Order of Phase Transition on a Simple Cubic Lattice
Determined by Cluster Variation Method
Naoya Kiyokane
1;*and Tetsuo Mohri
1;21
Division of Materials Science and Engineering, Graduate School of Engineering, Hokkaido University, Sapporo 060-8628, Japan 2Research Center for Integrative Mathematics, Hokkaido University, Sapporo 060-8628, Japan
Detailed analyses of the free energy behavior near the order-disorder transition temperature on a simple cubic lattice are attempted by cluster variation method (CVM). The entropy is formulated within a cubic approximation of the CVM and two kinds of nearest neighbor pair interaction energies are assumed in the internal energy; one is independent of an atomic distance,r, and the other depends onrthrough Lennard-Jones type pair potential. In both cases, it is found that the order of the transition is of second order. [doi:10.2320/matertrans.MBW200904]
(Received October 16, 2009; Accepted December 28, 2009; Published February 25, 2010)
Keywords: cluster variation method, order of phase transition, simple cubic lattice, correlation function
1. Introduction
In general, phase transition is classified into two catego-ries, first order and second order transitions. Among various criteria proposed to define the order of the transition, Ehrenfest’s criterion1)has been often employed. According
to this criterion, the order of the phase transition is defined by the discontinuity of a free energy or its derivative. When a series of derivatives of a free energy function with respect to an independent thermodynamic variable is continuous up to the (n1)-th order and the discontinuity appears at then-th order derivative, the transition is of then-th order. The other criterion which has been equally employed is due to Landau2)
who claims that the existence and non-existence of a meta-stable phase are a measure of the order of the transition, and the first (second) order transition is characterized by the existence of a meta-stable phase (state). The equivalence of these two criteria has been amply demonstrated by one of the present authors (TM)3,4)for order-disorder transitions based
on the temperature dependence of a free energy curve as a function of a long-range order parameter (hereafter abbre-viated as LRO parameter). As is shown in the latter, the free energy of an ordering system is generally a function of various configurational variables. By eliminating all those configurational variables except a LRO parameter, one can demonstrate that the existence (non-existence) of a meta-stable state is closely related to the variation of the curvature of the free energy curve with temperature.
In actual alloy systems, it is often observed that several ordering reactions proceed with decreasing/increasing the temperature at a same composition. One of the typical examples is found in Ni2MnGa and Ni2AlTi systems for
which L21 and B2 orderings take place in the low
temper-ature region, and these ordering reactions play a deterministic role in the functional and structural properties such as ferromagnetic shape memory effect, thermoelasticity etc. It is interesting to note that the L21to B2 transition for Ni2MnGa
is of the second order as was confirmed by the measurement of a LRO parameter,5)while it is of the first order for Ni2AlTi
which is claimed based on the co-existence of two phases in
the microstructural observation.6)The origin of the different
order of the transition between these two alloys has not been clarified yet.
In our previous work,7)the phase stability of L2
1 ordered
phase and the transition behavior from L21 to B2 ordered
phase were studied based on the cluster variation method (hereafter CVM).8) The entropy is calculated within the
tetrahedron approximation of the CVM9) and the internal energy is written by the pair interaction model including the nearest and next nearest neighbor pairs. It should be recalled that both the ordered phases degenerate energetically if only the nearest neighbor pair interactions are considered. We carefully examined the temperature dependencies of the grand potentials of the two ordered phases by changing the combination of the first and second nearest neighbor pair interaction energies, and concluded that the transition is of the second order since the two grand potentials merge into a single grand potential of B2 ordered phase without intersect-ing each other as approachintersect-ing the transition temperature. In these calculations, however, the pair interaction energies are given as fixed values and do not depend on an atomic distance, concentration and all other chemistry of constituent elements. Hence, the main objective of the present study is to examine the ordering behavior by introducing the depend-ence on the atomic distance into pair interaction energies.
It is anticipated, however, that the analysis of the ternary ordering behavior under the pair interaction energies which depend on an atomic distance is quite cumbersome. Then, by noting the fact that the L21to B2 transition of aA2BCternary
system can be regarded as the binary ordering of aBCsystem on a simple cubic lattice which constitutes a sublattice of the L21 ordered phase, the main focus is placed on the binary
ordering on a simple cubic lattice.
A free energy curve behaves very sensitively to the change of a LRO parameter around a transition temperature and the determination of the order of the transition requires both the accurate free energy model and careful numerical calcula-tions with high accuracy. In the present study, CVM is employed to describe the free energy together with Lennard-Jones type pair interaction energies. It has been amply demonstrated that such a scheme with a relatively small basic cluster of the CVM provides both the accurate transition
*Graduate Student, Hokkaido University
temperatures as well as the order of the transition for both fcc- and bcc-based systems.10,11)It should be noted that the
basic cluster which is a largest cluster considered in the
entropy formula in the present calculation for a cubic system is a cubic cluster in which numerous independent cluster variables are identified as will be demonstrated in the following section. Therefore, as compared with the tetra-hedron approximation which has been most often employed for fcc- and bcc-systems, the number of the configurational variables dealt in the present study is far larger and extra care should be taken in the numerical calculations.
The organization of the present report is as follows. In the next section, for the sake of completeness, the theoretical framework of the present study is briefly described. Prior to the main subject of the binary ordering under the pair interactions which depend on an atomic distance, the results for a simple constant pair interaction model are presented in the third section.
2. Calculation Method
2.1 Free energy
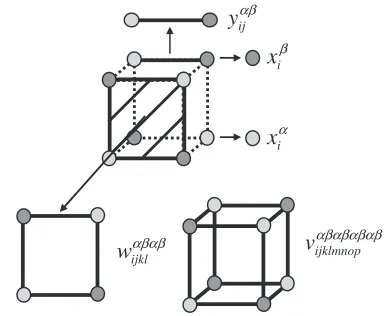
The ordered phase for a simple cubic lattice which is studied in the present report is shown in Fig. 1. The stoichiometric composition of this ordered phase is 50 at% and the entire lattice consists of two sublattices,and, as shown in Fig. 1. WhenBandCspecies arrange preferentially on and sublattices, respectively, which are surrounded by A species on the interpenetrating simple cubic lattice, L21 ordered phase is formed. The free energy is formulated
for theBCbinary system on a simple cubic lattice. In the following expression of the Helmholtz energy,F
F¼ETS; ð1Þ
whereEis the internal energy,Tthe temperature andSis the entropy. The internal energyEis described as
E¼ z
2
X
i;j
eijyij ; ð2Þ
where eij is the atomic pair interaction energy between
speciesi andj,yijis a pair cluster probability of finding an
atomic arrangement specified by subscripts on a nearest neighbor pair cluster and z is the first nearest neighbor coordination number which is six for a simple cubic lattice. For the sake of simplicity, only pair interaction energies are considered in the present study. This is believed to be quite reasonable since the dominant cohesive mechanism to maintain high symmetric cubic structure is described to central forces (pair interaction energy).
As is described in the introduction, the cubic approxima-tion of the CVM is employed to formulate the entropy term which is written as8)
S¼kB (
3X
i;j;k;l
Lðwijkl Þ þ1
2
X
i
LðxiÞ þX j
LðxjÞ !
X
i;j;;p
Lðvijklmnop Þ 3X
i;j
Lðyij Þ )
; ð3Þ
whereLðXÞis defined asXlnXX,xi,wijkl andvijklmnopare
cluster probabilities of finding atomic arrangements specified by subscript(s) on a point, square and cubic clusters, respectively, andkBis the Boltzmann constant. Each cluster
is illustrated in Fig. 2. Note that the superscript(s) in the cluster probabilities indicates a sublattice(s). Then, together with the internal energy given by eq. (2), the Helmholtz energy is expressed as
F¼3X
i;j
eijyij
kBT
(
3X
i;j;k;l
Lðwijkl Þ þ1
2
X
i
LðxiÞ þX j
LðxjÞ !
X
i;j;;p
Lðvijklmnop Þ 3X
i;j
Lðyij Þ )
: ð4Þ
It is essential to realize that the cluster probabilities appearing in eq. (3) are mutually dependent through normal-ization and geometrical conditions given by
1¼X
i
xi ¼X
j
xj ¼X
i;j
yij
¼X
i;j;k;l
wijkl ¼ X
i;j;;p
vijklmnop ð5Þ
and
xi ¼ X
j;k;l;;p
vijklmnop ; xj ¼ X i;k;l;;p
vijklmnop ;
yij ¼ X
k;l;;p
vijklmnop ; wijkl ¼ X m;n;o;p
vijklmnop ; ð6Þ
respectively. Under such constraints, the minimization of the Helmholtz energy is carried out by employing Natural Iteration Method.9) In the present study, however, cluster probabilities are transformed to a set of correlation
functions12–14) which forms independent configurational
α sublattice
β sublattice
Fig. 1 Ordered phase in the simple cubic structure.
α
i
x
β
i
x
αβ
ij
y
αβαβ
ijkl
w
v
αβαβαβαβijklmnop [image:2.595.326.518.73.231.2] [image:2.595.306.550.602.723.2]variables and Newton-Raphson procedure is employed to minimize the Helmholtz energy. The definition of the correlation functions has been amply demonstrated in the previous publication15)and is not reproduced in this report,
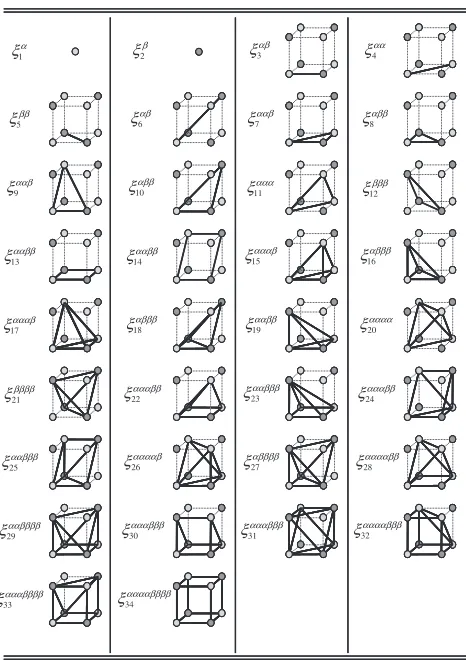
but it is noted that 34 kinds of independent correlation function are identified in the present study and these are tabulated in Table 1. The Helmholtz energy in eq. (4) is, therefore, formally rewritten as
F¼FðT :xi;xj;yij ;wijkl ;vijklmnop Þ
¼FðT :1; 2; 3; 4 ; 5 ; ; 34 Þ: ð7Þ
Among these 34 correlation functions, at1 : 1stoichiometric composition which is the main concern of the present study, the number of independent variables is reduced due to the following conditions originating from the symmetry (anti-symmetry) of exchanging the species on a cluster which consists of even (odd) number of lattice points,
1 ¼ 2; 4 ¼5; 7 ¼ 8 ; : ð8Þ
Then, the number of independent correlation functions is reduced to 21 and the Helmholtz energy is further rewritten as
F¼F T :
1;
3 ; 4 ;
6 ;
7 ;
9 ; 11 ;
13 ; 14 ; 15 ; 17 ;
19 ; 20 ;
22 ; 24 ; 26 ; 28 ; 30 ; 31 ; 32 ; 34 !
: ð9Þ
Among these correlation functions, the point correlation function,
1, on thesublattice, describes the difference of
the concentration of species A (or B) between the two sublattices and, therefore, serves as a LRO parameter, while others are short-range order (SRO) parameters. The unity of
1(1¼1:0) indicates a fully ordered state which is realized
only at 0 K, while a complete disordered state is described by null value (1¼0:0).
Then, the Helmholtz energy is minimized with respect to SRO parameters under a given set of temperature and a LRO parameter through Newton-Raphson procedure, which yields the Helmholtz energy written in the following form,
F ¼FðT;
1Þ: ð10Þ
By plottingFas a function of
1for various temperaturesT,
one can analyze the Helmholtz energy behavior to determine the order of the transition.
2.2 Pair interaction energy
In the pair interaction energyeijin eq. (2), the dependence
on the atomic distance, r, is introduced through Lennard-Jones type potential,
eijðrÞ ¼eij
r ij r m m n r ij r n
ð11Þ
where e ij and r
ij describe the depth of the potential and
equilibrium atomic separation, respectively, and the expo-nentsmandnare related with stiffness of the lattice against the compression and expansion. In the present study,m¼12
andn¼6are assigned and the values employed fore ijandrij
are summarized in Table 2. Becauseje
ABjis larger thanjeAAj
and je
BBj, ordering reaction can be expected. Then, the
internal energy is written as
E¼X
i;j
eijðrÞ y
ij
¼X
i;j
eij r
ij r m m n r ij r n
yij : ð12Þ
It is noted that due to the introduction of the dependence of the atomic distance into the Helmholtz energy, additional minimization of the Helmholtz energy should be carried out with respect to r, which yields the equilibrium atomic distance,r,
@E
@r
r¼r
[image:3.595.50.283.454.784.2]¼0: ð13Þ
Table 1 34 kinds of correlation functions in the simple cubic lattice.
ααβ
ξ9
αββ
ξ10
ααα
ξ11 ξ12βββ
ααββ ξ13 ααββ ξ14 αααβ ξ15 αβββ ξ16 α ξ1 αβ ξ3 αα ξ4 β ξ2 ββ ξ5 ααβ ξ7 αββ ξ8 αβ ξ6 αααβ ξ17 αβββ ξ18 ααββ ξ19 αααα ξ20 ββββ ξ21 αααββ ξ22 ααβββ
ξ23 ξ24αααββ
[image:3.595.305.550.733.786.2]ααβββ ξ25 ααααβ ξ26 αββββ ξ27 ααααββ ξ28 ααββββ ξ29 αααβββ ξ30 αααβββ ξ31 ααααβββ ξ32 ααααββββ ξ34 αααββββ ξ33
Table 2 The Lennard-Jones parameters employed in the present study.
ij e
ij rij
AA 0.65 0.95
AB 1.00 1.00
3. Results and Discussion
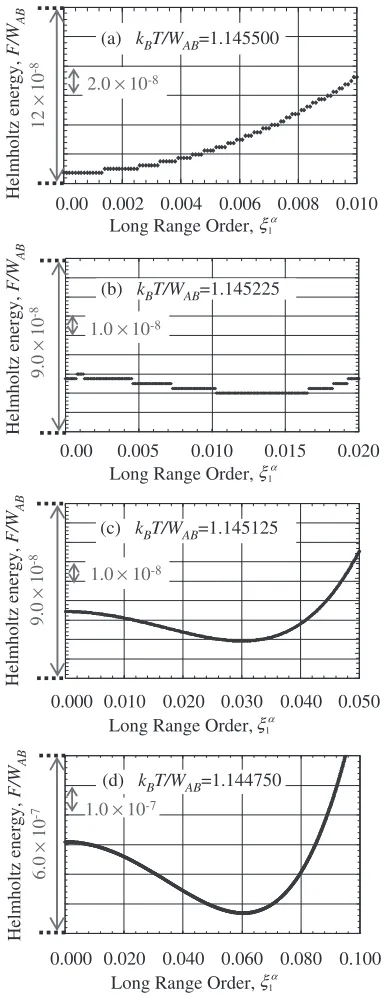
Prior to introducing the dependence of the atomic distance into the pair interaction energy, the Helmholtz energy behavior in the vicinity of the disordered state (
1 ¼0) is
analyzed under a set of constant pair interaction energies,
eAA¼eBB¼ eAB¼0:25. The dependencies of the
Helmholtz energy on the LRO parameter for various tem-peratures are depicted in Figs. 3(a)–(d). The Helmholtz energy and temperature are normalized with respect to the effective pair interaction energy which is defined byWAB¼ ðeAAþeBBÞ 2eAB. The change of the Helmholtz energy
shown in Figs. 3(a) and (b) are not smooth, which is caused by numerical processing within a very small scale as one can realize in the vertical axis. However, the discontinuous change of the Helmholtz energy is negligibly small and no significant effects are anticipated in determining the order of the transition.
As shown in Fig. 3(a), the Helmholtz energy minimum appears at the disordered state (
1 ¼0), and with decreasing
temperature, the minimum shifts in the right hand side direction, and at the lower temperatures (2(c) and (d)). Helmholtz energy minima are realized in the finite values of the LRO parameter. During this transition, the Helmholtz energy curves are kept to be flat without forming a local minimum at the disordered state, which confirms the second order nature of the order-disorder transition on a simple cubic lattice. Equivalently, as shown in Figs. 3(c) and (d), the equilibrium LRO parameter of an ordered state shifts towards the null value (1 ¼0) continuously with increasing the temperature, which is a different expression of the second order transition. The flatness of the Helmholtz energy curve which is mathematically described by the vanishing of the second order derivative suggests that the small amount of configurational fluctuation is amplified significantly. This is directly related to the short range order diffuse intensity scattering associated with the spinodal ordering.16–18)
Within the accuracy of the present calculation, the transition temperature is located between 1.145500 and 1.145225, which agrees quite well with previous result.19)
As was described in the previous section, in order to introduce the dependence on the atomic distance into the pair interaction energies, Lennard-Jones type pair potential given by eq. (12) is employed. The Lennard-Jones parameters are tabulated in Table 2. The behavior of the Helmholtz energy in the vicinity of the disordered state is calculated for four temperatures and demonstrated in Figs. 4(a)–(d). In these calculations, the equilibrium atomic distance is determined by eq. (13) for each temperature and LRO parameters. It was confirmed that the magnitude of the equilibrium lattice constants do not change appreciably within the narrow range of LRO parameters considered in Figs. 4(a)–(d). Note that both the Helmholtz energy and temperature are normalized with respect to e
AB. Throughout these temperatures, the
numerical convergence was a serious problem and the obtained Helmholtz energy curves are not smooth except
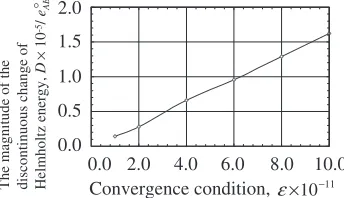
at kBT=eAB¼0:96100 (3(d)). The maximum discontinuity
(jump) of the Helmholtz energy is plotted as a function of the convergence parameter "c in Fig. 5. The convergence
criterion adopted in the present study is as follows; when
the value of "¼PjðiinputÞðioutputÞj, where i specifies a cluster and the sum is taken over 21 correlation functions, is smaller than "c, the convergence is attained. One can
confirm in Fig. 5 that the magnitude of the discontinuity tends to go to null value with smaller convergence parameter. The convergence parameter employed in the present study is"c¼1013which is strict enough to examine the detailed
behavior of the Helmholtz energy and to determine the order of the transition.
The behavior of the Helmholtz energy curve in the vicinity of a disordered state across the transition temperature
kBT=eAB¼0:9612is quite similar to the one with constant
12
×
10
-8
Helmholtz energy,
F/W
AB
0.00 0.002 0.004 0.006 0.008 0.010 Long Range Order, ξ1α
0.00 0.005 0.010 0.015 0.020
9.0
×
10
-8
Helmholtz energy,
F/W
AB
Long Range Order, ξ1α
Long Range Order, ξ1α
0.000 0.020 0.040 0.060 0.080 0.100
6.0
×
10
-7
Helmholtz energy,
F/W
AB
0.000 0.010 0.020 0.030 0.040 0.050
9.0
×
10
-8
Helmholtz energy,
F/W
AB
Long Range Order, ξ1α (a) kBT/WAB=1.145500
2.0×10-8
(b) kBT/WAB=1.145225
(c) kBT/WAB=1.145125
(d) kBT/WAB=1.144750
1.0×10-8
1.0×10-8
1.0×10-7
[image:4.595.331.524.71.568.2]pair interaction energies, and the order of the transition is determined to be of the second order. The equilibrium atomic distance at the transition temperature for the disordered state,
1 ¼0, is obtained asr=rAB ¼1:0011, accordingly the
effective pair interaction energy, WAB, is determined to be
0.892. The normalized transition temperature is, therefore, calculated as kBT=WAB¼1:08 which is somewhat smaller
than the one determined for the case of constant pair interaction energies.
Within the pair interaction model, the internal energy can be tacitly rewritten as,
E¼3X
i;j
eijyij
¼ 3
22 fðeAAþ2eABþeBBÞ
þ2ðeAAeBBÞ1þWAB2g ð14Þ
where 1 and 2 are point and nearest neighbor pair
correlation functions, respectively. In fact, eq. (14) is equivalent to cluster expansion20) of the internal energy.
When the random solid solution at 50 at% which is a main concern of the present study is taken as the energy reference state, the ordering energy,Eo, is written as
Eo ¼EEr¼
3
22 f2ðeAAeBBÞ 1þWAB2g ð15Þ
whereEris the internal energy of the random solid solution
and given as Er¼ ð3=22Þ ðeAAþ2eABþeBBÞ. By noting
that the point correlation function vanishes at1 : 1 stoichio-metric composition, the ordering energy is given as
Eo¼
3
22WAB2: ð16Þ
Equation (16) indicates that, in the case of a constant pair interaction energy model, two atomic speciesAandBcan not be energetically distinguished, sinceWAB is symmetric with
respect to the exchange ofAandB. While under the Lennard-Jones type pair potential, difference of atomic species is reflected in the effective pair interaction energyWABthrough
Lennard-Jones parameters,e
ijandrij. General arguments of
the order of the transition are based on the symmetry of the superlattice points without referring to the constituent elements. The constant pair interaction model falls into this category. Whereas the present study suggests that order-disorder transition on the simple cubic Lennard-Jones lattice is of the second order no matter what the constituent elements are, although the possibility that the different choice of the Lennard-Jones parameters may lead to the first order transition is not completely excluded.
The present calculations are carried out for the binary transition on a simple cubic lattice and these are regarded as the preliminary analysis to investigate more realistic ternary ordering on the L21lattice. It is stressed that the second order
transition confirmed in the present study for the simple cubic lattice is not directly applicable to L21to B2 transition on the
parent lattice. In particular, the experimental observation of the first order transition in Ni2AlTi implies that the order of
the transition on two interpenetrating lattices is seriously affected by the configurational interference between the two lattices through the chemistry of constituent elements. This
Convergence condition, ε×10−11
0.0 2.0 4.0 6.0 8.0 10.0 0.0
0.5 1.0 1.5 2.0
The magnitude of the discontinuous change of Helmholtz energy,
D
×
10
-5/ eAB
°
Fig. 5 Relationship between discontinuous Helmholtz energy change vs convergence condition of Newton-Raphson method.
0.00 0.010 0.020 0.030 0.040 0.050
14
×10
-7
Long Range Order, ξ1α
7
×10
-7
Long Range Order, ξ1α
0.00 0.010 0.020 0.030 0.040 0.050
0.0 0.020 0.040 0.060 0.080 0.100
14
×10
-7
Long Range Order, ξ1α
0.0 0.020 0.040 0.060 0.080 0.100
30
×
10
-7
Long Range Order, ξ1α (a) kBT/eAB°=0.96150
(b) kBT/eAB°=0.96120
(c) kBT/eAB°=0.96110
(d) kBT/eAB°=0.96100
2.0×10-7
1.0×10-7
2.0×10-7
2.0×10-7
Helmholtz energy,
F/e
AB
°
Helmholtz energy,
F/e
AB
°
Helmholtz energy,
F/e
AB
°
Helmholtz energy,
F/e
AB
°
Fig. 4 Helmholtz energy change as a function of LRO parameter under Lennard-Jones pair interaction energies at kBT=eAB¼0:96150 (a),
kBT=eAB¼0:96120 (b), kBT=eAB¼0:96110 (c) and kBT=eAB¼ 0:96100(d). The Helmholtz energy and temperature are normalized with respect toe
[image:5.595.76.269.70.566.2] [image:5.595.83.255.657.756.2]implication motivates the further investigation based on the first-principles investigation based on electronic structure total energy calculation which remains as a future inves-tigation.
4. Conclusions
In the present study, the order of phase transition of a binary system on a simple cubic lattice which undergoes the order-disorder transition was determined. We investigated two systems with different type of pair interaction energies; one is the system with constant pair interaction energies like Ising system and the other is Lennard-Jones system for which the pair interaction energies depend on an atomic distance. In both cases, it is confirmed that the order of the transition is of the second order.
REFERENCES
1) H. B. Callen:Thermodynamics and an introduction to thermostatistics, 2nd ed., (John Wiley & Sons Inc., 1985).
2) L. D. Landau and E. M. Lifshitz:Statistical Physics 3rd ed.; Course of Theoretical Physics, vol. 5, (Butterworth-Heinernann, 1984). 3) T. Mohri: Modelling Simul. Mater. Sci. Eng.8(2000) 239.
4) T. Mohri:Properties of Complex Inorganic Solid 2, ed. A. Meike, (Kluwer Academic/Plenum Publishers, 2000) p. 123.
5) R. W. Overholser and Manfred Wuttig: Sci. Mater.40(1999) 1095– 1102.
6) R. S. Polvani, Wen-shian Tzeng and P. R. Strutt: Metall. Trans.7A
(1976) 33–40.
7) N. Kiyokane and T. Mohri: Collected Abstracts of the 2008 Autumn Meeting of the Japan Inst. Metals (2008) p. 262.
8) R. Kikuchi: Phys. Rev.81(1951) 988–1003. 9) R. Kikuchi: J. Chem. Phys.60(1974) 1071–1080.
10) C.-S. Oh, T. Mohri and D. N. Lee: Mater. Trans., JIM35(1994) 445– 450.
11) T. Mohri, I. Yamagishi, T. Suzuki, C.-S. Oh, D. N. Loo, M. Yashima, M. Yoshimura and C. Ohno: Z. Metallk.86(1995) 353–358. 12) J. M. Sanchez and D. de Fontaine: Phys. Rev. B17(1978) 2926–2936. 13) J. M. Sanchez, F. Ducastelle and D. Gratias: Physica (Utrecht)128A
(1984) 33.
14) T. Mohri, J. M. Sanchez and D. de Fontaine: Acta Metal.33(1985) 1463–1474.
15) T. Mohri:Statistical Thermodynamics and Model Calculations in Alloy Physics Chapt. 10, ed. W. Pfeiler, (WILEY-VCH, 2007) pp. 525–588 (and references therein).
16) D. de Fontaine: Acta Metal.23(1975) 553–571.
17) T. Mohri, J. M. Sanchez and D. de Fontaine: Acta Metal.33(1985) 1171–1185.
18) J. M. Sanchez: Physica (Utrecht)111A(1982) 200. 19) D. de Fontaine: Solid State Phys.34(1979) 73–274.