Author for Correspondence: Sudip Das
Email: [email protected] Research Article
ISSN: - 2306 – 6091
Available Online at: www.ijphr.com An African Edge Journal
SJ Impact Factor (2015) – 5.546
Preparation and in-vitro evaluation of solid and liquid self emulsifying drug
delivery system of Ibuprofen
Sudip Das*, Dr. Shubhrajit Mantry, Anirban Sannigrahi, Debakanta Saha, Debangana
Pal, Jyotirmoy Thokdar, Kaushik Majumder
Department of Pharmaceutics, Himalayan Pharmacy Institute, Majhitar, East Sikkim-737136, INDIA
_____________________________________________________________________________________________ABSTRACT
The aim of the present study was to formulate the solid self-emulsifying drug delivery system (S-SEDDS) by using Cod liver oil, Olive oil as oil phase, Tween 20 as surfactant, Spam 80, PEG as surfactant, and Ethanol as co-solvent of poorly water soluble drug Ibuprofen. Aerosil 200 was used as a solid carrier and convert liquid form to solid form and develop S-SEDDS by adsorption to solid carrier technique. The main objective of study was to prepare S-SEDDS of Ibuprofen on the basis of preformulation study like solubility study, standard curve preparation in order to achieve a better dissolution rate which would further help in enhancing absorption and oral bioavailability. The solubility of Ibuprofen was determined in several oils, surfactants and co-surfactants using an UV-Spectroscopy method. Four formulation (i.e. F1 to F4) Solid and Liquid SEDDS were prepared and evaluated for their micromeritic property, drug content, in-vitro drug release study. From the solubility study better solubility was seen in Cod liver oil, Tween 20 (surfactant), Span 80 (co-surfactant) and ethanol (co-solvent). From the data we obtained the optimized F3 batch shows good Micromeritic properties. The formulation was found to show a significant improvement in the drug release from powdered drug Ibuprofen. Drug release from S-SEDDS (F3) was 98.153% in 6 hrs.
Keywords:
Ibuprofen, Cod liver oil, Olive oil, self-emulsifying drug delivery system._________________________________________________________________________________________
INTRODUCTION
Self-emulsifying drug delivery system is an isotropic mixture of oil, surfactant, co-surfactant that has able to self-emulsify rapidly and form oil in water emulsion in the presence of GI fluid with the help of gentle agitation and improve solubility and dissolution profile of poorly water soluble drug [1]. Lipophilic drug exhibit dissolution rate limited absorption [2]. But here prepared a formulation in which first pre dissolving the drug compound for this overcome the initial rate limiting step dissolution in
aqueous environment. 40% of new chemical compound (drug) are poorly water soluble or lipophilic in nature. For this reason, which leads to poor oral bioavailability, high intra and inter subject variability and lack of dose proportionality. Has some way to overcome this problem such as micronization, salt formation, solid dispersions, inclusion complexes with cyclodextrin and lipid based drug delivery system like self-emulsifying drug delivery systems. But every process has some difficulties. In salt formation salt may convert back to their original acid
or base form and lead to aggregation in GI tract. Particle size reduction may not be desirable in situations where handling difficulties and poor wettability are experienced for very fine powders [3]. Lipid based formulation are important, because of their some advantages like SEDDS has unique property that they are able to self-emulsify rapidly and form o/w emulsion and results in small droplets of oil dispersed in GI fluid that provide a large interfacial area and enhancing the activity, improved drug dissolution, increased intestinal epithelial permeability, increased tight junction permeability [4]. If liquid SEDDS are enclosed in hard or soft gelatin capsule to facilitate oral administration, but it produces some disadvantages such as stability, incompatibility, drug leakage, precipitation and capsule ageing for this reason liquid form convert to solid form and overcome this problem. For converting L-SEDDS to S-SEDDS has some way such as spray drying, adsorption into solid carriers, melt granulation, melt extrusion. Adsorption by solid carrier is simple method for converting the solid form. Solid carrier such as aerosil 200 mixed with required amount of liquid self-emulsifying drug delivery system by a blender and convert it to solid form. Approximately 40% of new drug candidate emerging from drug discovery process displays low
solubility in water, which leads to poor
bioavailability, high intra subject/inter subject variability and lack of dose proportionality. Among lipid based formulations, much attention has been focused on self microemulsifying drug delivery system which has successfully improved solubility and bioavailability of poorly water soluble drugs.
Self-micro emulsifying formulation comprises
isotropic mixtures of natural or synthetic oils with lipophilic or hydrophilic surfactants and co-surfactants, which spontaneously emulsify when exposed to the GIT fluids to form oil-in-water emulsions or microemulsions. SMEDDS is generally prepared as liquid dosage form which has some shortcomings such as incompatibility with the shells of hard or soft gelatin capsules, and complex process of manufacturing. So to overcome these problems, studies on solid self-micro emulsifying drug delivery system (SSMEDDS) have come into limelight. Emulsions are used as vehicles for the administration of drugs, especially due to its potential of enhancing the oral bioavailability of poorly absorbed drugs [5].
Microemulsions has got advantage like excellent thermodynamic stability, high drug solubilisation capacity, improvement in oral bioavailabity and protection against enzymatic hydrolysis. The only problem with microemulsion is poor palatability due to the lipid content leading to poor patient compliance, more over due to their water content, microemulsions cannot be encapsulated into soft gelatin or hard gelatine capsule. There is a need for converting it into an alternative formulation like anhydrous self emulsifying drug delivery system (SEDDS) etc., because of its low loading dose [6]. SEDDS are solid dosage form with a unique property, that is they are able to self emulsify rapidly into fine O/W emulsion in the gastrointestinal fluids,
under gentle agitation provided by the
gastrointestinal tract. This fine O/W emulsion results in small droplets of oil dispersed in the gastrointestinal fluids that provide a large interfacial area enhancing the activity and minimizing the irritation due to contact of drug in the gut wall [7‐10]. Self Emulsifying System (SES) can be formulated with little energy input and the shelf life is longer than conventional emulsions. Therefore, an SES can be an efficient vehicle for class II to IV molecules of the Biopharmaceutical Classification System (BCS) drugs [11,12]. Ibuprofen is a non-steroidal anti-inflammatory drug (NSAID) derived from propionic acid and it is considered the first of the propionics. The formula of Ibuprofen is 2-(4-isobutylphenyl) propionic acid and its initial development was in 1960 while researching for a safer alternative for aspirin. Ibuprofen was finally patented in 1961 and this drug was first launched against rheumatoid arthritis in the UK in 1969 and USA in 1974. It was the first available over-the-counter NSAID.
MATERIALS AND METHOD
METHODS
Preformulation study
Determination of saturation solubility of
Ibuprofen in different oils, surfactant, and
co-surfactant
The most important criterion for the screening
of components for self-emulsifying drug delivery system is the solubility of poorly soluble drug in oils, surfactants and co surfactants. The solubility of Ibuprofen in various oils was determined by adding an excess amount of drug.
10 ml of selected oils (Cinnamon oil, Clove oil,
Arachis oil, Sesame oil, Coconut oil, Cod-liver oil, Olive oil, Oleic acid) and surfactants (tween-20, tween-80, Cremophor RH 40) and co-surfactant (Span 20, Span80, PEG 200, PEG 600, PEG 400, PEG 4000) and co-solvent (Ethanol, Propylene glycol, glycerol) was taken in 50ml beaker.
An excess amount of drug was individually
mixed with selected every oils, surfactant, co-surfactant, co-solvents in the presence of heat.
Dissolve properly.
2 ml was withdrawn and put in 5 ml capacity
stopper vials and properly sealed the vials.
After sealing the mixture was heated at 40
degree centigrade in a water bath to facilitate the solubilisation. Mixtures were shaken for 48hrs. After reaching equilibrium, each vial was centrifuged at 3000 rpm for 15 min.
Excess insoluble Ibuprofen was discarded by
filtration using a membrane filter.
The concentration of Ibuprofen was determined
using UV- spectrophotometer at 221 nm
Preparation of liquid & solid self emulsifying
drug delivery system of Ibuprofen
The appropriate quantities of oil phase,
surfactant and co-surfactant were selected based on the result of solubility study.
Required quantity of Ibuprofen dispersed in
appropriate required quantity of co-surfactant in a beaker in the presence of heat
Accurately weighed quantity of oil and
surfactant blends ware taken in small portion in a beaker which was placed on a magnetic stirrer
The blends were mixed thoroughly.
This (drug + co-surfactant) mixture was added with (oil+surfactant) mixture.
At last, total (drug+co-surfactant+surfactant+oil phase) ware mixed properly with the help of magnetic stirrer
Prepared Liquid self-emulsifying drug delivery system.
The L-SEDDS of Ibuprofen was adsorbed onto Aerosil200 carrier by physical mixing in a small mortar and pestle. The resulting solid SEDDS was uniformly homogenized to ensure that the mixture was uniformly distributed. The damp mass was passed through sieve No.120 and was dried at ambient temperature.
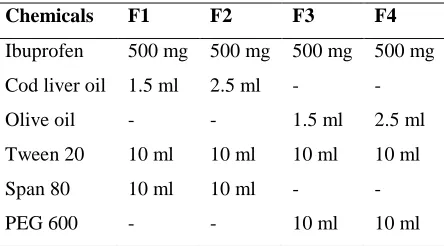
Table 1: Composition of SEDDS of Ibuprofen
Chemicals F1 F2 F3 F4
Ibuprofen 500 mg 500 mg 500 mg 500 mg
Cod liver oil 1.5 ml 2.5 ml - -
Olive oil - - 1.5 ml 2.5 ml
Tween 20 10 ml 10 ml 10 ml 10 ml
Span 80 10 ml 10 ml - -
Preparation method of Liquid & Solid Self-Emulsifying Drug Delivery System.
Ibufrofen added in co-surfactant
Accurately weighed quantity of oil was taken in a separate beaker
(Drug + co-surfactant) mixture was added with oil surfactant
Picture of liquid SEDDS
L-SEDDS was mixed with adsorbent Aerosil 200
S-SEDDS after Sieving and proper drying
Figure 1: Images of preparation of Liquid & Solid Self-Emulsifying Drug Delivery System.
RESULT AND DISCUSSION
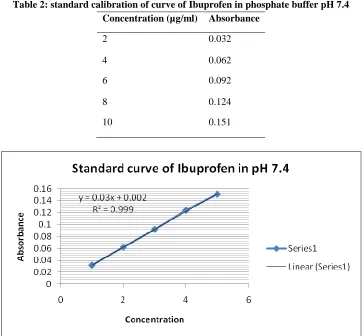
A standard calibration of curve of Ibuprofen in phosphate buffer pH 7.4.A calibration curve for Ibuprofen was successfully obtained by measuring
the absorbance of series of concentration of Ibuprofen at the λmax of 221nm. A corresponding standard curve generated by linier regression analysis is shown in figure.
Table 2: standard calibration of curve of Ibuprofen in phosphate buffer pH 7.4 Concentration (µg/ml) Absorbance
2 0.032
4 0.062
6 0.092
8 0.124
10 0.151
Solubility Study
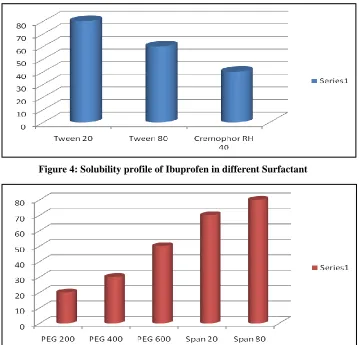
Figure 3: Solubility profile of Ibuprofen in different oil phase
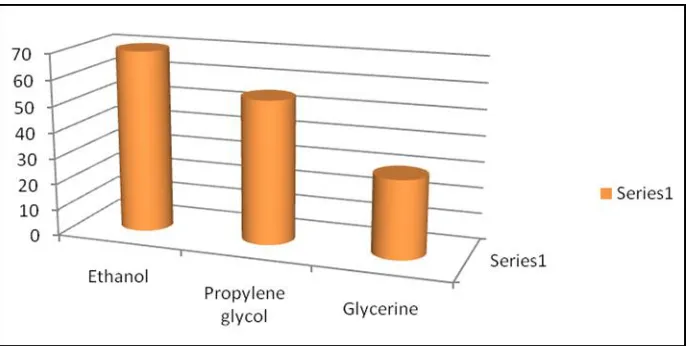
Different oils, surfactant, surfactant, co-solvent were screened for the solubility of Ibuprofen. Maximum solubility was determined in cod liver oil from all types of oil phase, Tween 20 from all types
of surfactant, Span 80 from all types of co-surfactant, ethanol from all types of co-solvents and all four
reagent were selected for self-emulsifying
formulation.
Figure 4: Solubility profile of Ibuprofen in different Surfactant
Figure 6: Solubility profile of Ibuprofen in different Co-Solvent
Micromeritic properties and drug content for Solid self-emulsifying drug delivery system
All formulations hausner’s ratio value less than 1.25, angle of repose value <25, Carr’s index below 15 in formulation 1, indicates good flowability.
From the data obtained, the optimized F3 batch shows good Micromeritic properties.
Table 4: Results of micromeritic properties and drug content for Solid self-emulsifying drug delivery system. Formulation Code Angle of Repose
(ϴ)
Bulk Density (gm/cm3)
Tap Density (gm/cm3)
Drug Content
F1 33.12 0.469 0.61 85.632
F2 30.23 0.473 0.64 87.289
F3 23.63 0.481 0.59 93.648
F4 24.01 0.467 0.63 91.214
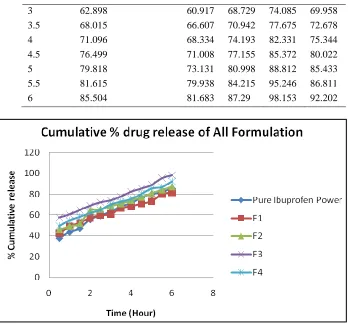
In-vitro drug release of S-SEDDS and Plain
Ibuprofen in pH 7.4
Drug release from the all SEDDS formulation was found to be significantly higher as compared with plain powdered Ibuprofen drug. Ibuprofen loaded S-SEDDS formulation (F3) has greater drug released, above 98% within 6hr. It could be suggested that the SEDDS formulation resulted in spontaneous formation of a micro-emulsion, which
permitted a faster rate of drug release into the aqueous phase, much faster than that of plain Ibuprofen and adsorbing of SEDDS in the presence of Aerosil 200 may not affect the progress of emulsification as well as the release of drug. Almost all the formulations followed first order release kinetics model due to its highest regression coefficient value.
Table 5: In-vitro drug release value of S-SEDDSs and Plain Ibuprofen after 6 hr in pH 7.4 Time (hr) Pure Ibuprofen Power F1 (%) F2 (%) F3 (%) F4 (%)
0.5 37.674 42.502 45.857 57.674 49.205
1 43.442 49.184 49.271 60.64 54.598
1.5 47.183 52.514 51.942 64.809 58.116
2 55.484 57.254 64.278 68.897 61.651
3 62.898 60.917 68.729 74.085 69.958
3.5 68.015 66.607 70.942 77.675 72.678
4 71.096 68.334 74.193 82.331 75.344
4.5 76.499 71.008 77.155 85.372 80.022
5 79.818 73.131 80.998 88.812 85.433
5.5 81.615 79.938 84.215 95.246 86.811
6 85.504 81.683 87.29 98.153 92.202
Figure :In-vitro drug release value of S-SEDDSs and Plain Ibuprofen after 6 hr in pH 7.4
CONCLUSION
Ibuprofen is one of the propionic acid derivatives of non-steroidal anti-inflammatory drugs (NSAID) with analgesic and antipyretic effects. A self-emulsifying drug delivery system was prepared which enhance the solubility of poorly water soluble drug Ibuprofen. S-SEDDS formulations of a poorly water-soluble drug Ibuprofen were formulated for direct filling into hard gelatin capsules for oral administration.The conclusion was confirmed from the value of angle of repose less than 25, drug content in F3 was 98.153%. The formulations after reconstitution formed good emulsion with drug
solubilisation. Drug release from S-SEDDS (F1, F2, F3, F4) was found to be significantly higher as compared with that of plain powdered Ibuprofen. Ibuprofen loaded S-SEDDS formulation (F3) has greater drug released, above 98% within 6hr. Almost all the formulations followed first order release kinetics model due to its highest regression coefficient value. Thus the solubility and the dissolution rate of BCS Class –II type drug Ibuprofen was enhanced which would prove a promising result
of increased absorption and increased oral
bioavailability of conventional release Ibuprofen formulation.
REFERENCES
[1]. York P, The design of dosage forms, In: Aulton M.E. (eds.), Pharmaceutics The science of dosage form design. Churchill Livingstone, Edinburgh; 1988, 1‐13.
[3]. Investigation of formulation approaches to improve the dissolution of SB‐210661, a poorly water soluble 5‐lipooxygenase inhibitor. Int. J. Pharma, 176, 1998, 31‐38.
[4]. Nazzal S, Guven N, Reddy IK, Khan MA. Preparation and characterization of Coenzyme Q10 – Eudragit solid
dispersation. Drug Dev. Ind. Pharm. 28, 2002, 49‐57.
[5]. Pouton CW, Self‐emulsifying drug delivery system; assessment of the efficiency of emulsification. Int. J. Pharm. 27, 1985, 335‐ 348.
[6]. Patravale VB, Date AA, Kale AA. Oral self micro emulsifying system: potential in drug delivery system. Pharma Technology Express Pharma Pulse special feature, 29, 2003, 44‐48.
[7]. O’Driscoll CM. Lipid based formulation for intestinal lymphatic delivery. Eur. J. Pharm. Sci. 15, 2002, 405‐415.
[8]. Charman SA, Charman WN, Rogge MC, Wilson TD, Dutko FJ, Pouton C W. Self emulsifying drug delivery
system: formulation and biopharmaceutical evaluation of an investigational lipophilic compound. Pharm. Res. 9, 1992, 87‐93.
[9]. Shah NH, Carvajal M T, Patel CI, Infiled M H, Malick AW. Selfemulsifying Drug Delivery systems (SEDDS)
with polyglycolized glycerides for improving in‐vitro dissolution and oral absorption of lipophilic drugs. Int. J. Pharm. 106, 1994, 15‐23.
[10]. Khoo S M, Humberstone A J, Porter CJH, Edward GA, Charman WN. Formulation Design and bioavailability
assessment of lipidic self‐emulsifying formulation of halofantrine. Int. J. Pharm. 167, 1998, 155‐164.
[11]. Amidon GL, Lennernas H, Shah VP, Crison JRA. Therotical basis for abiopharmaceutical classification: The
correlation of invitro drug product dissolution and in vivo bioavailability. Pharm Res, 12, 1995, 413‐420. [12]. Franceschinis E, Voinovich D, Grassi M, PerissuttiFilipovic‐Grcic BJ, Martinac
A, Meriani‐Merlo F. Self‐emulsifying pellets prepared by wet granulation in high‐shear mixer: influence of formulation variables and preliminary study on the in vitro absorption. Int. J. Pharm. 291, 2005, 87–97.