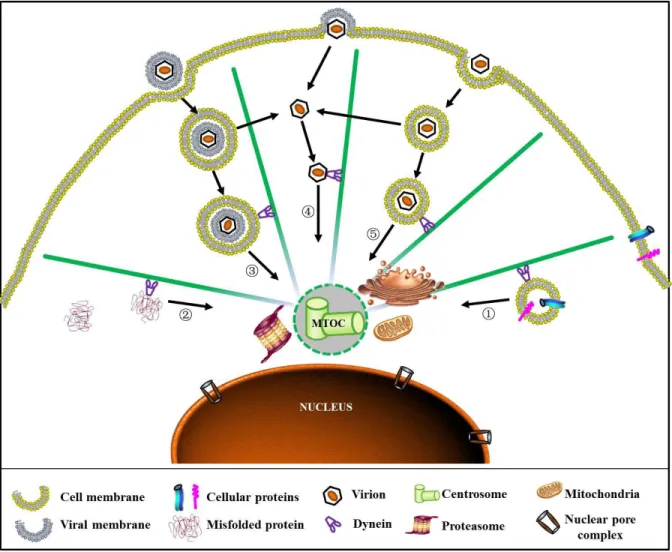
It has been documented that AAV2 enters the nucleus as an intact particle. Studies using nuclear injection of AAV monoclonal antibody (A20) can block viral transduction (Xiao et al. 2002; Sonntag et al. 2006). Sonntag and colleagues further demonstrated that most AAV genomes, unlike other parvoviruses such as MVM, are shuttled into nucleus by intact virions and subsequently uncoated there (Sonntag et al. 2006). Several nuclear localization signal (NLS) motifs including BR1-4 have been identified across the entire AAV capsid proteins VP1, VP2 and VP3. BR1-3 motifs at the N-terminus of VP1 protein have been shown to be critical for the nuclear targeting of post-entry virions (Grieger et al. 2006). Although some of these motifs have sufficient NLS activity to allow a protein to penetrate the nuclear envelope into the nucleus, the nuclear entry of AAV virions has been noted to be highly inefficient (Lux et al. 2005; Grieger et al. 2006; Sonntag et al. 2006; Xiao et al. 2011). These findings suggest that there may be another barrier besides nuclear envelope that limits the nuclear entry of AAV and successful transduction.
Strikingly, during the early stage of infection, many incoming viruses are also delivered to and retained at a perinuclear site after entering the cells (Sodeik et al. 1997; Dohner et al. 2002; Suikkanen et al. 2002; Mani et al. 2006; Yea et al. 2007; Boisvert et al. 2010; Liu et al. 2012). This perinuclear site has been suggested to be co-localized with the MTOC region and as observed for cellular proteins, transport of these viruses is facilitated by dynein motors and transport along MTs (Fackler et al. 2006; Greber et al. 2006; Wileman 2007). Although this perinuclear retention of incoming virions seems to be a common phenomenon among many viruses, especially those that enter the nucleus, it remains unclear whether this accumulation is beneficial or inhibitory to viral infection (Wileman 2007).
CHAPTER 2
QUANTITATIVE 3D TRACING OF GENE-DELIVERY VIRAL VECTORS IN
HUMAN CELLS AND ANIMAL TISSUES
Summary
Introduction
Therapeutic gene delivery is becoming one of the major strategies to improve human health. For example, both viral and non-viral vectors (e.g. adenovirus, adeno-associated virus(AAV), liposomes and nanospheres) have recently been adapted to deliver therapeutic agents in an endeavor to treat human disorders(Warrington et al. 2006; Rissanen et al. 2007). Encapsulation of the intracellularly-acting materials (i.e. nucleotides, proteins) into specialized delivery vehicles (i.e. viral capsids, nanospheres) that can deliver these agents to specific organelle in a controlled fashion is critical to attain efficient and selective pharmacological effects (Torchilin 2006; Breunig et al. 2008). To achieve such functions, these vectors have to partially or fully transverse a biological maze, which is mainly a multi-step trafficking process from cell surface binding to nuclear entry(Bartlett et al. 2000; Brandenburg et al. 2007). Consequently, development of therapeutic delivery vectors has concentrated on pharmacological reagents and vector variants that affect these pathways(Duan et al. 2000; Maheshri et al. 2006; Asokan et al. 2009; Yang et al. 2009). Effective achievement of such efforts requires quantitatively evaluating the biological effect(s) of those reagents or vector variants on these complex trafficking routes and bio-distribution of delivery vehicles.
differentially traffic through late and recycling endosomes in a dose dependent manner(Ding et al. 2006), while TIRF microscopy assisted live cell imaging helped to show that actin disruption drugs could block the retrograde flow of Human papillomavirus on cell surface(Schelhaas et al. 2008).
a sub-resolutional region like vesicles to give a single fluorescent spot, as is common for nanoparticle trafficking. As a result, no method to date is available for quantitatively determining the bio-distribution of nano-scaled vectors in three dimensions (such as outside or inside nucleus, or traversing nuclear membrane).
Materials and Methods
Production and purification of viruses. Virus was produced in HEK-293 cells as previously described(Xiao et al. 1998). Briefly, using polyethylenimine (linear molecular weight, ~25,000), cells were triple transfected with pXR2, the pXX680 helper plasmid, and pTR-CMV-GFP containing the GFP reporter transgene flanked by inverted terminal repeats. At 60hrs post-transfection, cells were harvested and nuclei were isolated as previously described(Grieger et al. 2006). The nuclear pellet from 10 plates was resuspended in 10ml PBS with 0.5% Deoxycholate (DOC) and then sonicated for 1min. This suspension was incubated at 37℃ for 45 minutes in the presence of 100μg/ml DNase. Virus suspension was
subjected to one round of cesium chloride (CsCl) step gradient density (1.3g/cm3 and 1.5g/cm3) fractionation. The viral fraction that resided in the interface between the two gradients was collected and subjected to another round of fractionation using CsCl continuous gradient density. Fractions that contained peak virus titers as determined by both slot dot blots and SDS-PAGE electrophoresis were dialyzed against 1× phosphate-buffered saline (PBS) supplemented with 5% sorbitol. Viral titers were determined by both dot blot(Grieger et al. 2006) and qPCR. The infectivity of AAV is determined to about 1 transduction unit per 100 particles.
sorbitol and stored at -80℃ as small aliquots. The degree of labeling (DOL) was determined by spectophotometry using DOL= Amax / ([virus] ×Edye), with Amax =absorbance of dye at
absorbance maximum, [virus] =virus concentration, and Edye = extinction coefficient of the
dye at its absorbance maximum. Please refer to the manufacturer’s instructions for further details. Labeled viral titers were determined by both dot blot(Grieger et al. 2006) and qPCR.
Vector administration and animal studies. Housing and handling of BALB/c mice used in the current study were carried out in compliance with National Institutes of Health guidelines and approved by the IACUC at the University of North Carolina-Chapel Hill. Recombinant AAV2 vectors packaging GFP transgenes were administered through the intramuscular (2E+09vgs into the hind limb) in a volume of 20μl PBS. At 0.5, 2, 4 hours and 6 days after intramuscular injection, animals received an overdose of pentobarbital (100mg/kg intraperitoneally) and were perfused transcardially with ice-cold 100mM sodium PBS (pH 7.4), followed by 4% paraformaldehyde in PBS (pH 7.4). After muscle was post-fixed for 24hrs at 4°C in paraformaldehyde/PBS (Xiao et al. 2007), 15μm cross sections were cut using a cyrosection microtome. Then the slides were directly sealed with mounting medium (Prolong Antifade Gold with DAPI [4’,6’- diamidino-2-phenylindole]; Molecular Probes).
Immunofluorescence. Similarly to what we have previously described(Johnson et al. 2008), HeLa cells (3E+04 per well) were plated on 12-mm glass coverslips at 24hrs before infection. Next day, after incubation in DMEM containing 20mM HEPES at 4℃ for 5min, cells were incubated with Cy5-labeled virions (5,000 or 25,000 vgs/cell) at 4℃ for another
PBS and then fixed with 4% paraformaldehyde (PFA) for 15min at room temperature (RT). The cells were then permeabilized with 0.2% Triton X-100 in PBS for 5min at RT. Following four washes with PBS, the permeabilized cells were blocked with immunofluorescence buffer (IFB) (5% normal goat serum in PBS containing 0.05% Tween-20) for 1hr at RT. The cells were incubated with primary antibody to detect Lamp1 (monoclonal from Santa Cruz Biotechnology Inc.) diluted in 50% IFB for overnight at 4°C. The cells were then incubated in secondary antibody, diluted 1:2,000 in 50% IFB (anti-mouse Alexa-Fluor 488 [Molecular Probes]), for 1hr at RT. After six washes with PBS, coverslips were mounted cell side down on glass slides with mounting medium (Prolong Antifade Gold with DAPI [4’,6’- diamidino-2-phenylindole]; Molecular Probes).
3D confocal fluorescence microscopy and 3D blind deconvolution. The labeled Hela cells were examined by use of a Zeiss LSM710 laser scanning confocal microscope equipped with a Zeiss Plan-Apochromat 63×/NA 1.40 oil objective. The confocal pinhole aperture was set to the diameter of the first Airy disk. Stacks of 20-30 focal planes were captured at 0.31μm z-intervals through the depth of the cell. 3D images of the cells were reconstructed by using the image stacks. The Nyquist theorem, which utilizes the limitation of the microscope optics [full-width half-maximum (FWHM)] to dictate adequate sampling, was used to determined that pixel dimensions of 0.13×0.13×0.31μm (X, Y, Z) were required to properly sample the data.
objective, refractive index of the medium, excitation wavelength, emission wavelength, confocal pinhole radius, pixel size, z-axis interval, microscope type (i.e., wide field, confocal), and number of excitation photons. A new adjusted adaptive PSF derived from the previous deconvolution round was used to generate next adaptive PSF that fits the real imaging data better than the previous one (termed as one iteration or deconvolution round(Pawley 2006)). The number of iterations may serve as a regularization factor. In general, the remaining restoration error decreases with an increasing number of iterations. At the same time, the error due to noise amplification increases. The procedure should be stopped at an iteration number in which the sum of both errors is minimal(Vandervoort et al. 1995). To determine the optimal number of iteration, intracellular Cy5-AAV2 particles were deconvolved up to 50 rounds and the resulting images were saved every 5 rounds. The resulting images from 10 deconvolution rounds (number of iteration) displayed the highest SNR, best spatial resolution and most closely resemble the fluorescence signal from a point light source (Fig.II-2). This iteration number (10 rounds) was then used to deconvolve all the confocal images in this paper.
isosurface rendering was thresholded at the fluorescence intensity of 2500a.u.. The parameters (volume, MFI, TFI) for these isosurface coated Cy5-AAV2 objects were extracted from the IMARIS program and analyzed as described in result section. The localization of Cy5-AAV2 in nucleus or lysosomes was analyzed as described in result section. Aberrations caused by refractive index mismatch results in a suboptimal z resolution. FWHM changes of the point spread functions of the two channels (DAPI and Cy5) differed one voxel (0.13 × 0.13 × 0.31 μm3
) in XY- and Z-directions, while no significant changes was observed between AF488 and Cy5. The channel registration difference between DAPI and Cy5 was fixed by adjusting the DAPI channel using channel shift module in IMARIS to avoid potential false-positive localization results.
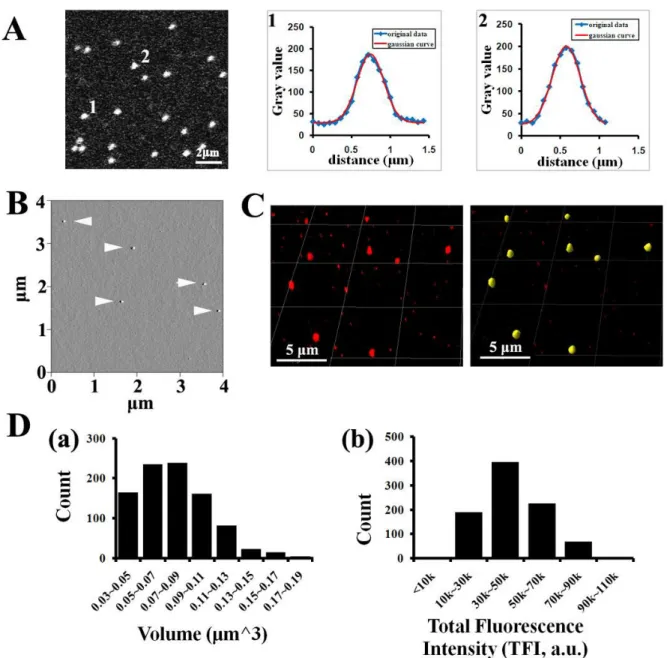
AFM. After imaging with confocal microscope, the coverslips were removed from glass slides and gently rinsed with PBS. The coverslips were quickly rinsed in ddH2O, blotted dry and then slightly dried under a stream of nitrogen. The images were captured in air with a Nanoscope IIIa (Digital Instruments) microscope in tapping mode. Pointprobe tapping mode silicon probes (Molecular Imaging Corporation) with spring constants of ~50 N/m and resonance frequencies ~170 kHz were used for all imaging. The images were collected at a speed of 4Hz, a size of 4μm × 4μm, and a resolution of 512 × 512 pixels.
Results
Quantitative 3D distribution microscopy. To precisely tracking and localize labeled particles in cells and tissues, we developed an object-based quantitative 3D distribution microscopy which is composed of two steps: 1) assess the localization of particles, 2) quantitate the number of particles in each cellular structure. Here we used localization of particles in nucleus as an example to explain this method. As diagramed in Figure II-1A-a, five nanoparticles (P1-P5 in four groups G1-G4) are differentially localized in a cell, with P1 inside the nucleus, P2 transversing nuclear membrane, P3-P5 outside of the nucleus, and especially P4 and P5 associated with a sub-resolutional region (i.e. vesicles with diameter less than 200nm). Confocal microscope is used to create a z-stack sectioning through the entire cell and generate 3D fluorescence images. Image distortions are a natural aspect of an optical microscope (noise, scatter, glare and blur) and diminish the contrast and resolution of raw images, thus impairing the accuracy of quantitative image analysis. To counteract these problems, restorative 3D deconvolution is used to reverse the optical distortions, negating the effects of the optical system that are represented by its point spread function (PSF, Fig.II-2A). After 3D deconvolution, the fluorescence images are visualized by 3D volume rendering as shown in Figure II-1A-b, with four fluorescence spots (#1-#4) formed by corresponding particles.
group (Fig. II-1A-a: G1-G4). The localization of each particle is then determined by the amount of DAPI signal within its VOI (Fig. II-1A-c: V1-V4). Specifically, the mean fluorescence intensity (MFI) of DAPI in V1 is the same as that in nucleus, demonstrating that P1 is inside the nucleus; the MFI of DAPI in V3 and V4 is the same as that in the cytoplasm, but much lower than that in nucleus, demonstrating that P3-P5 are outside the nucleus; and the MFI of DAPI in V2 is between those in nucleus and in cytoplasm, demonstrating that P2 is traversing the nuclear membrane.
To count the number of particles, classical centroid counting method fails when several particles associate with a sub-resolutional region and display as a single fluorescent spot (Fig. II-1A-b: spot #4), which is very common in nanoparticle trafficking. In addition, the amount of dyes (or dye-labeled particles) should be calculated by the number of emitted photon or fluorescence intensity instead of fluorescence volume(Akita et al. 2004; Chen et al. 2008), since there is no linear correlation between the amount of dyes and fluorescence volume ((Pawley 2006) and Fig.II-3). To quantitate particles including those within sub-resolutional regions, we calculate such number using the total fluorescence intensity (TFI) of each fluorescence spot by the formula: number of particles in structure-X =∑TFIstrX/mTFIs,
in which TFIstrX denotes the TFI of fluorescence spots formed by labeled particles in cellular
structure X, and mTFIs denotes the mean value of TFI experimentally calculated from