Abstract
COKER, JEFFREY SCOTT. The systemic response to fire damage in tomato plants: A case study in the development of methods for gene expression analysis using sequence data. (Under the direction of Dr. Eric Davies.)
Fire is a natural component of most terrestrial ecosystems and can act as a local
wound stimulus to plants. The ultimate goal of this work was to characterize the array of
transcripts which systemically accumulate in plants after fire damage. Before this could be
accomplished, substantial development of methods for gene expression analysis using
sequence data was necessary. This involved developing methods for identifying
contamination in DNA sequence data (Chapter 2), identifying over 78,000 false sequences in
GenBank and several thousand more in the indica rice genome (Chapter 2), developing a
novel method for identifying housekeeping controls using sequence data (Chapter 3),
performing relative expression analyses for 127 potential housekeeping control transcripts
(Chapter 3), and characterizing 23 transcripts which encode all 13 subunits of vacuolar H+ -ATPases in tomato plants (Chapter 4). A subtractive cDNA library served as a starting point
to identify and characterize 9 novel tomato transcripts systemically up-regulated in leaves in
the first hour after a distant leaf is flame wounded (Chapters 5). Real-time RT-PCR using
leaf RNA isolated at different times after flaming showed that the most common pattern of
transcript accumulation was an increase within 30 to 60 minutes, followed by a return to
basal levels within 3 hours. Expression analyses also showed that most up-regulated
transcripts were already present in unwounded tissues. A total of 46 different transcripts
were identified from the subtractive cDNA library (Chapters 6). Compared with the entire
majority fell into 5 classes: enzymes of general metabolism; protein synthesis, modification,
and transport; transcription; membrane transport; and photosynthesis and respiration. At
least half of the transcripts have been previously associated with wounding or stress,
suggesting that the systemic response to fire damage has components similar to those of other
wound and stress responses. On the other hand, 30% of transcripts were associated with
photosynthesis and respiration, suggesting that part of the response to fire damage is notably
different from other wound and stress responses. Conclusions and future directions are
THE SYSTEMIC RESPONSE TO FIRE DAMAGE IN TOMATO
PLANTS: A CASE STUDY IN THE DEVELOPMENT OF METHODS
FOR GENE EXPRESSION ANALYSIS USING SEQUENCE DATA
by
JEFFREY SCOTT COKER
A dissertation submitted to the Graduate Faculty of North Carolina State University
in partial fulfillment of the requirements for the Degree of
Doctor of Philosophy
DEPARTMENT OF BOTANY
Raleigh 2004
APPROVED BY:
___________________________ ____________________________
Dr. Judy Thomas Dr. Jack Wheatley
Advisory committee member Advisory committee member
___________________________ ____________________________
Dr. Dominique Robertson Dr. Chris Brown
Advisory committee member Advisory committee member
___________________________
Dr. Eric Davies
Dedication
The dissertation of Jeffrey S. Coker, which completes the Degree of Doctor of Philosophy, is dedicated to the educators of Plymouth, North Carolina.
Leafie Bryant Julia Towe Rita Rhodes Frances Callander Ann Bland Doris Downing Ruth Pharr Beth Thompson Shirley Thomas Sally Woolard Glenda Smith Bea Waters Judy Wynn Ms. Wilkins Senya Norman Roxanna Brown Judy Bragg
Biography
Jeffrey Scott Coker was born the son of Jerry and Debra Coker in the small town of
Plymouth, North Carolina. His interest in plants is probably due to a family of gardeners,
pulp and paper engineers, and wood-workers, as well as a community where farms, forests,
ball fields, and swamps are plentiful. Jeffrey attended Davidson College, where he studied
biology and ancient Greek and Roman civilizations, and played baseball. After graduation,
he worked for one year at the Helen Paesler School in Raleigh, NC, teaching high school
biology, chemistry, and calculus, as well as middle school science/math. It was during this
year that he found a passion for teaching science and decided to pursue it at the college level.
Jeffrey entered graduate school at N.C. State University in 1999 as an RA/TA in the
Botany Department, where he taught laboratories in Botany and Biotechnology, and
co-taught a new Whole Plant Physiology course. He earned a M.Ed. in Science Education in the
spring of 2001, and formally became a Ph.D. student in Botany (under Dr. Eric Davies)
shortly thereafter. He has been recognized for his teaching at N.C. State by receiving the
CALS Outstanding Teaching Assistant Award, the Martha Sue Sebastian Memorial Award
for Excellence in Teaching, a GSA Outstanding Teaching Award, an Alcoa Teaching
Fellowship, and a NACTA Graduate Student Teaching Award. Student researchers under his
supervision have been recognized locally and nationally for their work.
While in Raleigh, Jeffrey met a wonderful girl named Beth, and they were married on
December 20, 2003, in Greenville, N.C. Beginning in August of 2004, Jeffrey will be an
Assistant Professor in the Biology Department at Elon University. He looks forward to a
Acknowledgements
There are many people who have supported me over the last five years in various
capacities. My committee members have been extremely supportive, and for that I am most
grateful. Dr. Eric Davies has been an outstanding research advisor in every sense. His
openness to new ideas, support of my work, willingness to integrate scientific and
educational pursuits, careful review of manuscripts, daily friendliness, and general guidance
have all been invaluable. Perhaps the most distinct impression Eric has left on me is the
amount of effort he spends helping to advance the lives and careers of his students and
colleagues. I cannot think of a more admirable quality. We have had many conversations
about how many students do not fully appreciate a teacher or mentor until years later. Let me
assure you that I am fully aware of what an outstanding advisor I have had. Dr. Judy Thomas
has been an excellent mentor and friend, and was an instrumental part of my success in
graduate school. She believed in me when others were skeptical, and set me on the right path
more times than I can count. Dr. Chris Brown has been a role model for me in terms of
professionalism, teaching, and the leadership of research and teaching collaborations. He
introduced me to concepts of Space Biology which changed the way I look at my own
discipline. I credit Dr. Niki Robertson with shaping my earliest thoughts about
biotechnology, and value her thoughts very highly. Her enthusiastic and insightful
approaches to science and life are contagious among her students. Dr. Jack Wheatley’s
presence on my committee is especially meaningful because he represents good teaching and
educational scholarship. I am thankful for his guidance, patience, and insightful reviews of
A number of people worked alongside me in the laboratory, and provided daily
assistance for which I am thankful. Dr. Raul Salinas was especially helpful and patient.
Most of my “co-workers” were high school and undergraduate student researchers who
always made the lab a more enjoyable place. In particular, I am thankful to have worked
with Derek Jones regarding vacuolar ATPases and enjoyed both his enthusiasm and
friendship. Other student researchers included Katie Grant, Jessica Staley, Holly Cline, Ryan
Parks, Ashwynn Stanger, John Pollard, and Turqouise Ross.
Dr. Gerald Van Dyke has been an invaluable teaching mentor and friend. His
excitement about teaching and commitment to students have inspired me to seek excellence
in the classroom. My time at N.C. State would not have been the same without the friendship
and conversation of Dr. Isaac Bruck. I am also thankful for the Botany administrative staff,
especially Sue Vitello and Vicki Lemaster, who dealt with many issues on my behalf.
I am blessed with a loving family which has provided support in many forms. Mom,
Dad, Grandmother, Chris, Eric, Laura, Sheila, Mike, Josh, and Debbie have all played
important roles in my life. On at least two occasions, family members (Eric and Mom)
helped me to overcome significant research difficulties.
Finally, I could not dream of having a more supportive wife. Beth has been at my
side through virtually every step of my dissertation research. She has assisted me in the field,
in the laboratory, and in the classroom. She has read my papers, inspected tables and figures,
listened to whole lectures just so I could practice and, perhaps most importantly, encouraged
me to work long hours when deadlines approached or I became really excited about
something (which happens frequently). She must be, as we joke, the “best chemical
Part of the research and travel associated with this dissertation was funded by grants
from the Plant Molecular Biology Consortium, Sigma Xi, and the American Society of Plant
Biologists. Acknowledgements of a more technical nature are provided at the end of each
Table of Contents
List of Tables... xi
List of Figures... xiv
1. Introduction... 1
2. Sequence quality control... 6
A. Identifying adaptor contamination when mining DNA sequence data ... 7
Abstract ... 7
Acknowledgments... 11
References... 12
B. Cleaning data mined from the indica rice genome... 16
Abstract ... 16
SmaI-linearized pUC18 plasmid... 16
Regions of other cloning vector(s)... 18
Phytophthora ... 19
Conclusions... 20
References... 21
C. Correction of the 5’ end of the human com1/p8 gene... 26
Letter ... 26
References... 26
3. Selection of candidate housekeeping controls in tomato plants using EST data... 28
Abstract ... 29
Introduction... 29
Materials and methods ... 30
Data mining... 30
Calculation of relative expression levels ... 30
Calculation of fold ranges and transcript variation... 30
Results and discussion ... 31
Acknowledgements... 33
References... 33
4. Identification, conservation, and relative expression of V-ATPase cDNAs in tomato plants... 34
Abstract ... 35
Introduction... 35
Identification of V-ATPase ESTs ... 37
Relative expression analyses... 37
Gene nomenclature ... 37
Results and discussion ... 40
23 V-ATPase genes identified in tomato... 40
Hexamer rings are highly conserved... 40
Relative expression levels in different tissues ... 41
V-ATPase relative expression increases during fruit ripening ... 45
Conclusion ... 46
Acknowledgements... 46
References... 47
5. Identification, accumulation, and functional prediction of novel tomato transcripts systemically up-regulated after fire damage... 49
Abstract ... 50
Introduction... 51
Results... 55
Discussion ... 59
CSWR-1 Acyl carrier protein ... 60
CSWR-2 Adenylyl-sulfate reductase... 60
CSWR-3 Unknown protein... 61
CSWR-4 Photosystem II oxygen-evolving complex protein 3... 61
CSWR-5 Putative anion:sodium symporter... 61
CSWR-6 Unknown wound/stress protein... 62
CSWR-7 Chloroplast-specific ribosomal protein ... 63
CSWR-8 Alpha/beta fold family protein ... 63
CSWR-9 Histidine triad family protein ... 63
Materials and Methods... 65
Plant material, growth conditions, and tissue collection... 65
Subtractive cDNA library construction, screening and sequencing ... 65
DNA sequence analysis and data mining... 66
Verification of consensus sequences ... 67
Real-time RT-PCR assays... 67
Relative expression analyses... 68
Polypeptide sequence analysis... 69
Acknowledgements... 69
Literature cited ... 70
6. Fire damage causes the systemic up-regulation of a set of highly conserved transcripts in tomato plants... 84
Abstract ... 85
Introduction... 86
Materials and methods ... 89
Subtractive cDNA library construction, screening and sequencing ... 89
DNA sequence analysis ... 90
Comparisons with the Arabidopsis genome ... 90
Results... 92
Overview of the subtractive cDNA library... 92
Library validation... 94
Conservation between tomato and Arabidopsis... 95
Discussion ... 97
Transcripts common to other wound and stress responses ... 97
Transcripts not common to other wound and stress responses ... 101
References... 103
7. Conclusions and future directions... 108
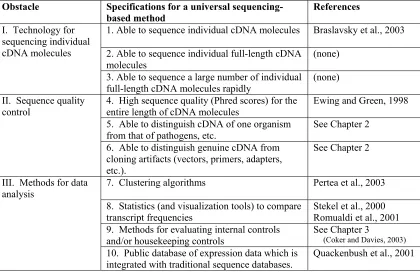
Conclusions and future directions regarding the development of methods for gene expression analysis using sequence data: Blueprint for a universal sequencing-based method of gene expression analysis... 109
Abstract ... 109
Disadvantages of binding-radiation methods... 110
Advantages of sequencing methods... 112
Obstacles and specifications for a universal sequencing-based method... 117
References... 120
Conclusions and future directions regarding the biology of systemic responses to fire damage ... 121
Appendices... 124
Appendix 1: V-ATPase amino acid alignments... 125
Appendix 2: Annotated sequences for novel tomato transcripts/proteins ... 141
Appendix 3: Perspectives on student research experiences in plant biology... 152
Overview... 152
A. Involvement of plant biologists in undergraduate and high school student research ... 153
Abstract ... 153
Introduction... 153
Methods... 153
Member participation... 153
Advantages and disadvantages of research training ... 154
References... 156
B. A national perspective on mentoring student researchers in plant biology... 157
Introduction... 158
Materials and methods ... 161
Results and discussion ... 164
Acknowledgements... 176
References... 177
C. Evaluation of teaching and research experiences undertaken by botany majors at N.C. State University... 185
Abstract ... 185
Introduction... 186
Methods... 188
Results and discussion ... 188
Acknowledgements... 195
List of Tables
Chapter 2-A
Table 1. Sequences and search parameters to identify entries in GenBank contaminated by 7 commercial adaptor sequences ... 14
Chapter 2-B
Table 1. Matches in the indica genome with the pUC18 SmaI site... 22 Table 2. Examples of internal pUC18 artifacts (≥14 bp) in indica scaffolds ... 24 Table 3. Examples of phytophthora-like sequences in the indica genome... 25
Chapter 3
Table 1. Summary of tentative consensus sequences (TCs) from the TIGR TGI that were analyzed for their potential as housekeeping control genes... 30 Table 2. Highest-ranking housekeeping control genes in various tomato plant tissues ... 31
Chapter 4
Table 1. V-ATPase genes in Arabidopsis and tomato ... 38
Chapter 5
Table 1. Sequence extension and polypeptide deduction for unidentifiable tomato cDNA fragments that are "candidates for the systemic wound response" (CSWR) ... 76 Table 2. PCR primers specific to 9 novel tomato cDNAs that were used to verify putative open reading frame sequences and perform real-time RT-PCR experiments... 77
Chapter 6
Table 1. Summary of a subtractive cDNA library containing transcripts systemically up-regulated in the hour after fire damage ... 93
Chapter 7
Table 1. Specifications for a universal sequencing-based method of gene expression
... 181
Appendix 1 Table 1. Subunit c amino acid identities... 127
Table 2. Subunit c” amino acid identities ... 128
Table 3. Subunit d amino acid identities... 129
Table 4. Subunit e amino acid identities... 130
Table 5. Subunit A amino acid identities... 132
Table 6. Subunit B amino acid identities ... 134
Table 7. Subunit C amino acid identities ... 135
Table 8. Subunit D amino acid identities... 135
Table 9. Subunit E amino acid identities ... 137
Table 10. Subunit F amino acid identities ... 138
Table 11. Subunit G amino acid identities... 139
Table 12. Subunit H amino acid identities... 140
Appendix 3-A Table 1. ASPB member involvement and satisfaction with supporting undergraduate and high school research... 154
Table 2. Frequencies of ASPB member comments regarding the potential advantages of supporting undergraduate (UG) and high school (HS) research... 154
Table 3. Frequencies ofASPB member comments regarding the potential disadvantages of supporting undergraduate (UG) and high school (HS) research... 155
List of Figures
Chapter 1
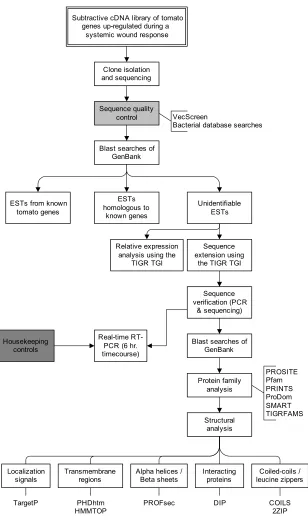
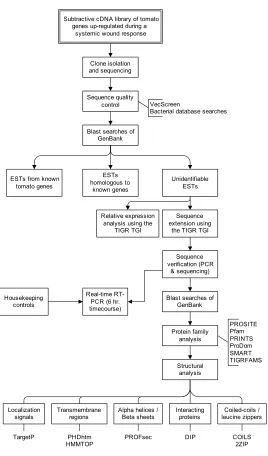
Figure 1. Strategy to identify and analyze cDNAs up-regulated in tomato leaf tissue during a systemic wound response to fire damage... 5
Chapter 2-A
Figure 1. The path from sequencing a cDNA to an improperly edited sequence... 15
Chapter 2-B
Figure 1. Matches of 20 bp, 19 bp, 18 bp, etc. in the indica genome corresponding to the pUC18 SmaI site ... 23
Chapter 3
Figure 1. Percentage of tomato cDNA libraries (n = 27) which contain ESTs for given genes within various fold ranges of relative expression ... 32
Chapter 4
Figure 1. Amino acid identity of tomato V-ATPase subunits compared to Arabidopsis... 42 Figure 2. Relative expression levels of V-ATPase ESTs in different cDNA libraries of the TIGR TGI... 43 Figure 3. Relative expression levels of individual V-ATPase cDNAs... 44 Figure 4. Cumulative relative expression levels of tomato V-ATPase subunits ... 44 Figure 5. Similarity between ATPase relative expression in developing tomatoes and V-ATPase activity in developing grapes (grape data from Terrier et al., 2001)... 46
Chapter 5
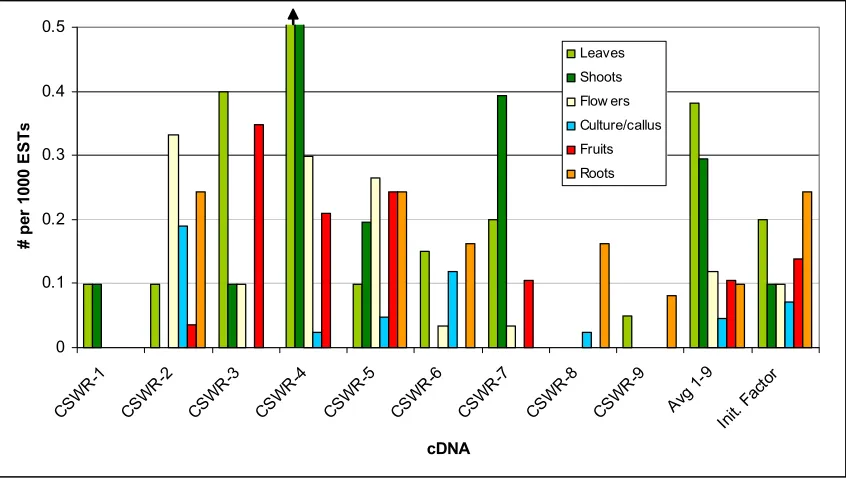
Figure 4. Organ-specific relative abundance of CSWR-1 through CSWR-9 in unwounded
tomato plants... 81
Figure 5. Systemic transcript accumulation of 9 tomato cDNAs (CSWR-1 through CSWR-9) in leaf 4 after flame wounding leaf 3 ... 82
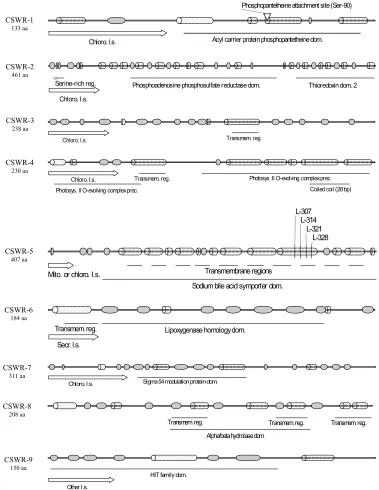
Figure 6. Structural and functional prediction of 9 tomato proteins, encoded by CSWR-1 through CSWR-9 ... 83
Chapter 6 Figure 1. Conservation of transcript sequences between tomato and Arabidopsis... 95
Figure 2. Phenylpropanoid biosynthesis from phenylalanine... 98
Figure 3. The methyl cycle and ethylene synthesis ... 99
Chapter 7 Figure 1. Comparisons that can be made between 2 transcript populations using binding-radiation (a) and sequencing (b) methods... 114
Figure 2. Theoretical blueprint for a universal sequencing-based method of gene expression analysis... 119
Appendix 1 Figure 1. Alignment of c subunits in tomato ... 125
Figure 2. Alignment of c subunits in tomato and Arabidopsis... 126
Figure 3. Alignment of c” subunits in tomato... 127
Figure 4. Alignment of c” subunits in tomato and Arabidopsis... 128
Figure 5. Alignment of d subunits in tomato and Arabidopsis... 129
Figure 6. Alignment of e subunits in tomato ... 130
Figure 7. Alignment of e subunits in tomato and Arabidopsis... 130
Figure 8. Alignment of A subunits in tomato and Arabidopsis... 131
Figure 10. Alignment of B subunits in tomato and Arabidopsis... 133
Figure 11. Alignment of C subunits in tomato and Arabidopsis... 134
Figure 12. Alignment of D subunits in tomato and Arabidopsis... 135
Figure 13. Alignment of E subunits in tomato... 136
Figure 14. Alignment of E subunits in tomato and Arabidopsis... 136
Figure 15. Alignment of F subunits in tomato and Arabidopsis... 137
Figure 16. Alignment of G subunits in tomato ... 138
Figure 17. Alignment of G subunits in tomato and Arabidopsis... 138
Figure 18. Alignment of H subunits in tomato and Arabidopsis... 139
Appendix 3-A Figure 1. ASPB member comments regarding potential advantages of supporting undergraduate researchers... 155
Figure 2. ASPB member comments regarding potential advantages of supporting high school researchers... 155
Figure 3. Number of ASPB member comments regarding undergraduate and high school research ... 155
Appendix 3-B Figure 1. Percentages of plant biologists who mentored various numbers of undergraduates in different “length of their mentoring career” categories ... 183
Figure 2. Total number of undergraduates mentored by plant biologists of different academic ranks at land-grant universities, other research universities, and primarily undergraduate institutions (PUIs) ... 184
Figure 3. Percentages of plant biologists of different academic rank at land-grant universities, other research universities, and primarily undergraduate institutions (PUIs) who perceive institutional incentives for mentoring undergraduate researchers ... 184
Figure 2. Average levels of student involvement in typical research-related activities ... 198 Figure 3. Student perceptions of their research and/or teaching experience ... 199
Chapter 1
The ultimate goal of this dissertation was to identify transcripts that are systemically
up-regulated in response to fire damage in tomato plants. In order to accomplish this task,
several advances for sequencing-based methods of gene expression analysis had to be
developed and refined before meaningful analysis of a subtractive cDNA library could be
achieved. In Chapter 2, methods for improving sequence quality control and identifying
false sequences are presented. A method for identifying adaptor contaminants was
developed and used to identify over 78,000 false sequences in GenBank. One of the many
contaminated sequences was from the human p8/com1 gene, which has implications for
research on breast cancer. Other types of sequence contamination include sequences from
vectors and foreign organisms (pathogens, etc.), which were found in several thousand
locations in the indica rice genome. In Chapter 3, a novel method for identifying and
evaluating housekeeping genes using sequence data is presented. Using this method with
tomato sequences, relative expression analyses for 127 potential housekeeping control
transcripts were performed. These analyses provided potential housekeeping transcripts
which were used for real-time RT-PCR experiments later in the dissertation (Chapter 5).
In order to characterize the array of transcripts which systemically accumulate in
plants after fire damage, a subtractive cDNA library was used for their isolation and
identification, and these are described in Chapters 4-6. Chapter 4 (with Appendix 1) presents
the identification and characterization of 23 transcripts which encode all 13 subunits of
vacuolar H+-ATPases in tomato plants. This study stemmed from the discovery that one of
the transcripts from the library encoded a c subunit of vacuolar H+-ATPase. In Chapter 5
(with Appendix 2), the library served as a starting point to identify and characterize 9 novel
flame wounded. Real-time RT-PCR using leaf RNA isolated at different times after flaming
showed that the most common pattern of transcript accumulation was an increase within 30
to 60 minutes, followed by a return to basal levels within 3 hours. Expression analyses also
showed that most up-regulated transcripts were already present in unwounded tissues.
Structural and functional predictions were also performed for each of the 9 novel transcripts.
In Chapter 6, a total of 46 different transcripts are described which were identified from the
subtractive cDNA library. Compared with the entire tomato transcriptome, these 46
wound-up-regulated transcripts are very highly conserved. The vast majority fell into 5 classes:
enzymes of general metabolism; protein synthesis, modification, and transport; transcription;
membrane transport; and photosynthesis and respiration. At least half of the transcripts have
been previously associated with wounding or stress, suggesting that the systemic response to
fire damage has components similar to those of other wound and stress responses. On the
other hand, 30% of transcripts were associated with photosynthesis and respiration,
suggesting that part of the response to fire damage is notably different from other wound and
stress responses. In addition to furthering knowledge on systemic responses to fire damage,
Chapters 4-6 (and Appendices 1 and 2) demonstrate how sequence data can be used
simultaneously for gene discovery and expression analyses.
In Chapter 7, conclusions and future directions are provided for gene expression
analyses using sequence data and for the biology of systemic responses to fire damage.
Future directions include a universal sequencing-based method of gene expression analysis,
as well as experiments to address whether or not the 46 transcripts lead to proteins which
Appendix 3 presents several educational studies on how to involve undergraduates
and high school students in research projects such as the ones presented in this dissertation.
The overall flow of work for this dissertation is shown in Figure 1. Work began with
a subtractive cDNA library containing tomato transcripts up-regulated during a systemic
response to flame wounding. From the subtractive cDNA library, tomato cDNA fragments
were isolated and sequenced. The sequences were then screened for various types of
contamination (using methods developed in Chapter 2). Blast searches of GenBank
databases allowed the sequences to be divided into 3 classes based on their similarity to
known genes: known tomato genes, homologous to known genes (but not known in tomato),
and unidentifiable. The cDNA fragments which were unidentifiable were then analyzed in
much more detail. Using expressed sequence tags (ESTs) in public databases, the full-length
open reading frames of the transcripts were pieced together with the aid of bioinformatics
tools. These full-length sequences were then checked experimentally by building PCR
primers, amplifying them from a cDNA sample, and sequencing. The ESTs from public
databases were also used to perform expression analyses. Using the full-length open reading
frame sequences, extensive bioinformatics work was performed to predict the structures and
functions of the putative proteins. Finally, real-time RT-PCR was performed over a 6 hour
time course after flame wounding to better understand the kinetics of transcript
accumulation. Housekeeping controls which were used in real-time RT-PCR experiments
Subtractive cDNA library of tomato genes up-regulated during a
systemic wound response
Clone isolation and sequencing
Sequence quality control
Blast searches of GenBank ESTs homologous to known genes Sequence extension using
the TIGR TGI ESTs from known
tomato genes
Sequence verification (PCR
& sequencing)
Blast searches of GenBank Protein family analysis Structural analysis Unidentifiable ESTs PROSITE Pfam PRINTS ProDom SMART TIGRFAMS Transmembrane regions Localization
signals Alpha helices /Beta sheets Interactingproteins
PHDhtm HMMTOP
TargetP DIP
Real-time RT-PCR (6 hr. timecourse)
PROFsec Housekeeping
controls
VecScreen
Bacterial database searches
Coiled-coils / leucine zippers
COILS 2ZIP Relative expression
analysis using the TIGR TGI
Figure 1. Strategy to identify and analyze cDNAs up-regulated in tomato leaf tissue during a systemic wound
Chapter 2
Sequence Quality Control
Jeffrey S. Coker and Eric Davies
Eric Davies provided guidance and editorial assistance.
This chapter is divided into three separate papers. Data associated with the first paper were reported to the National Center for Biotechnology Information in 2001, leading to the correction of numerous RefSeqs (curated gene sequences). The first paper has been accepted
for publication in Biotechniques, and the second will be submitted. The third paper was
published in 2002 in the journal Cancer Research 62, 4164-4165, and led to the correction of
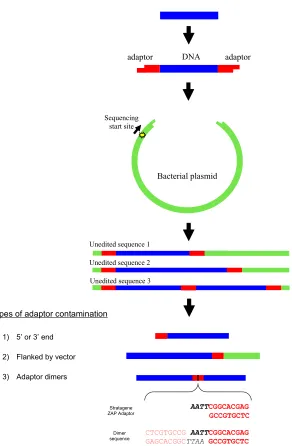
Identifying adaptor contamination when mining DNA sequence data
Jeffrey S. Coker and Eric Davies
Department of Botany, North Carolina State University, Campus Box 7612, Raleigh, North Carolina 27695. email: [email protected]
Abstract
Meaningful analysis of DNA sequences depends on the accuracy of the sequences
themselves, and so false sequences in public databases are a major concern for bioinformatics
research. We describe a simple screen which has identified adaptor contamination in over
78,000 eukaryotic sequences in GenBank. Most of these entries were found in the GenBank
EST databases, but 4,528 were found in the GenBank/EMBL/DDBJ/PDB “nr” database. Out
of a subset of 210 contaminated “nr” database entries, adaptor sequence was present in 82
(39%) as part of a gene or cDNA and in 11 (5%) as part of an open reading frame. Adaptor
contamination was found to extend beyond public databases since 108 of the 210 “nr” entries
are linked to peer-reviewed publications. Bioinformatics work which uses data mined from
public sequence databases should include a simple check for adaptor contamination.
Detection of adaptor sequence contamination is made far easier by knowing that over 99% of
adaptor contaminants appear near the ends of sequences, are flanked by vector, or involve
Analysis of DNA sequences can only be as correct as the sequences themselves, and
so contamination in public databases is a major concern for bioinformatics research. Here
we describe a simple screen which identified adaptor contamination in over 78,000
eukaryotic sequences in GenBank. Awareness that over 99% of adaptor contaminants appear
near the ends of sequences, are flanked by vector, or involve adaptor dimerization allows the
detection of 99% of these sequences (Fig. 1).
A contaminated sequence is defined as “one that does not faithfully represent the
genetic information from the biological source organism/organelle because it contains one or
more sequence segments of foreign origin” (http://www.ncbi.nlm.nih.gov/VecScreen/contam.html). Sources
of contamination for nuclear DNA and cDNA include vector sequence (1-6), plasmid vector
insertion sequences (7), impure tissue sources (8), faulty laboratory protocols (9-10),
mitochondrial DNA (11), and ribosomal DNA/RNA (12). There is one published account of
contamination due to adaptor sequences, where it was shown that commercial adaptor
sequences matched the 5’ or 3’ end of 728 GenBank and EMBL sequences (13). Strategies
to decrease contamination in database sequences have emphasized vector sequences (4-6, 8)
and given little attention to adaptor contamination.
An adaptor is a short oligonucleotide that is ligated to the ends of cDNAs for
incorporation into a vector cloning site (Fig. 1). Usually adaptors consist of several
restriction sites, one blunt end (for ligation to cDNA) , and one cohesive end (for ligation to a
vector). Adaptors are frequently used in the construction of cDNA libraries and in
The presence of adaptor sequences in organismal sequences in public databases has
the potential to cause many different errors of interpretation (14,15) which include the
following:
False hits for others using public databases.
Added difficulties in identifying genes and joining contigs. Misconstruction of PCR primers, microarrays, probes, etc.
Incorrect conclusions regarding evolution and differences between organisms. Incorrect conclusions about gene structure, mRNA splicing, and mRNA transport. Incorrect conclusions about protein sequence, structure, transport, and function.
To investigate adaptor contamination in public databases, BLASTn searches of
GenBank (release 140.0; Feb. 15, 2004) eukaryotic sequences were performed using the
search parameters shown in Table 1. The search parameters returned perfect matches (100%
identity) with the respective adaptor sequences (Table 1). It should be noted that 3 separate
searches of the EST databases were performed for Stratagene Zap and Clontech P1/PN1
adaptors (human, mouse, and non-human/mouse ESTs were searched separately using the
E-values in Table 1) because searching all ESTs simultaneously returned more hits than the
server could process. Manual review of individual GenBank entries, literature review, and
personal communications were used to investigate several hundred matches further.
GenBank entries with adaptor contamination were also screened for vector contamination
using VecScreen (www.ncbi.nlm.nih.gov/VecScreen/VecScreen.html), the tool commonly
used to screen GenBank submissions.
The searches and subsequent analyses identified over 78,000 contaminated sequences
in GenBank (Table 1). Most contaminated sequences were found in the GenBank expressed
sequence tag (EST) database, but the “nr” database (which contains annotated genes, etc.)
with adaptors that were not included when using the search parameters in Table 1, making it
evident that the actual number of contaminated sequences is much higher than shown in
Table 1. Simply increasing the E-value will return these shorter matches.
Within the contaminated GenBank sequences, over 99% of adaptors were within 50
bp of an end, connected to vector sequence match as shown by VecScreen, or involved in
dimerization (Fig. 1). The majority of matches not near the 5’ or 3’ end involved
dimerization of Stratagene’s ZAP adaptor as shown in Figure 1. We performed BLASTn
searches using the full sequences of many GenBank entries that included putative dimer
sequences in the gene or cDNA sequence. These searches typically resulted in some
GenBank entries matching the query on one side of the dimer, but had totally different entries
matching the other side, suggesting that the query sequences actually contained two unrelated
sequences that were joined via dimerization. Obviously, this has the potential to create
significant errors, especially since the dimer is often in the middle of sequences where it is
more likely to be interpreted as part of the open reading frame.
A subset of 210 matches (from the “nr” database) with Clontech’s Marathon primer
adaptors were examined more closely. These adaptors are part of Clontech’s suppression
subtractive hybridization procedure (U.S. patents 5,565,340 and 5,759,822) used originally to
make cDNA libraries and probes (16,17). Currently, a single 44 bp adaptor (P1/PN1) is used
in both Marathon and PCR-Select products. The first guanine residue in P1 has been
changed to a cytosine in recent Clontech kits.
STAATACGACTCACTATAGGGC TCGAGCGGCCGCCCGGGCAGGT
In the first Clontech libraries utilizing this technology, a second adaptor (P2/PN2) was also
used (16).
TGTAGCGTGAAGACGACAGAA AGGGCGTGGTGCGGAGGGCGGT
P2 PN2
Of 210 matches with Clontech Marathon adaptors, at least 82 (39%) are contaminated in
regions designated as gene or cDNA sequence, including 11 open reading frames (5%).
Through literature review and personal communications, we confirmed that Clontech
protocols had been used. Published literature shows these false sequences appearing in
transposons, protein sequences, regions used to join contigs, and other biologically relevant
regions. In fact, we found published accounts of (unrecognized) contaminated sequence in
most major journals of genetics and molecular biology.
The recognition of adaptor contamination has the potential to resolve many problems
in the literature (14,15). It is expected that removing adaptor contamination will clarify
many gene sequences as individual labs reinterpret their own sequences, and will prevent
those mining data from amplifying such errors.
Acknowledgments
We thank the scientists who corresponded with us regarding their GenBank entries,
Sophia Clotho for advice, Ron Sederoff for critical review, and staff at NCBI for their
References
1. Lamperti, E.D., J.M. Kittelberger, T.F. Smith, and L. Villakomaroff. 1992. Corruption of genomic databases with anomalous sequence. Nucl. Acids Res. 20:2741-2747.
2. Lopez, R., T. Kristensen, and H. Prydz. 1992. Database contamination. Nature 355:211.
3. Reynolds, T.L. 1994. Vector DNA artifacts in the nucleotide-sequence database. Biotechniques 16:1124-1125.
4. Harger, C., M. Skupski, J. Bingham, A. Farmer, S. Hoisie, P. Hraber, D. Kiphart, L. Krakowski, et al. 1998. The Genome Sequence DataBase (GSDB): improving data quality and data access. Nucl. Acids Res. 26:21-26.
5. Miller, C., J. Gurd, and A. Brass. 1999. A RAPID algorithm for sequence database comparison: application to the identification of vector contamination in the EMBL databases. Bioinformatics 15:111-121.
6. Seluja, G.A., A. Farmer, M. McLeod, C. Harger, and P.A. Schad. 1999. Establishing a method of vector contamination identification in database sequences. Bioinformatics 15:106-110.
7. Binns, M. 1993. Contamination of DNA database sequence entries with Escherichia coli insertion sequences. Nucl. Acids Res. 21:779-779.
8. White, O., T. Dunning, G. Sutton, M. Adams, J.C. Venter, and C. Fields. 1993. A quality-control algorithm for DNA-sequencing projects. Nucl. Acids Res. 21:3829-3838.
9. Gersuk, V.H. and T.M. Rose. 1993. Database contamination. Science 260:606.
10. Dean, M. and R. Allikmets. 1995. Contamination of cDNA libraries and expressed-sequence-tags databases. Am. J. Hum. Genet. 57:1254-1255.
11. Wenger, R.H. and M. Gassmann. 1995. Mitochondria contaminate databases. Trends Genet. 11:167-168.
12. Gonzalez, I.L. and J.E. Sylvester. 1997. Incognito rRNA and rDNA in databases and libraries. Genome Res. 7:65-70.
13. Yoshikawa, T., A.R. Sanders, and S.D. Detera Wadleigh. 1997. Contamination of sequence databases with adaptor sequences. Am. J. Hum. Genet. 60:463-466.
15. Forster, P. 2003. To err is human. Annals of Human Genetics 67: 2-4.
16. Diatchenko, L., Y-F. Chris Lau, A.P. Campbell, A. Chenchik, F. Moqadam, B. Huang, S. Lukyanov, K. Lukyanov, et al. 1996. Suppression subtractive hybridization: A method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc. Natl. Acad. Sci. USA 93:6025-6030.
Table 1. Sequences and search parameters to identify entries in GenBank contaminated by 7 commercial adaptor sequences.
Adaptor Sequence to search
Filter E-value Word size Identity nr database EST database
Clontech P1/PN1 TCGAGCGGCCGCCCGGGCAGGT Yes none 1 7 100 255 11655 Clontech P2/PN2 AGGGCGTGGTGCGGAGGGCGGT No none 1 7 100 13 705 Clontech EcoRI AATTCGCGGCCGCGTCGAC Yes none 0.05 7 100 156 15071
Promega EcoRI AATTCCGTTGCTGTCG No none 5 7 100 120 1167
Stratagene/Amersham Pharmacia EcoRI/NotI AATTCGCGGCCGC No none 150 7 100 765 16196 Stratagene ZAP AATTCGGCACGAG No none 150 7 100 3166 28830 Stratagene ZAP (dimer) CTCGTGCCGAATTCGGCACGAG No none 0.005 7 100 (778) (24106) Life Technologies 3' RACE GGCCACGCGTCGACTAGTAC Yes none 10 7 100 53 66
4528 73690 =78218 Matches in Eukaryota
Detected by VecScreen?
Search Parameters
cDNA
c
3 types of adaptor contamination
1) 5’ or 3’ end
2) Flanked by vector
3) Adaptor dimers
AATT
CTCGTGCCG AATT
GAGCACGGCTTAA Stratagene
ZAP Adaptor
Dimer sequence
Unedited sequence 3 Unedited sequence 2 Unedited sequence 1
Sequencing start site
DNA adaptor
adaptor
Bacterial plasmid
CGGCACGAG GCCGTGCTC CGGCACGAG GCCGTGCTC
Cleaning data mined from the
indica
rice genome draft
Jeffrey S. Coker and Eric Davies
Department of Botany, North Carolina State University, Campus Box 7612, Raleigh, North Carolina 27695. email: [email protected]
Filtering out false sequences is a challenge for every genome project. Because the
Oryza sativa L. ssp. indica genome draft (1) is a major resource for efforts to improve the
world food supply, its accuracy is of paramount importance and thus needs to be
scrutinized very closely. The analysis presented here is intended especially for those
mining data from the indica genome, and indicates false sequences of three different
types: short (< 21 bp) remnants of SmaI-linearized pUC18 plasmid, regions of other
cloning vector(s), and genomic sequence from an unidentified species of Phytophthora.
Recommendations are given for how to identify each type of false sequence when using
data mined from the indica genome draft. Removal of false sequences is necessary to
avoid errors in calculating polymorphism rates, gene discovery, estimating lateral gene
transfer, and many other forms of bioinformatics research.
SmaI-linearized pUC18 plasmid
It was reported that a SmaI-linearized pUC18 plasmid was used for cloning rice
genomic fragments (1), and thus it follows that each rice sequence would have been
flanked by pUC18 before the sequence was “cleaned”. We have found that short
remnants of pUC18 are still scattered throughout the indica genome. As shown in Table
1, 98% of matches with the pUC18 SmaI site (≥14 bp) in both the unassembled data and
unassembled data and one fully masked read are within 15 bp of an end. This suggests
that the vast majority of matches with the pUC18 SmaI site derive from cloning vector
and are not genuine rice sequences. Peripheral contaminants in unassembled data are not
a problem as long as they are removed before assembly.
A much more significant problem occurs when these contaminants become
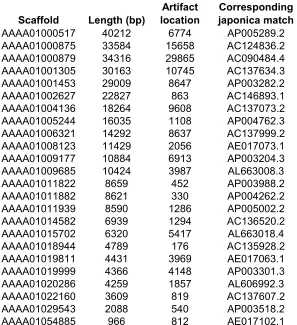
internalized as sequences are joined together. Table 2 shows examples of internalized
pUC18 artifacts which were found in the scaffolds listed in Table 1. The ratio of
internalized contaminants to total contaminants leads us to conclude that 5-7% of
peripheral contaminants were internalized during contig/scaffold construction. Each
scaffold in Table 1 matches japonica rice entries in GenBank directly before and after the
short region in question but not within it, proving that each is a false sequence. For
example, Scaffold 9177 (GenBank acc. no. AAAA01009177) contains a pUC18 fragment
at 6913 bp, and matches japonica sequences on both sides of the fragment (Table 2).
Although the pUC18 fragment is only 20 bp long, the “hole” in the indica sequence
(compared to japonica) is 517 bp long. There are many examples of such holes which
are clearly not biological in origin. From a comparison of Chromosome 4 between
indica and japonica, it has been suggested that japonica sequence may be “larger”
because of insertions of transposable elements, and the average frequency of
single-nucleotide polymorphisms is 1 SNP per 268 bp (3). However, since many apparent
insertions and SNPs are due to the presence of false sequences and holes in the indica
draft, such conclusions about differences between indica and japonica may be premature.
Since contamination by 14-20 bp fragments is present, a much larger number of
random chance would furnish only 4.5 matches with the 13 nucleotide sequence
preceding the SmaI site (CTAGAGGATCCCC), but indica scaffolds have 1274 matches,
while japonica has only 10 (2). Comparing the number of possible pUC18 artifacts (7-20
bp) with the number of matches one would expect by chance (E-values) leads to a
prediction of over 13,000 contaminants (Fig. 1), or .029% of the total contig length. The
7-20 bp pUC18 fragments alone (not including 1-6 bp fragments and the “holes” they
often represent) could account for 14% of the SNPs (1 SNP per 269 bp) between indica
and japonica (3).
For those mining data from the indica rice genome, we recommend the following
steps: 1) Search all sequences for fragments of the pUC18 SmaI site
(GTCGACTCTAGAGGATCCCC) 2) Remove the pUC18 sequences when they occur at
the end(s) 3) For internal pUC18 matches, take 200-500bp of sequence surrounding each
possible pUC18 artifact and Blast it against japonica and/or other rice sequences in
GenBank. If the region is not genuine rice sequence, the sequences may match on either
side of the SmaI site, but will not match indica in the SmaI site. Closer examination
usually reveals a “hole” in the indica sequence ranging from 10bp to several thousand
base pairs. Data miners should also be aware that every pUC18 contaminant that is at
least 12 bp contains a potential false “STOP” site (TAG) from base 10 to 12.
Regions of other cloning vector(s)
It appears that vectors other than pUC18 were also used for indica library
construction. In some cases, matches with a particular vector appear on both ends of a
Life Technologies pZL1 from Lambda ZipLox (or a similar vector) is at the ends of at
least 25 scaffolds (e.g. Scaffold 89563) (4). In other cases such as Scaffolds 39078 (1276
bp), 45670 (1105 bp), and 82154 (691 bp), entire indica scaffolds are 99-100% identical
to several dozen common vectors but match no rice sequences in GenBank or Syd (2). In
other more ambiguous cases (e.g. Scaffold 101296), scaffolds are near perfect matches
with both vectors and rice ESTs in GenBank, but still match nothing in Syd. Judging by
the large size of these matches, it is unlikely that all vectors used in library construction
were accounted for in decontamination screens.
For those mining data from the indica genome, we recommend that sequences of
particular interest are compared to the VecScreen database (4) and/or bacterial databases.
Phytophthora
Phytophthora are well-known stramenopiles that commonly parasitize a wide
variety of plant species. There are several dozen indica scaffolds that match
Phytophthora sequences but do not closely match sequences either in japonica or any
other higher plant (Table 3). For example, Scaffold 45690 (Contig 77125) has 99.7%
identity with 1107 bp of P. infestans mitochondrial DNA coding for three ribosomal
proteins, but has no significant match with any plant sequence. Searches of indica
identified 226 scaffolds that match GenBank Phytophthora sequences with an E-value of
1x10-10 or lower (5). Many of these may be highly conserved rice sequences and not
from Phytophthora. Even so, since it is evident that there are sequences from
Phytophthora present (Table 3) and no Phytophthora genome has been completely
There are three possible explanations for Phytophthora-like sequences in the
indica genome: pathogen-infected tissue, cross-contamination of libraries, and lateral
gene transfer. It is quite possible that pathogen-infected rice tissue was used for DNA
isolation since pathogens are notoriously prevalent in plant tissue. The more exciting
explanation would be lateral gene transfer after the divergence of indica from japonica.
However, we are unaware of any example of simultaneous lateral gene transfer of nuclear
genes encoding mRNA (e.g. ric1 and actA) and rRNA (e.g. 18S), and mitochondrial
genes encoding mRNA (e.g. rp12, rps19, and rps3) and rRNA (e.g. 16S rRNA), all of
which seem to be present in indica (Table 3).
For those mining data from the indica genome, we recommend that sequences of
particular interest are compared to Phytophthora and japonica sequences (including
ESTs). Contaminants will be nearly identical to Phytophthora sequences (if they have
been sequenced in Phytophthora). On the other hand, if the indica sequence is nearly
identical to a japonica sequence, then it is not likely to be a contaminant.
Conclusions
The indica rice genome draft has already been used to evaluate monocot and
eudicot divergence (6), sequence variation between varieties of rice (3, 7), single
nucleotide polymorphisms in rice varieties (3, 8), characteristics of various gene families
(9, 10), and many other important topics. It serves as an important resource for
improving world food supply and will be used extensively in the future, and so it is
References
1. J. Yu et al., Science 296, 79 (2002); http://210.83.138.53/rice/.
2. S.A. Goff et al., Science 296, 92 (2002); http://portal.tmri.org/rice/.
3. Q. Feng et al., Nature 420, 316 (2002).
4. Kitts, P.A., Madden, T.L., Sicotte, H. & Ostell, J.A. Manuscript in preparation; http://www.ncbi.nlm.nih.gov/VecScreen/VecScreen.html.
5. All GenBank Phytophthora sequences (including ESTs) were searched against the
indica genome using MegaBlast. Scaffolds with significant matches were then used to search all GenBank sequences (BLASTn).
6. M. Vincentz et al., Plant Physiol. 134, 951 (2004).
7. C. Li et al., Theor. Appl. Genet. 108, 392 (2004).
8. S. Nasu et al., DNA Res. 9, 163 (2002).
9. S. Griffiths et al., Plant Physiol. 131, 1855 (2003).
10. L. Jia et al., Plant Physiol. 134, 575 (2004).
Table 1. Matches in the indica genome with the pUC18 SmaI site
(GTCGACTCTAGAGGATCCCC). Matches shown are at least 14 bp long (Expect ≤
5.7). pUC18 sequences are typically on an end (within 5 bp) of raw genomic sequences such as those in the unassembled data and fully masked reads, but became internalized as contigs and scaffolds were pieced together.
Sequence type Matches Matches at a 5’ or 3’ end
Unassembled data 1990 98.1% (1953)
Fully masked reads 4342 98.0% (4255)
Contigs 944 85.9% (811)
0 5000 10000 15000 20000 25000 30000 35000
20 19 18 17 16 15 14 13 12 11 10 9 8 7
Length of match with pUC18 SmaI site (bp)
N
u
mb
er
o
f
seq
u
en
ce
s
Matches in indica genome
Expect value
Fig. 1. Matches of 20 bp (GTCGACTCTAGAGGATCCCC), 19 bp (TCGACTCTAGAGGATCCCC), 18 bp
(CGACTCTAGAGGATCCCC), etc. in the indica genome corresponding to the pUC18 SmaI site. The expect values
approximate the number of hits one would expect by chance, assuming a random genome sequence. This leads to a prediction of over 10,000 contaminants of 7 bp or longer.
Table 2. Examples of internal pUC18 artifacts (≥14 bp) in indica scaffolds. In each case
shown, the corresponding japonica sequence matches the indica scaffold directly before
and after the artifact. The “holes” in the indica sequences range from 14 to several
thousand bp long. All artifacts shown are more than 100 bp from a scaffold end and from any unfilled gaps within scaffolds (designated by a stretch of N's in GenBank). Scaffolds are listed as their GenBank accession numbers (AAAA01 + scaffold number) to facilitate further review.
Scaffold Length (bp)
AAAA01000517 40212 6774 AP005289.2
AAAA01000875 33584 15658 AC124836.2
AAAA01000879 34316 29865 AC090484.4
AAAA01001305 30163 10745 AC137634.3
AAAA01001453 29009 8647 AP003282.2
AAAA01002627 22827 863 AC146893.1
AAAA01004136 18264 9608 AC137073.2
AAAA01005244 16035 1108 AP004762.3
AAAA01006321 14292 8637 AC137999.2
AAAA01008123 11429 2056 AE017073.1
AAAA01009177 10884 6913 AP003204.3
AAAA01009685 10424 3987 AL663008.3
AAAA01011822 8659 452 AP003988.2
AAAA01011882 8621 330 AP004262.2
AAAA01011939 8590 1286 AP005002.2
AAAA01014582 6939 1294 AC136520.2
AAAA01015702 6320 5417 AL663018.4
AAAA01018944 4789 176 AC135928.2
AAAA01019811 4431 3969 AE017063.1
AAAA01019999 4366 4148 AP003301.3
AAAA01020286 4259 1857 AL606992.3
AAAA01022160 3609 819 AC137607.2
AAAA01029543 2088 540 AP003518.2
AAAA01054885 966 812 AE017102.1
Corresponding japonica match Artifact
Table 3. Examples of phytophthora-like sequences in the indica genome. "Closest" matches are defined as those with the lowest E-value (E<10) in GenBank databases. In all cases shown here, the Phytophthora match spanned the majority of the scaffold and had an effective E-value of 0. Short regions (18-80 bp) on the ends of 8 of these scaffolds are also contaminated by plasmid sequences.
Indica scaffold
Identity Acc. No. Description Identity Acc. No. Description
AAAA01045690 1104/1107 (99%) U17009.2 P. infestans rib. prot. L2, S19, and S3 --- ---
---AAAA01065444 838/844 (99%) AJ238654.1 P. undulata18S rRNA gene 536/617 (86%) AP004778.3 Genomic DNA, chromosome 2 AAAA01078719 705/709 (99%) X54265.1 P. megasperma 16S rRNA 613/715 (85%) AP004778.3 Genomic DNA, chromosome 2 AAAA01076286 639/647 (98%) BE776357.1 P. infestansunidentified cDNA --- ---
---AAAA01070180 630/633 (99%) BE776214.1 P. infestans unidentified cDNA --- --- ---AAAA01084216 630/636 (99%) BE777367.1 P. infestansunidentified cDNA --- ---
---AAAA01070144 581/584 (99%) BE775905.1 P. infestansunidentified cDNA 381/437 (87%) AK063121.1 cDNA clone:001-111-E07
AAAA01090700 579/587 (98%) AJ133023.1 P. infestans ric1 gene --- ---
---AAAA01091080 556/557 (99%) U50844.1 P. infestans host-specific elicitor inf1 gene --- --- ---AAAA01082659 556/559 (99%) BE776610.1 P. infestansunidentified cDNA --- --- ---AAAA01086249 557/567 (98%) BE776104.1 P. infestans unidentified cDNA --- ---
---AAAA01055069 555/584 (95%) BE776247 P. infestansunidentified cDNA 832/904 (92%) AK060330.1 cDNA clone:001-008-B01 AAAA01049644 489/498 (98%) BE777164 P. infestansunidentified cDNA --- ---
---AAAA01063300 444/445 (99%) M59715.1 P.infestans actin (actA) gene 387/457 (84%) AK059967.1 cDNA clone:006-211-F12 AAAA01102792 237/237 (100%) AF339424.1 P. infestans 5.8S rRNA (and spacer) --- ---
---Closest match in all organisms Closest match in japonica genome
Chapter 3
Selection of Candidate Housekeeping Controls in Tomato Plants using EST Data
Jeffrey S. Coker and Eric Davies
Eric Davies provided guidance and editorial assistance.
This chapter was published in 2003 in the journal Biotechniques 35, 740-748. It is
currently being considered for a patent under the title “Method for Identifying Constantly Expressed Genes Using Nucleic Acid Sequence Data” (NCSU Disclosure File Number
Chapter 4
Identification, Conservation, and Relative Expression of V-ATPase cDNAs in Tomato Plants
Jeffrey S. Coker, Derek Jones, and Eric Davies
Derek Jones assisted in mining data for c subunit cDNAs. Eric Davies provided guidance and editorial assistance.
This chapter was published in 2003 in the journal
Chapter 5
Identification, Accumulation, and Functional Prediction of Novel Tomato Transcripts Systemically Up-regulated after Fire Damage
Jeffrey S. Coker, Alan Vian, and Eric Davies
Alan Vian constructed the subtractive cDNA library. Eric Davies provided guidance and editorial assistance.
Abstract
Despite the major impacts of fire on plants, responses to fire damage have not
been closely studied on the level of gene expression. Here we present analyses of novel
transcripts from tomato (Lycopersicon esculentum) which are systemically up-regulated
in leaves after a distant leaf is wounded by flame. Nine cDNA fragments were isolated
from a subtractive cDNA library of leaf tissue 1 hour after flaming. Using data mining
and PCR, full-length open reading frames were predicted, amplified, and then sequenced.
Comparisons with the Arabidopsis genome suggested that 8 of the encoded proteins are
slow-evolving. Real-time RT-PCR using leaf RNA after flaming confirmed the systemic
accumulation of 4 and 7 transcripts within 30 and 60 minutes, respectively, before
returning to basal levels within 3 hours. During this same time course, proteinase
inhibitor I levels gradually increased over 30-fold in 6 hours. Expression analyses also
showed that 8 of the transcripts are present in unwounded leaf, stem, and root tissues.
The predicted proteins include an acyl carrier, adenylyl sulfate reductase, PS II
oxygen-evolving complex protein 3, anion:sodium symporter, chloroplast-specific ribosomal
protein, a histidine triad family protein, and an unknown wound/stress-related protein.
Homologues of several of these proteins have been associated with other types of wound
and stress responses. It appears that within an hour after being damaged by fire, plants
systemically up-regulate a variety of genes involved with basic cell metabolism and
Introduction
Plants must cope with a wide variety of natural wounding stimuli such as fire,
herbivory, wind, rain, hail, UV radiation, sand, and trampling. Because plants are sessile
and cannot escape these stimuli, to ensure survival they often respond to tissue damage
by changes in gene expression (Graham et al., 1986; Braam and Davis, 1990; Schaller
and Ryan, 1996; León et al., 2001) in both damaged tissues (local responses) and in
undamaged tissues (systemic responses). Many “systemic wound response proteins”
(Schaller and Ryan, 1996), which are expressed in undamaged tissues following the
intercellular transmission of a wound signal, have been previously identified in tomato
plants. These include proteinase inhibitors (Green and Ryan, 1972), systemin (Pearce et
al., 1991), an aspartic protease (Schaller and Ryan, 1996), chloroplast mRNA-binding
protein (Vian et al. 1999), a bZIP DNA-binding protein (Stanković et al., 2000), allene
oxide synthase and fatty acid hydroperoxide lyase (Howe et al., 2000), and others.
Further characterization of the array of systemically up-regulated genes is necessary to
better understand plant defense and stress response mechanisms.
Knowledge of systemically up-regulated genes is also necessary to characterize
the intercellular signals that move from wounded to unwounded tissue. Systemic signals
that have been proposed include proteinase inhibitor-inducing factor (Ryan, 1974),
systemin (Pearce et al., 1991), abscisic acid (Peña-Cortés et al., 1991), oligosaccharides
(Ryan and Farmer, 1991), methyl jasmonate (Herde et al., 1996), action potential
(Stanković and Davies, 1996), and variation potential (Wildon et al., 1992; Vian et al.,
1996). It is clear that the systemic wound response is a complex network(s) induced by
many different signals, and that the extent and timing of these signals may vary
significantly depending on the plant species and the precise nature of the wound. For
example, evidence from Arabidopsis microarray experiments suggests that there are
fundamental differences in gene expression in response to mechanical wounding and
insect feeding (Reymond et al., 2000). On the other hand, there is clear evidence for
cross-talk between defense responses such as those that are herbivore- and
pathogen-directed (Stennis et al., 1998). Much about how responses to fire damage compare with
other types of wound responses is unknown.
Fire impacts most terrestrial ecosystems, and plants have evolved mechanisms to
survive fire (Bond and van Wilgen, 1996; DeBano et al., 1998). For example, in the
southeastern United States, shrubs and herbaceous plants in savannas, forests, evergreen
shrub bogs, wire grass sand-hills, swamps, and other ecosystems often survive fires and
are able to resprout and reproduce in future years (Bond and van Wilgen, 1996; DeBano
et al., 1998; Wells, 2002). In fact, some of the most species-rich plant ecosystems (i.e.
the herbaceous groundcover of longleaf pine savannahs) require fire to persist (Platt et
al., 1988; Drewa et al., 2002). A common misconception is that all wildfires kill all
plants in the burned area. The National Parks Service has used a 5-tiered “burn severity
class” system to describe vegetation damage following a wildfire which includes
undamaged (tier 1), scorched (tier 2; leaf litter is singed and foliage is slightly yellowed),
and low severity (tier 3; leaf litter is partly/mostly consumed but foliage remains intact)
classes (USDI, 1992). Resprouting after fire damage can occur from partially burned
above-ground organs or from roots after complete destruction of above-ground organs.
Despite the major impacts of fire on plants, responses to fire damage have not
been closely studied on the level of gene expression. From an experimental standpoint,
flame causes severe, yet reproducible, damage without moving the plant. Leaf flaming
has already proven useful for identifying novel components of the systemic wound
response to fire such as Pin 1 (Wildon et al., 1992; Stanković and Davies, 1996),
chloroplast mRNA-binding protein (Vian et al., 1999) and a bZIP DNA-binding protein
(Stanković et al., 2000).
To study the impacts of fire damage (flame wounding), tomato plants have
several advantages. First, since extensive work with other wound stimuli has been done
using tomato plants, it is possible to compare flame-induced gene expression with this
previous work. Second, a substantial amount is known about wound signaling events in
tomato plants which will facilitate understanding of the timing of the response. Finally,
like many species in the Solanaceae, tomato plants (both wild and cultivated) possess
many characteristics which typically allow many herbaceous plants to survive fires.
These characteristics include being a perennial (Taylor, 1986), having carbohydrate
reserves stored in underground organs (Peres et al., 2001; Verdaguer and Ojeda, 2002),
and the ability to regenerate shoots from hypocotyls, roots, or other tissues (Takashina et
al., 1998; Bertram and Lercari, 2000; Peres et al., 2001). It has been found that smoke
extract stimulates the growth of tomato roots in vitro (Taylor and van Staden, 1998), and
that growth of species within the Solanaceae can be regulated by fire regimes (Preston
and Baldwin, 1999). Also, a bZIP gene similar to the one we found to be up-regulated by
flame-wounding (Stankovic et al., 2000) has also been associated with adventitious shoot
regeneration (Low et al., 2001). Thus, tomato plants are the preferred model system for
work on the systemic wound responses to fire damage.
For genes previously examined, the most common pattern of transcript
accumulation in leaf 4 of three-week old tomato plants following a flame wound on leaf 3
is an increase that peaks within an hour, followed by a rapid decrease (Davies et al.,
1997; Vian et al., 1999). These rapid changes are then followed by a more gradual period
of increased, decreased, or unchanged transcript accumulation. This has been shown
most vividly for Pin 1 (Stanković and Davies, 1997), CMBP (Vian et al., 1999), and a
bZIP DNA-binding protein (Stanković et al., 2000). The complexity of responses to
wounding for individual transcripts (rapid increases and decreases) and the variation
between transcripts (different time points for increase/decrease) suggests that different
genes are being up-regulated by different systemic signals, or combinations of signals.
This cannot be deciphered without characterizing a wider array of transcripts that
accumulate systemically following flame wounding.
Here we present analyses of 9 previously unidentified tomato cDNAs which are
systemically up-regulated after a distant leaf is wounded by flame. These cDNAs were
isolated from a subtractive cDNA library (wound minus control) from tissue harvested
one hour after flaming.
Results
Our strategy for identifying and characterizing clones from a subtractive cDNA
library of wound-induced transcripts is shown in Figure 1. Clones from the library were
labeled as “candidates for the systemic wound response” (CSWR). The 9 clones initially
isolated from the cDNA library ranged from 59 to 647 bp and had an average length of
292 bp (Table 1). Attempts to identify them using Blast searches of GenBank were
inconclusive and/or ambiguous. Therefore, we searched expressed sequence tags (ESTs)
in the TIGR Tomato Gene Index (TGI) to identify identical matches and extend the
cDNA sequences using consensus sequence information (Table 1). The resulting putative
cDNAs ranged from 596 to 1830 and had an average length of 1048 bp (Table 1). These
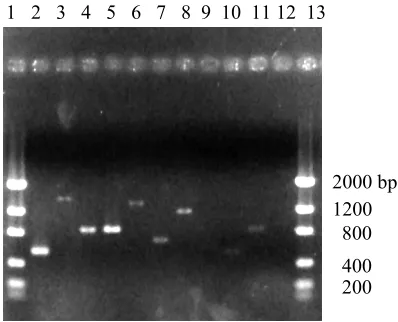
putative cDNAs were confirmed by performing PCR (Fig. 2) and sequencing the PCR
products using the primers in Table 2.
Blast searches using the extended sequences returned matches with protein
sequences in GenBank ranging from 43% to 83% identical (Table 1). The putative
translations of all 9 cDNAs suggested full-length proteins which were approximately the
same size as their respective GenBank matches. Therefore, all 9 cDNAs encode proteins
similar to those sequenced in other plants, although the exact functions of most are still
unknown.
By comparing tomato Unigenes in the TIGR TGI with the Arabidopsis genome
(using tBlastx), Van der Hoeven et al. (2002) divided tomato ESTs into “not
homologous” (E value ≥ 0.1), “fast-evolving” (1.0E-15 < E value < 0.1), “intermediate
evolving” (1.0E-50 < E value < 1.0E-15), and “slow-evolving” (E value < 1.0E-50)
classes. Only about 22% of all Unigenes fell into the “slow-evolving” class. By