organic papers
Acta Cryst.(2005). E61, o2945–o2946 doi:10.1107/S1600536805025717 Getsis and Mudring C
16H31N2+BrH2O
o2945
Acta Crystallographica Section E
Structure Reports Online
ISSN 1600-5368
1-Dodecyl-3-methylimidazolium bromide
monohydrate
Anna Getsis and Anja-Verena Mudring*
Institut fu¨r Anorganische Chemie, Universita¨t zu Ko¨ln, Greinstrasse 6, D-50939 Ko¨ln, Germany
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study T= 263 K
Mean(C–C) = 0.003 A˚ Disorder in main residue Rfactor = 0.026 wRfactor = 0.069
Data-to-parameter ratio = 21.9
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography
Printed in Great Britain – all rights reserved
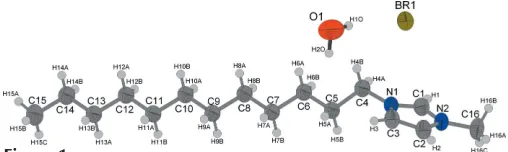
The asymmetric unit of the title compound, C16H31N2+Br -H2O, contains one crystallographically independent 1-do-decyl-3-methylimidazolium cation, one bromide anion, to counterbalance the charge, and one solvent water molecule. The halide anion forms hydrogen bonds with the H atoms of the imidazole ring and with the water H atoms.
Comment
Many 1-alkyl-3-methylimidazolium salts show thermotropic liquid crystalline behaviour (Seddonet al., 1996; Gordonet al., 1998; Hardacreet al., 2001; Bradleyet al., 2002). Aside from a large mesophasic range, many of these salts also have a melting point below 373 K and thus belong not only to the class of liquid crystals but also to the class of ionic liquids (Wasserscheid, 2002).
The liquid crystalline behaviour of 1-dodecyl-3-methyl-imidazolium bromide, [C12mim]Br, and its monohydrate, [C12mim]BrH2O, (I), has recently been a topic of investiga-tion although the crystal structures had not been established. We were now able to obtain crystals of the monohydrate which were of sufficient quality for structure analysis by recrystallizing the compound from ethyl acetate.
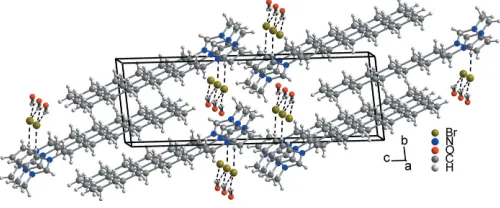
[C12mim]BrH2O crystallizes isotypic with [C12mim]ClH2O (Guillet et al., 2004). The bond lengths and angles are well within the expected ranges; the imidazolium head group is planar and the alkyl chain adopts an all-transconformation. The 1-dodecyl-3-methylimidazolium cations form lamellar bilayers parallel to the (022) plane. The orientation of the cations (due to their amphiphilic character) alternates from layer to layer so that separate hydrophobic (alkyl tail groups) and hydrophilic (the imidazolium head group, bromide and water) regions are formed. The alkyl tails of cations with different orientation are interdigitated. The [C12mim] cations
[image:1.610.205.459.630.706.2]Received 8 August 2005 Accepted 11 August 2005 Online 17 August 2005
Figure 1
themselves are held together by electrostatic interactions with bromide anions and water molecules. Within each layer a water molecule and a bromide anion are located between two 1-dodecyl-3-methylimidazolium cations. The water–imidazo-lium O H1 distance of 2.34 A˚ is clearly shorter than the mean value for a hydrogen bond between an (N,N)Csp2—H group and a water O atom (Steiner, 2002), whereas the interaction of the imidazolium C3—H3 with Briof 2.75 A˚ is about 0.1 A˚ above the commonly observed mean values. The water hydrogen–bromide distances of 2.61 (2) A˚ for H1O Br and 2.57 (2) A˚ for H2O Br are also in the typical range for water hydrogen–bromide hydrogen bonds, whereas unlike in the chloride the ring methyl (C16)–hydrogen halide distances are too long for a significant hydrogen bond (Steiner, 2002); the shortest contact is 2.98 (2) A˚ . Furthermore, each water molecule is located above or below an imidazolium ring with an oxygen ring centre distance of 3.699 (8) A˚ .
Experimental
1-Methylimidazole (16.7 ml, 0.21 mol) was added to 1-bromo-dodecane (72.0 ml, 0.30 mol) (both were freshly distilled before use). After heating the reaction mixture at 373 K for 2 h, a yellow liquid was obtained which was degassed under vacuum at 393 K. After cooling to room temperature, the reaction product solidified. The crude product was washed with ethyl acetate until the ester phase became colourless. After recrystallization from ethyl acetate, white transparent needle-shaped crystals of 1-dodecyl-3-methylimidaz-olium bromide monohydrate were obtained. Crystals suitable for structure analysis were sealed in glass capillaries (0.3 mm diameter) and their quality checked by Laue photographs. A complete data set was taken of the best specimen.1H NMR: (300 MHz, DMSO-d6):
0.85 (t, 3H, H-15), 1.24 (br. s, 20H, H-5–14), 3.85 (s, 3H, H-16), 4.15 (t, 2H, H-4), 7.71 (s, 1H, H-2), 7.78 (s, 1H, H-3), 9.15 (br. s, 1H, H-1).
13C{1H} NMR: (75.5 MHz, DMSO-d
6):13.85 (s, C-15), 21.97, 25.38,
28.26, 28.60, 28.71, 28.83, 28.90, 29.27, 31.18, 35.65 (s, C-5–14), 38.60 (s, C-16), 48.65 (s, C-4), 122.15 (s, C-3), 123.48 (s, C-2).
Crystal data
C16H31N2+Br
H2O
Mr= 349.34
Triclinic,P1 a= 5.500 (5) A˚ b= 7.794 (5) A˚ c= 22.961 (5) A˚
= 81.893 (5)
= 83.761 (5)
= 78.102 (5)
V= 950.3 (11) A˚3
Z= 2
Dx= 1.221 Mg m 3
MoKradiation Cell parameters from 4273
reflections
= 2.8–28.3
= 2.16 mm1 T= 263 (2) K Needle, white 0.30.20.1 mm
Data collection
Stoe IPDS-I diffractometer
’scans
Absorption correction: numerical [X-RED32(Stoe & Cie, 2002) andX-SHAPE(Stoe & Cie, 2001)]
Tmin= 0.585,Tmax= 0.794
14912 measured reflections
4167 independent reflections 3523 reflections withI> 2(I) Rint= 0.034
max= 27.3
h=6!7 k=9!9 l=29!29
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.026
wR(F2) = 0.069
S= 1.04 4167 reflections 190 parameters
H atoms treated by a mixture of independent and constrained refinement
w= 1/[2
(Fo2) + (0.0397P)2
+ 0.1463P]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.001
max= 0.24 e A˚
3
min=0.29 e A˚
3
Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
O—H2O Bri
0.86 (2) 2.57 (2) 3.386 (3) 159 (3) O—H1O Br 0.84 (2) 2.61 (2) 3.435 (3) 166 (3) C3—H3 Bri 0.93 2.75 3.661 (3) 167 C1—H1 Oii
0.93 2.34 3.193 (4) 152
Symmetry codes: (i)x1;y;z; (ii)xþ1;y1;z.
C-bound H atoms were positioned geometrically (C—H = 0.93 (methine), 0.99 (methylene) and 0.98 A˚ (methyl) and refined with fixed isotropic displacement parameters using a riding model [Uiso(H)
= 1.5 (methyl C) or 1.2 (methylene, methine C)Ueq]. Water H atoms
were located in a difference map and refined isotropically with their O—H bond lengths restrained to 0.84 (1) A˚ . The H atoms of the methyl group bonded to the imidazole ring are equally disordered over two positions. This group was allowed to rotate but not to tip.
Data collection: X-AREA (Stoe & Cie, 2002); cell refinement: X-AREA; data reduction: X-AREA; program(s) used to solve structure:SHELXS97(Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: DIAMOND(Brandenburg, 1996); software used to prepare material for publication:SHELXL97.
References
Bradley, A. E., Hardacre, C., Hobrey, J. D., Johnston, S., McMath, S. E. J. & Nieuwenhuyzen (2002).Chem. Mater.14, 629–635.
Brandenburg, K. (1996).DIAMOND. Release 2.1. Crystal Impact GbR, Bonn, Germany.
Gordon, C. M., Holbrey, J. D., Kennedy, A. R. & Seddon, K. R. (1998).J. Mater. Chem.8, 2627–2636.
Guillet, E., Imbert, D. Scopelliti, R. & Bu¨nzli, J.-C. G. (2004).Chem. Mater.16, 4063–4070.
Hardacre, C., Hobrey, J. D., McCormac, P. B., McMath, S. E., Nieuwenhuyzen & Seddon, K . R. (2001).J. Mater. Chem.11, 346–350.
Seddon, K. R., Bowlas, C. J. & Bruce, D. W. (1996).Chem. Commun.pp. 1625– 1626.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of Go¨ttingen, Germany.
Steiner, T. (2002).Angew. Chem.114, 50–80.
Stoe & Cie (2001).X-SHAPE. Version 2.01. Stoe & Cie, Darmstadt, Germany. Stoe & Cie (2002). X-AREA and X-RED32. Version 1.03. Stoe & Cie,
Darmstadt, Germany.
[image:2.610.46.296.73.177.2]Wasserscheid, P. (2002).Ionic Liquids in Synthesis. Weinheim: VCH–Wiley.
Figure 2
supporting information
sup-1
Acta Cryst. (2005). E61, o2945–o2946
supporting information
Acta Cryst. (2005). E61, o2945–o2946 [https://doi.org/10.1107/S1600536805025717]
1-Dodecyl-3-methylimidazolium bromide monohydrate
Anna Getsis and Anja-Verena Mudring
1-Dodecyl-3-methylimidazolium bromide monohydrate
Crystal data
C16H31N2+·Br−·H2O
Mr = 349.34 Triclinic, P1 Hall symbol: -P 1 a = 5.500 (5) Å b = 7.794 (5) Å c = 22.961 (5) Å α = 81.893 (5)° β = 83.761 (5)° γ = 78.102 (5)° V = 950.3 (11) Å3
Z = 2 F(000) = 372 Dx = 1.221 Mg m−3
Melting point: unknown K Mo Kα radiation, λ = 0.71073 Å Cell parameters from 4273 reflections θ = 2.8–28.3°
µ = 2.16 mm−1
T = 263 K Needle, white 0.3 × 0.2 × 0.1 mm
Data collection
Stoe IPDS-I diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ scans
Absorption correction: numerical
[X-RED32 (Stoe & Cie, 2002) and X-SHAPE (Stoe & Cie, 2001)]
Tmin = 0.585, Tmax = 0.794
14912 measured reflections 4167 independent reflections 3523 reflections with I > 2σ(I) Rint = 0.034
θmax = 27.3°, θmin = 1.8°
h = −6→7 k = −9→9 l = −29→29
Refinement
Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.026
wR(F2) = 0.069
S = 1.04 4167 reflections 190 parameters 2 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(F
o2) + (0.0397P)2 + 0.1463P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.001
Δρmax = 0.24 e Å−3
Special details
Experimental. A suitable single-crystal was carefully selected under a polarizing microscope and mounted in a glass capillary. The scattering intensities were collected with an imaging plate diffractometer (Stoe IPDS-I) equipped with a fine focus sealed tube X-ray source (Mo Kα, λ = 0.71073 Å) operating at 50 kV and 30 mA. Intensity data were collected at 263 K by φ scans in 360 frames (0 < φ < 180° exposure time of 10 min) in the 2Θ range 1.91 - 54.78 °·Structure solution and refinement were carried out using the program SHELXL97 (Sheldrick, 1997). A numerical absorption correction (X-RED (Stoe & Cie, 2001) was applied after optimization of the crystal shape (X-SHAPE (Stoe & Cie, 2001)). The final difference maps were free of any chemically significant features. The refinement was based on F2 for
ALL reflections.
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. Carbon-bound H atoms were positioned with idealized geometry (methine d(C—H) = 93 pm, methylene: d(C—H) = 99 pm; methyl: d(C—H) = 98 pm) and refined with fixed isotropic displacement parameters [Uiso(H)=1.5Ueq (for methyl C) and
Uiso(H)=1.2Ueq (for methylene C)] using a riding atom model. The methyl groups were idealized, then refined as a rigid
group allowed to rotate but not tip. The water H atoms were located in a difference map and their bond length restrained.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq Occ. (<1)
Br 0.73175 (3) 0.32730 (3) 0.383956 (8) 0.05446 (8)
N1 0.4615 (3) −0.05054 (19) 0.36555 (6) 0.0442 (3)
N2 0.6005 (3) −0.21059 (19) 0.44379 (6) 0.0463 (3)
O 0.1671 (4) 0.5835 (3) 0.34933 (13) 0.1026 (7)
H1O 0.315 (4) 0.528 (4) 0.3517 (16) 0.128 (13)*
H2O 0.084 (5) 0.500 (3) 0.3521 (15) 0.110 (11)*
C1 0.6543 (3) −0.1654 (2) 0.38675 (8) 0.0472 (4)
H1 0.8025 −0.2072 0.3652 0.057*
C2 0.3659 (3) −0.1219 (2) 0.45970 (8) 0.0506 (4)
H2 0.2822 −0.1296 0.4971 0.061*
C3 0.2797 (3) −0.0214 (3) 0.41077 (8) 0.0505 (4)
H3 0.1252 0.0538 0.4082 0.061*
C4 0.4544 (4) 0.0394 (3) 0.30472 (8) 0.0525 (4)
H4A 0.6050 −0.0089 0.2817 0.063*
H4B 0.4522 0.1639 0.3053 0.063*
C5 0.2312 (3) 0.0207 (2) 0.27513 (7) 0.0469 (4)
H5A 0.0796 0.0754 0.2965 0.056*
H5B 0.2278 −0.1035 0.2758 0.056*
C6 0.2431 (4) 0.1079 (3) 0.21164 (8) 0.0505 (4)
H6A 0.2505 0.2312 0.2116 0.061*
H6B 0.3959 0.0524 0.1909 0.061*
C7 0.0246 (3) 0.0978 (3) 0.17806 (7) 0.0498 (4)
H7A −0.1281 0.1563 0.1980 0.060*
H7B 0.0143 −0.0253 0.1788 0.060*
supporting information
sup-3
Acta Cryst. (2005). E61, o2945–o2946
H8A 0.0524 0.3049 0.1137 0.060*
H8B 0.1981 0.1242 0.0946 0.060*
C9 −0.1699 (4) 0.1712 (3) 0.07940 (8) 0.0502 (4)
H9A −0.3246 0.2284 0.0990 0.060*
H9B −0.1783 0.0480 0.0796 0.060*
C10 −0.1486 (3) 0.2563 (3) 0.01589 (8) 0.0503 (4)
H10A 0.0062 0.1991 −0.0036 0.060*
H10B −0.1401 0.3794 0.0157 0.060*
C11 −0.3622 (4) 0.2466 (3) −0.01946 (8) 0.0507 (4)
H11A −0.5172 0.3038 0.0000 0.061*
H11B −0.3708 0.1235 −0.0194 0.061*
C12 −0.3394 (4) 0.3321 (3) −0.08305 (8) 0.0506 (4)
H12A −0.3312 0.4552 −0.0831 0.061*
H12B −0.1840 0.2751 −0.1023 0.061*
C13 −0.5513 (4) 0.3224 (3) −0.11872 (8) 0.0513 (4)
H13A −0.5584 0.1992 −0.1190 0.062*
H13B −0.7069 0.3782 −0.0991 0.062*
C14 −0.5312 (4) 0.4089 (3) −0.18186 (8) 0.0579 (5)
H14A −0.5254 0.5323 −0.1817 0.070*
H14B −0.3755 0.3535 −0.2015 0.070*
C15 −0.7430 (5) 0.3974 (3) −0.21700 (10) 0.0725 (6)
H15A −0.7183 0.4551 −0.2564 0.109*
H15B −0.8978 0.4543 −0.1985 0.109*
H15C −0.7473 0.2756 −0.2185 0.109*
C16 0.7654 (4) −0.3362 (3) 0.48298 (9) 0.0606 (5)
H16A 0.6680 −0.3808 0.5168 0.091* 0.50
H16B 0.8842 −0.2775 0.4956 0.091* 0.50
H16C 0.8518 −0.4325 0.4622 0.091* 0.50
H16D 0.9347 −0.3464 0.4662 0.091* 0.50
H16E 0.7184 −0.4496 0.4875 0.091* 0.50
H16F 0.7509 −0.2947 0.5209 0.091* 0.50
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Br 0.05039 (11) 0.05782 (12) 0.05377 (11) −0.00501 (8) −0.01003 (8) −0.00571 (8)
N1 0.0451 (7) 0.0469 (7) 0.0403 (7) −0.0070 (6) −0.0101 (6) −0.0025 (6)
N2 0.0477 (8) 0.0453 (7) 0.0441 (7) −0.0008 (6) −0.0135 (6) −0.0043 (6)
O 0.0575 (10) 0.0631 (11) 0.175 (2) −0.0021 (9) 0.0001 (12) 0.0037 (12)
C1 0.0429 (8) 0.0494 (9) 0.0477 (9) −0.0024 (7) −0.0071 (7) −0.0084 (7)
C2 0.0489 (9) 0.0588 (10) 0.0413 (9) −0.0022 (8) −0.0060 (7) −0.0069 (8)
C3 0.0438 (9) 0.0561 (10) 0.0476 (9) 0.0023 (8) −0.0089 (7) −0.0065 (8)
C4 0.0568 (11) 0.0591 (11) 0.0434 (9) −0.0174 (9) −0.0123 (8) 0.0027 (8)
C5 0.0492 (9) 0.0526 (9) 0.0396 (8) −0.0115 (8) −0.0081 (7) −0.0026 (7)
C6 0.0525 (10) 0.0569 (10) 0.0432 (9) −0.0158 (8) −0.0099 (8) 0.0021 (8)
C7 0.0517 (10) 0.0584 (10) 0.0406 (9) −0.0176 (8) −0.0089 (7) 0.0036 (7)
C8 0.0508 (10) 0.0597 (10) 0.0412 (9) −0.0165 (8) −0.0083 (7) 0.0024 (7)
C10 0.0511 (10) 0.0584 (10) 0.0428 (9) −0.0160 (8) −0.0084 (8) 0.0009 (8)
C11 0.0537 (10) 0.0579 (10) 0.0421 (9) −0.0168 (8) −0.0092 (8) 0.0008 (8)
C12 0.0527 (10) 0.0574 (10) 0.0432 (9) −0.0165 (8) −0.0088 (8) 0.0006 (8)
C13 0.0548 (10) 0.0564 (10) 0.0439 (9) −0.0148 (8) −0.0094 (8) −0.0002 (8)
C14 0.0631 (12) 0.0675 (12) 0.0443 (9) −0.0183 (10) −0.0104 (9) 0.0023 (8)
C15 0.0807 (15) 0.0894 (16) 0.0504 (11) −0.0223 (13) −0.0224 (11) 0.0028 (11)
C16 0.0626 (12) 0.0563 (11) 0.0569 (11) 0.0072 (9) −0.0230 (9) −0.0005 (9)
Geometric parameters (Å, º)
N1—C1 1.327 (2) C9—C10 1.517 (2)
N1—C3 1.370 (2) C9—H9A 0.9700
N1—C4 1.472 (2) C9—H9B 0.9700
N2—C1 1.325 (2) C10—C11 1.518 (3)
N2—C2 1.370 (2) C10—H10A 0.9700
N2—C16 1.467 (2) C10—H10B 0.9700
O—H1O 0.842 (18) C11—C12 1.521 (2)
O—H2O 0.859 (18) C11—H11A 0.9700
C1—H1 0.9300 C11—H11B 0.9700
C2—C3 1.347 (2) C12—C13 1.515 (3)
C2—H2 0.9300 C12—H12A 0.9700
C3—H3 0.9300 C12—H12B 0.9700
C4—C5 1.508 (3) C13—C14 1.513 (2)
C4—H4A 0.9700 C13—H13A 0.9700
C4—H4B 0.9700 C13—H13B 0.9700
C5—C6 1.520 (2) C14—C15 1.511 (3)
C5—H5A 0.9700 C14—H14A 0.9700
C5—H5B 0.9700 C14—H14B 0.9700
C6—C7 1.517 (3) C15—H15A 0.9600
C6—H6A 0.9700 C15—H15B 0.9600
C6—H6B 0.9700 C15—H15C 0.9600
C7—C8 1.520 (2) C16—H16A 0.9600
C7—H7A 0.9700 C16—H16B 0.9600
C7—H7B 0.9700 C16—H16C 0.9600
C8—C9 1.513 (3) C16—H16D 0.9600
C8—H8A 0.9700 C16—H16E 0.9600
C8—H8B 0.9700 C16—H16F 0.9600
C1—N1—C3 108.37 (15) C9—C10—H10B 108.7
C1—N1—C4 125.22 (15) C11—C10—H10B 108.7
C3—N1—C4 126.26 (15) H10A—C10—H10B 107.6
C1—N2—C2 108.71 (14) C10—C11—C12 114.00 (16)
C1—N2—C16 125.31 (16) C10—C11—H11A 108.8
C2—N2—C16 125.98 (16) C12—C11—H11A 108.8
H1O—O—H2O 103 (3) C10—C11—H11B 108.8
N2—C1—N1 108.66 (15) C12—C11—H11B 108.8
N2—C1—H1 125.7 H11A—C11—H11B 107.6
supporting information
sup-5
Acta Cryst. (2005). E61, o2945–o2946
C3—C2—N2 106.90 (16) C13—C12—H12A 108.7
C3—C2—H2 126.5 C11—C12—H12A 108.7
N2—C2—H2 126.5 C13—C12—H12B 108.7
C2—C3—N1 107.36 (16) C11—C12—H12B 108.7
C2—C3—H3 126.3 H12A—C12—H12B 107.6
N1—C3—H3 126.3 C14—C13—C12 114.63 (16)
N1—C4—C5 113.05 (15) C14—C13—H13A 108.6
N1—C4—H4A 109.0 C12—C13—H13A 108.6
C5—C4—H4A 109.0 C14—C13—H13B 108.6
N1—C4—H4B 109.0 C12—C13—H13B 108.6
C5—C4—H4B 109.0 H13A—C13—H13B 107.6
H4A—C4—H4B 107.8 C15—C14—C13 114.08 (18)
C4—C5—C6 110.22 (15) C15—C14—H14A 108.7
C4—C5—H5A 109.6 C13—C14—H14A 108.7
C6—C5—H5A 109.6 C15—C14—H14B 108.7
C4—C5—H5B 109.6 C13—C14—H14B 108.7
C6—C5—H5B 109.6 H14A—C14—H14B 107.6
H5A—C5—H5B 108.1 C14—C15—H15A 109.5
C7—C6—C5 114.32 (16) C14—C15—H15B 109.5
C7—C6—H6A 108.7 H15A—C15—H15B 109.5
C5—C6—H6A 108.7 C14—C15—H15C 109.5
C7—C6—H6B 108.7 H15A—C15—H15C 109.5
C5—C6—H6B 108.7 H15B—C15—H15C 109.5
H6A—C6—H6B 107.6 N2—C16—H16A 109.5
C6—C7—C8 113.41 (16) N2—C16—H16B 109.5
C6—C7—H7A 108.9 H16A—C16—H16B 109.5
C8—C7—H7A 108.9 N2—C16—H16C 109.5
C6—C7—H7B 108.9 H16A—C16—H16C 109.5
C8—C7—H7B 108.9 H16B—C16—H16C 109.5
H7A—C7—H7B 107.7 N2—C16—H16D 109.5
C9—C8—C7 114.58 (16) H16A—C16—H16D 141.1
C9—C8—H8A 108.6 H16B—C16—H16D 56.3
C7—C8—H8A 108.6 H16C—C16—H16D 56.3
C9—C8—H8B 108.6 N2—C16—H16E 109.5
C7—C8—H8B 108.6 H16A—C16—H16E 56.3
H8A—C8—H8B 107.6 H16B—C16—H16E 141.1
C8—C9—C10 113.90 (16) H16C—C16—H16E 56.3
C8—C9—H9A 108.8 H16D—C16—H16E 109.5
C10—C9—H9A 108.8 N2—C16—H16F 109.5
C8—C9—H9B 108.8 H16A—C16—H16F 56.3
C10—C9—H9B 108.8 H16B—C16—H16F 56.3
H9A—C9—H9B 107.7 H16C—C16—H16F 141.1
C9—C10—C11 114.33 (16) H16D—C16—H16F 109.5
C9—C10—H10A 108.7 H16E—C16—H16F 109.5
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
O—H2O···Bri 0.86 (2) 2.57 (2) 3.386 (3) 159 (3)
O—H1O···Br 0.84 (2) 2.61 (2) 3.435 (3) 166 (3)
C3—H3···Bri 0.93 2.75 3.661 (3) 167
C1—H1···Oii 0.93 2.34 3.193 (4) 152