Malignant Pediatric Pathologies: LMP1 Sequence Characterization and
Linkage with Other Viral Gene Polymorphisms
Mario Alejandro Lorenzetti,aMagdalena Gantuz,aJaime Altcheh,bElena De Matteo,cPaola Andrea Chabay,a and María Victoria Preciadoa
Molecular Biology Laboratory, Pathology Division, Ricardo Gutiérrez Children Hospital, Gallo 1330 Buenos Aires, Argentinaa; Parasitology and Chagas Disease Laboratory, Ricardo Gutiérrez Children Hospital, Gallo 1330 Buenos Aires, Argentinab; and Pathology Division, Ricardo Gutiérrez Children Hospital, Gallo 1330 Buenos Aires, Argentinac
The ubiquitous Epstein-Barr virus (EBV) is related to the development of lymphoma and is also the etiological agent for infectious mononucleosis (IM). Sequence variations in the gene encoding LMP1 have been deeply studied in different pa-thologies and geographic regions. Controversial results propose the existence of tumor-related variants, while others ar-gued in favor of a geographical distribution of these variants. Reports assessing EBV variants in IM were performed in adult patients who displayed multiple variant infections. In the present study, LMP1 variants in 15 pediatric patients with IM and 20 pediatric patients with EBV-associated lymphomas from Argentina were analyzed as representatives of benign and malignant infections in children, respectively. A 3-month follow-up study of LMP1 variants in peripheral blood cells and in oral secretions of patients with IM was performed. Moreover, an integrated linkage analysis was performed with variants of EBNA1 and the promoter region of BZLF1. Similar sequence polymorphisms were detected in both pathological conditions, IM and lymphoma, but these differ from those previously described in healthy donors from Argentina and Brazil. The results suggest that certain LMP1 polymorphisms, namely, the 30-bp deletion and high copy number of the 33-bp repeats, are associated with EBV-related pathologies, either benign or malignant, instead of just being tumor re-lated. Additionally, this is the first study to describe the Alaskan variant in EBV-related lymphomas that previously was restricted to nasopharyngeal carcinomas from North America.
E
pstein-Barr virus (EBV) is a member of theLymphocryptovirusgenus, which is part of theGammaherpesvirinaesubfamily of
the largeHerpesviridaefamily. EBV commonly is transmitted
be-tween individuals through saliva and results in the lifelong latent infection of memory B cells (26). In developing regions, serocon-version arising after primary infection usually occurs during early childhood after the disappearance of maternal antibodies and is not associated with severe clinical symptoms. In Argentina, a typ-ical developing region profile is observed, since EBV antibodies can be detected in more than 80% of children by the third year of age (3). On the other hand, less than 40% of the population be-comes infected with EBV during childhood in developed regions or in groups with high socioeconomic status. When primary in-fection is delayed until adolescence or early adulthood, it can cause a more severe case of infectious mononucleosis (IM) in a proportion of individuals (26, 39).
Besides being the etiological agent of IM, EBV has been linked to a variety of lymphoid and epithelial malignancies, such as Burkitt (BL), Hodgkin (HL), and nasal NK/T lymphomas, naso-pharyngeal carcinoma (NPC), and gastric carcinoma (GC) (31, 42). Given that the list of EBV-related malignancies continues to increase, in 1997 the World Health Organization classified EBV as a carcinogenic agent (10). The involvement of EBV in the etiology, progression, and/or outcome of these malignancies is not yet fully understood, but it is likely that factors (RNAs or proteins) derived from the viral genome as well as their interaction with cellular proteins determines the susceptibility to infection of different cell types and/or contributes to the development of neoplasia. For these reasons EBV is still a candidate for analysis, and this fact
impels the quest for viral factors which are inherent to tumors and differ from the ones present in healthy carriers.
Based on polymorphisms in nuclear antigens EBNA3A, EBNA3B, and EBNA3C, two different EBV types, EBV1 and EBV2, can be dis-tinguished (33). Given that these polymorphisms are not enough to describe the entire EBV natural variation, variants and subvariants have been described based on polymorphisms in other viral antigens, such as BZLF1, EBNA1, and latent membrane protein 1 (LMP1) (ex-tensively reviewed in reference 6).
LMP1, encoded by the BNLF1 gene, was the first EBV onco-protein to be described, mainly due to its capacity to transform
rodent fibroblastsin vitro, rendering them tumorigenic in nude
mice, and is probably the most widely studied protein of the virus (41). LMP1 consists of 386 amino acids subdivided into three domains: a short amino-terminal domain (amino acids 1 to 23), six transmembrane hydrophobic segments (amino acids 24 to 186), and a long carboxy-terminal (C-ter) domain (amino acids 187 to 386) which possesses most of the signaling activity of the protein (42). The C-ter domain of LMP1 inter-acts with cellular proteins through C-terminal activation re-gion 1 (CTAR1) and CTAR2, activating several signaling
path-Received16 September 2011Returned for modification5 November 2011 Accepted14 December 2011
Published ahead of print28 December 2011
Address correspondence to M. A. Lorenzetti, [email protected].
Copyright © 2012, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JCM.05778-11
on May 16, 2020 by guest
http://jcm.asm.org/
Downloaded from
on May 16, 2020 by guest
http://jcm.asm.org/
Downloaded from
on May 16, 2020 by guest
http://jcm.asm.org/
ways, including nuclear factorB (NF-B), mitogen-activated protein kinases (MAPKs), and phosphatidylinositol 3-kinase (PI3K) pathways in a ligand-independent manner, leading to apoptosis inhibition and primary B-cell transformation (42). Recently, a CTAR3 region was identified and shown to regulate signal transduction pathways that regulates pheno-typic changes associated with oncogenesis (1).
Given that LMP1 C-ter sequences manifest a high degree of heterogeneity compared to other EBV genes, recent studies based on direct sequencing have reported conflicting results regarding tumor association, geographical distribution, and compartmen-talization, mainly because of the nonrandom sampling of individ-uals, geographic regions, health conditions, and the various types of specimens (e.g., blood, saliva, and tumor tissue) tested (re-viewed in reference 6). Furthermore, the use of different classifi-cation schemes has prevented direct comparison between studies (12, 34, 40). In addition, most of these studies were carried out in adults, where various percentages of coinfection due to subse-quent reinfections during adult life impair the identification of the first infective EBV variant, and only a few reports assessed pediat-ric samples (2), where coinfection is not frequent.
The most commonly reported LMP1 genetic polymorphisms within the C-ter include a 30-bp deletion (del30) along with a 15-bp insertion (ins15) encoding a Janus kinase 3 (JAK3) signal-ing motif, various numbers of 33-bp tandem repeats (rep33), and several mutation hotspots (12, 34, 40).
A number of reports have focused on the del30 LMP1 variants, which are found frequently in some neoplastic conditions and also in healthy populations (reviewed in reference 6). Moreover, it is still unclear whether the high prevalence of del30 variants detected in some EBV-related disorders merely reflects the predominance of these variants in some geographic areas or is due to peculiar virus-host interactions that lead to the enhanced production and/or selection of LMP1 deletion mutants.
How EBV contributes to the pathogenesis of different neoplas-tic processes remains unclear. The characterization of gene vari-ants which are preferentially tumor associated, and the identifica-tion of structural changes that might affect the funcidentifica-tionality of genes specifically related to oncogenic development, could con-tribute some answers to this question.
In the present study, LMP1 C-ter sequence variations from our geographic region were characterized in peripheral blood mono-nuclear cells (PBMCs) and oral secretions (OS) of pediatric pa-tients with IM as a representative of a replicative yet benign
con-dition and in EBV⫹tumor biopsy specimens as a representative of
latent infection in a malignant condition. In this approach, both acute infection and its convalescence were considered, allowing the study of variant dynamics between body compartments in the replicative state. Moreover, samples at the time of diagnosis from pediatric patients with IM allowed the identification of the origi-nally infecting variant.
MATERIALS AND METHODS
Patients and samples.This study was conducted on 35 pediatric patients. Fifteen had IM, had a median age of 4 years (range, 1 to 17 years), and were 47% male. Twenty patients had EBV-positive lymphomas (14 Hodgkin [HL] and 6 non-Hodgkin [NHL]), had a median age of 7 years (range, 3 to 18 years), and were 70% male. Hospital ethics committees reviewed and approved this study, which is in accordance with the human
experimen-tation guidelines of our institution. Written informed consent was ob-tained from all patients’ parents.
Lymph node biopsy specimens were collected for lymphoma diagnosis before therapy. EBV presence was assessed on formalin-fixed, paraffin-embedded tissue sections byin situhybridization (ISH) for Epstein-Barr encoded RNAs (EBERs) according to the manufacturer’s instructions (Novocastra Laboratories Ltd., United Kingdom). Those cases with pos-itive nuclear staining in tumor cells without staining in infiltrating lym-phocytes were included.
Peripheral blood (6 ml) and oral secretion (OS) samples were obtained from patients with presumptive acute IM at the time of diagnosis (D0), at 1 month (D30), and at 3 months after diagnosis (D90). OS samples were ob-tained by pharyngeal swabbing. Follow-up samples were not available for all patients due to patients being lost to follow-up. IM was identified on clinical grounds and confirmed by indirect immunofluorescent assay (IFA), and those patients with IgM and with or without IgG antibodies against virus capsid antigen (VCA) at D0 were included in the study.
DNA extraction.EDTA-peripheral blood lymphocytes (PBLs) were separated from whole blood (6 ml) with Ficoll-Paque plus (GE Health-care, Sweden). Genomic DNA was extracted from PBLs, OS samples, and lymph node biopsy specimens using a QIAamp DNA minikit (Qiagen, Germany) by following the manufacturer’s instructions.
EBV typing.EBV type was determined as described by Sample et al. (33).
LMP1 carboxy-terminal region analysis.The LMP1 C-ter region was amplified by seminested PCR of each sample. Primers used in the first round were 5=-TGATTAGCTAAGGCATTCCCA-3=(positions 168075 to 168095 of the B95.8 prototype EBV genome, GenBank accession num-berV01555) and 5=-TTCCTTCTCTAACGCACTTTCTC-3=(positions 169163 to 169186). PCR was performed in 25l using 200 ng of genomic DNA, 1 mM MgCl, 0.2 mM deoxynucleoside triphosphates (dNTPs), 0.6
M each primer, and 0.375 U PlatinumTaqDNA polymerase (Invitro-gen). Onel of this product was reamplified with primers 5=-TGATTAG CTAAGGCATTCCCA-3=(positions 168075 to 168095) and 5=-TCTGGA TGTATTACCATGGAC-3= (positions 168756 to 168776), yielding a 701-bp fragment for B95.8 EBV. The reaction was performed under the same conditions as those for the first round. PCR products were separated by electrophoresis in 2% agarose gel stained with ethidium bromide and purified with a QIAEXII gel extraction kit (Qiagen, Germany) according to the manufacturer’s instructions. These purified PCR products were directly sequenced using a BigDye Terminator 3.1 kit (Applied Biosys-tems) in an automated Genetic Analyzer 3130xl (Applied BiosysBiosys-tems). At least two independent sequencing reactions were performed with the in-ner primers to confirm each sequence. A direct sequencing scheme (sense and antisense) was applied instead of establishing lymphoblastoid cell lines to avoid potential selection against variants with poorer transform-ing capabilities.
Sequence analysis.Sequences were aligned and analyzed with Bioedit 7.0.1 software (17). LMP1 sequence variants then were classified according to the classification scheme introduced by Edwards et al. (12, 13). Briefly, seven LMP1 main sequence variants, termed Alaskan (AY337725.1), China 1 (AY337723.1), China 2 (AY337724.1), China 3, Mediterranean⫹(Med⫹)
(AY337722.2), Med⫺ (AY337721.2), and North Carolina (NC)
(AY337726.2), which are distinguished by various signature amino acid changes from the prototypic LMP1 (B95.8), have been isolated from clin-ical specimens (12, 27). Recently, another two related variants, termed SEA1 and SEA2, were described (32). Sequences were compared to the B95.8 reference sequence (GenBank accession numberV01555) and to other isolates from the GenBank database. The China 3 variant was recon-structed with Bioedit software according to the substitution profiles described previously (6). The most appropriate model of evolution for this 701-bp seg-ment was inferred using Modeltest 3.7 (30), and the phylogenetic tree was calculated with the previously defined evolutionary model using PAUP* 4.0.b10 (37) and MEGA 5 (38). Linkage between the different polymor-phisms was inferred with DnaSP 5.10.01 (23).
on May 16, 2020 by guest
http://jcm.asm.org/
Statistical analysis.Statistical analysis was performed using Graph-Pad InStat software, version 3.05 (Graphpad, San Diego, CA). For the univariate analysis, Fisher’s exact test or chi-squared test was used to assess the association between categorical variables. All tests were two
sided, and aPvalue of less than 0.05 was considered statistically sig-nificant.
Nucleotide sequence accession numbers.The sequences determined in the course of this work have been deposited in GenBank under acces-sion numbersJN820220toJN820314.
RESULTS
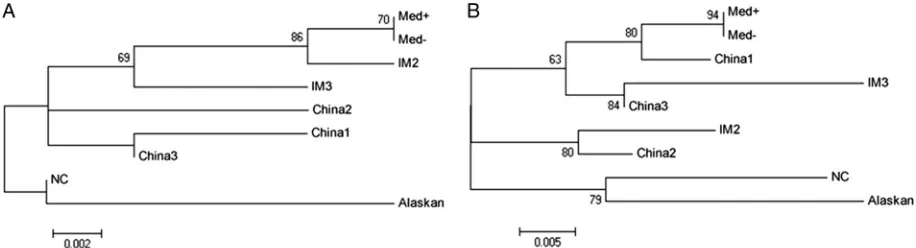
LMP1 variants were characterized according to the pattern of sub-stitutions detected and then confirmed by phylogenetic recon-struction with prototype sequences from GenBank (Fig. 1).
LMP1 variant characterization in IM patients.Among pa-tients with IM, the China 1 variant was the most prevalent in our geographic region. This variant was detected in 7/15 (46.7%) of
the cases, followed by Med⫺in 4 cases (26.7%) and China 2 in 2
cases (13.3%). The remaining 2 patients (IM2 and IM3) each
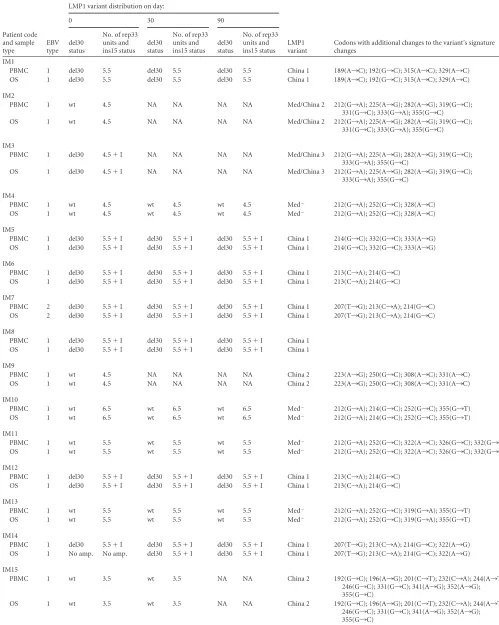
dis-played a different recombinant LMP1 variant. In both cases the 5=
end of the C-ter of LMP1 corresponded to the Med strain, while
the 3=end was characterized as the China 2 strain in patient IM2
and the China 3 strain in patient IM3. The site of recombination was determined for both patients within the rep33 zone, which was previously described as very favorable for recombination events to occur (14). The recombinant variants detected in these two patients were confirmed by phylogenetic reconstruction, but
on this occasion the 5=end and the 3=end were analyzed
indepen-dently with the corresponding fragments from prototype se-quences, and the rep33 region was excluded from the analysis.
Phylogenetic reconstruction proved that the 5=fragments from
LMP1 variants in both patients segregated together with Med
pro-totype variants (Fig. 2A), while the 3=end from patient IM2
clus-tered together with the China 2 variant and the 3=end from patient
IM3 clustered together with the China 3 variant (Fig. 2B). Given
that Med⫹and Med⫺variants differ only in the presence of the
del30 deletion (which is present in the Med⫹variant) in the 3=end
after the rep33 zone and only the 5=region is present in these two
recombinant isolates, it was impossible to discriminate
exten-sively between Med⫹and Med⫺in these two cases.
Curiously, all IM patients for whom follow-up samples were available each presented the same LMP1 variant in PBMC and OS at the time of diagnosis, and the same variant was detected as the only one in each compartment during the follow-up period. As a consequence, no LMP1 variant was statistically associated with a preferential distribution among the different compartments
stud-FIG 1Phylogenetic tree obtained from the alignment of the complete amplifica-tion fragments of the C-ter region of the LMP1 gene from patients with IM and EBV⫹lymphomas. Prototype sequences for each LMP1 variant were obtained from GenBank. Bootstrap values obtained after 1,000 resamplings are indicated. Asterisks denote recombinant variants. The bar indicates the number of substitu-tions.
FIG 2Phylogenetic reconstruction for recombinant variants. (A) Tree obtained from the alignments of the 5=portion of the C-ter region of the LMP1 gene from patients IM2 and IM3. The regions comprising rep33 and the 3=portion of each sequence were omitted from the alignment. Bootstrap values obtained after 1,000 resamplings are indicated. The bar indicates the number of substitutions. (B) Tree obtained from the alignments of the 3=portion of the C-ter region of the LMP1 gene from patients IM2 and IM3. The regions comprising rep33 and the 5=portion of each sequence were omitted from the alignment. Bootstrap values obtained after 1,000 resamplings are indicated. The bar indicates the number of substitutions.
on May 16, 2020 by guest
http://jcm.asm.org/
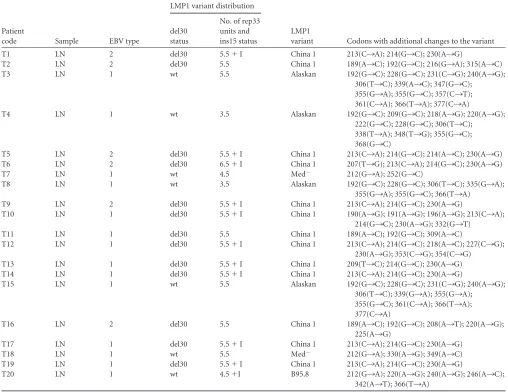
[image:3.585.44.283.64.401.2] [image:3.585.63.521.550.674.2]TABLE 1EBV type and LMP1 variant distribution in IM patientsa
Patient code and sample type
EBV type
LMP1 variant distribution on day:
LMP1 variant
Codons with additional changes to the variant’s signature changes
0 30 90
del30 status
No. of rep33 units and ins15 status
del30 status
No. of rep33 units and ins15 status
del30 status
No. of rep33 units and ins15 status IM1
PBMC 1 del30 5.5 del30 5.5 del30 5.5 China 1 189(A¡C); 192(G¡C); 315(A¡C); 329(A¡C) OS 1 del30 5.5 del30 5.5 del30 5.5 China 1 189(A¡C); 192(G¡C); 315(A¡C); 329(A¡C)
IM2
PBMC 1 wt 4.5 NA NA NA NA Med/China 2 212(G¡A); 225(A¡G); 282(A¡G); 319(G¡C); 331(G¡C); 333(G¡A); 355(G¡C) OS 1 wt 4.5 NA NA NA NA Med/China 2 212(G¡A); 225(A¡G); 282(A¡G); 319(G¡C);
331(G¡C); 333(G¡A); 355(G¡C)
IM3
PBMC 1 del30 4.5⫹I NA NA NA NA Med/China 3 212(G¡A); 225(A¡G); 282(A¡G); 319(G¡C); 333(G¡A); 355(G¡C)
OS 1 del30 4.5⫹I NA NA NA NA Med/China 3 212(G¡A); 225(A¡G); 282(A¡G); 319(G¡C); 333(G¡A); 355(G¡C)
IM4
PBMC 1 wt 4.5 wt 4.5 wt 4.5 Med⫺ 212(G¡A); 252(G¡C); 328(A¡C) OS 1 wt 4.5 wt 4.5 wt 4.5 Med⫺ 212(G¡A); 252(G¡C); 328(A¡C)
IM5
PBMC 1 del30 5.5⫹I del30 5.5⫹I del30 5.5⫹I China 1 214(G¡C); 332(G¡C); 333(A¡G) OS 1 del30 5.5⫹I del30 5.5⫹I del30 5.5⫹I China 1 214(G¡C); 332(G¡C); 333(A¡G)
IM6
PBMC 1 del30 5.5⫹I del30 5.5⫹I del30 5.5⫹I China 1 213(C¡A); 214(G¡C) OS 1 del30 5.5⫹I del30 5.5⫹I del30 5.5⫹I China 1 213(C¡A); 214(G¡C)
IM7
PBMC 2 del30 5.5⫹I del30 5.5⫹I del30 5.5⫹I China 1 207(T¡G); 213(C¡A); 214(G¡C) OS 2 del30 5.5⫹I del30 5.5⫹I del30 5.5⫹I China 1 207(T¡G); 213(C¡A); 214(G¡C)
IM8
PBMC 1 del30 5.5⫹I del30 5.5⫹I del30 5.5⫹I China 1 OS 1 del30 5.5⫹I del30 5.5⫹I del30 5.5⫹I China 1
IM9
PBMC 1 wt 4.5 NA NA NA NA China 2 223(A¡G); 250(G¡C); 308(A¡C); 331(A¡C) OS 1 wt 4.5 NA NA NA NA China 2 223(A¡G); 250(G¡C); 308(A¡C); 331(A¡C)
IM10
PBMC 1 wt 6.5 wt 6.5 wt 6.5 Med⫺ 212(G¡A); 214(G¡C); 252(G¡C); 355(G¡T) OS 1 wt 6.5 wt 6.5 wt 6.5 Med⫺ 212(G¡A); 214(G¡C); 252(G¡C); 355(G¡T)
IM11
PBMC 1 wt 5.5 wt 5.5 wt 5.5 Med⫺ 212(G¡A); 252(G¡C); 322(A¡C); 326(G¡C); 332(G¡C) OS 1 wt 5.5 wt 5.5 wt 5.5 Med⫺ 212(G¡A); 252(G¡C); 322(A¡C); 326(G¡C); 332(G¡C)
IM12
PBMC 1 del30 5.5⫹I del30 5.5⫹I del30 5.5⫹I China 1 213(C¡A); 214(G¡C) OS 1 del30 5.5⫹I del30 5.5⫹I del30 5.5⫹I China 1 213(C¡A); 214(G¡C)
IM13
PBMC 1 wt 5.5 wt 5.5 wt 5.5 Med⫺ 212(G¡A); 252(G¡C); 319(G¡A); 355(G¡T) OS 1 wt 5.5 wt 5.5 wt 5.5 Med⫺ 212(G¡A); 252(G¡C); 319(G¡A); 355(G¡T)
IM14
PBMC 1 del30 5.5⫹I del30 5.5⫹I del30 5.5⫹I China 1 207(T¡G); 213(C¡A); 214(G¡C); 322(A¡G) OS 1 No amp. No amp. del30 5.5⫹I del30 5.5⫹I China 1 207(T¡G); 213(C¡A); 214(G¡C); 322(A¡G)
IM15
PBMC 1 wt 3.5 wt 3.5 NA NA China 2 192(G¡C); 196(A¡G); 201(C¡T); 232(C¡A); 244(A¡T); 246(G¡C); 331(G¡C); 341(A¡G); 352(A¡G); 355(G¡C)
OS 1 wt 3.5 wt 3.5 NA NA China 2 192(G¡C); 196(A¡G); 201(C¡T); 232(C¡A); 244(A¡T); 246(G¡C); 331(G¡C); 341(A¡G); 352(A¡G); 355(G¡C)
aNA, not available; No amp., no amplification; del30, del30 present; wt, del30 not present; No. of rep 33 units, number of rep33 units detected;⫹I, presence of ins15. Accession numbers to the sequences in the table are JN820220 to JN820294.
on May 16, 2020 by guest
http://jcm.asm.org/
ied (P⬎0.05). All variants and their polymorphisms, including single-nucleotide substitutions which are extra to the signature changes of each variant, detected in IM patients during the entire study are described in Table 1.
While the presence of del30 along with other signature point mutations defines a variant, both variable numbers of rep33 and the presence or absence of ins15 were observed among different isolations which clustered together in the phylogenetic analysis. Hence, these polymorphisms are not suitable to define a variant.
Variants from patients IM4, IM10, IM11, and IM13 were Med⫺;
however, the variant in IM4 contained 4.5 rep33 units while pa-tients IM11 and IM13 presented 5.5 rep33 units and patient IM10 contained 6.5 rep33 units. It was also observed that the variant from patient IM1 was the only one characterized as China 1, which did not contain ins15 within the rep33 zone. Given that great controversy exists regarding the association of del30 and the num-ber of rep33 units with EBV-associated diseases, the relationship between these two polymorphisms together with the presence of ins15 were investigated. The number of rep33 units ranged be-tween 3.5 and 6.5 units (3.5 in 1 case, 4.5 in 4/15 cases, 5.5 in 9/15 cases, and 6.5 in 1 case). The 30-bp deletion was detected in a high proportion of IM cases (8/15 [53.3%]), while the remaining 7 cases (46.7%) contained the wild-type (wt) sequence for this re-gion. Additionally, 7/15 cases (46.7%) contained ins15 in the rep33 zone (Table 1). This JAK3 signaling motif insertion was previously described by other authors within the third repeated
unit of rep33 and is also present in strain B95.8 (14, 28, 40), but in the IM samples studied here ins15 was detected either within the third or the fourth repetitive unit; i.e., in patients IM3 and IM7 in the third one and in patients IM5, IM6, IM8, IM12, and IM14 in the fourth (Fig. 3).
LMP1 variant characterization in lymphoma patients. Simi-larly to that observed for IM cases, the most prevalent variant detected in the lymphoma samples was China 1, which was de-tected in 13/20 (65%) patients, followed by the Alaskan variant in
4 patients (20%), Med⫺in 2 cases (10%), and the B95.8 variant in
1 case (5%) (Table 2). On the other hand, no recombinant vari-ants were detected among these samples. Even though the Alaskan variant was only detected in lymphoma biopsy specimens, this variant was not statistically associated with this malignant
condi-tion (P⬎0.05).
The occurrence of del30 in these patients was higher than that of wt sequences, 65 and 35%, respectively, and the number of rep33 units ranged between 3.5 and 6.5 units. As described for IM cases, variations in the number of rep33 units and the presence of ins15 varied within similarly characterized variants; for example, LMP1 variants in patients T7 and T18 were both characterized as
Med⫺but contained 4.5 and 5.5 rep33 units, respectively (Table
2). Similarly, the Alaskan variants detected in patients T4 and T8 contained 3.5 rep33 units while patients T3 and T15 contained 5.5 units (Table 2). Although China 1 variants almost always har-bored 5.5 rep33 units (except for T6, which had 6.5 rep33 units),
FIG 3Alignment of the sequences corresponding to the third and fourth repetitive unit contained in the rep33 region from patients with IM and EBV⫹ lymphomas. Black boxes indicate the location of the 15-bp insertion (ins15) in the third or fourth repeat. Dots indicate homology with the consensus sequence (B95.8), and short dashes indicate the absence of sequence. Codon numbers referring to the B95.8 sequence and repeat numbers are indicated at the top of the alignment.
on May 16, 2020 by guest
http://jcm.asm.org/
[image:5.585.84.501.65.377.2]the variants in patients T2, T11, and T16 did not contain ins15. Within lymphoma samples, T20 was the only case with ins15 pres-ent within the third repeat of rep33, while all the other samples in which ins15 was detected contained it in the fourth repetitive unit (Fig. 3).
Association between polymorphisms in the C-ter of LMP1.
Additionally, the number of rep33 units was divided into two
categories: low (ⱕ4.5 repeats) and high (ⱖ5.5 repeats) numbers
of rep33 units. In this way, the presence of del30, ins15, or high numbers of rep33 in lymphomas turned out not to be significant
compared to results for IM cases (P⬎0.05).
To further characterize any possible association between the different polymorphisms in the C-ter domain of LMP1, sequences obtained from IM and those obtained from lymphoma patients were combined. In this way, a significant association between the
presence of del30 and a high number of rep33 units (ⱖ5.5 repeats)
(P⫽ 0.0009) and between del30 and ins15 (P⫽0.0001) was
determined, but the association between a high number of rep33
units and the presence of ins15 was not significant (P⬎0.05).
Association between polymorphisms in the C-ter of LMP1 with other EBV gene polymorphisms.In a way similar to that previously described, associations with other EBV genetic poly-morphisms were analyzed using combined LMP1 sequences.
The association of a particular variant with viral type was
assessed and found not to be significant (P⬎ 0.05). On the
other hand, the EBV2 genome always contained a del30 variant of LMP1, and the association between these two
polymor-phisms was significant (P⫽ 0.0272). In contrast, EBV1
dis-played both del30 and wt sequences evenly. No statistical
asso-ciation (P⬎0.05) was found between viral type and/or rep33
number and ins15.
[image:6.585.39.547.79.471.2]In previous reports, the naturally occurring variations in the C-ter domain of EBNA1 and in the promoter region of the BZLF1 gene were assessed and characterized in a proportion of pediatric patients from this series (24, 25). Gene variants char-acterized in IM cases during the whole follow-up period and in lymphoma patients are summarized in Table 3. With this in-formation, all of the genomic arrangements observed in both
TABLE 2EBV type and LMP1 variant distribution in EBV⫹lymphoma patientsa
Patient
code Sample EBV type
LMP1 variant distribution
LMP1
variant Codons with additional changes to the variant del30
status
No. of rep33 units and ins15 status
T1 LN 2 del30 5.5⫹I China 1 213(C¡A); 214(G¡C); 230(A¡G)
T2 LN 2 del30 5.5 China 1 189(A¡C); 192(G¡C); 216(G¡A); 315(A¡C)
T3 LN 1 wt 5.5 Alaskan 192(G¡C); 228(G¡C); 231(C¡G); 240(A¡G);
306(T¡C); 339(A¡C); 347(G¡C); 355(G¡A); 355(G¡C); 357(C¡T); 361(C¡A); 366(T¡A); 377(C¡A)
T4 LN 1 wt 3.5 Alaskan 192(G¡C); 209(G¡C); 218(A¡G); 220(A¡G);
222(G¡C); 228(G¡C); 306(T¡C); 338(T¡A); 348(T¡G); 355(G¡C); 368(G¡C)
T5 LN 2 del30 5.5⫹I China 1 213(C¡A); 214(G¡C); 214(A¡C); 230(A¡G)
T6 LN 2 del30 6.5⫹I China 1 207(T¡G); 213(C¡A); 214(G¡C); 230(A¡G)
T7 LN 1 wt 4.5 Med⫺ 212(G¡A); 252(G¡C)
T8 LN 1 wt 3.5 Alaskan 192(G¡C); 228(G¡C); 306(T¡C); 335(G¡A);
355(G¡A); 355(G¡C); 366(T¡A)
T9 LN 2 del30 5.5⫹I China 1 213(C¡A); 214(G¡C); 230(A¡G)
T10 LN 1 del30 5.5⫹I China 1 190(A¡G); 191(A¡G); 196(A¡G); 213(C¡A);
214(G¡C); 230(A¡G); 332(G¡T)
T11 LN 1 del30 5.5 China 1 189(A¡C); 192(G¡C); 309(A¡C)
T12 LN 1 del30 5.5⫹I China 1 213(C¡A); 214(G¡C); 218(A¡C); 227(C¡G);
230(A¡G); 353(C¡G); 354(C¡G)
T13 LN 1 del30 5.5⫹I China 1 209(T¡C); 214(G¡C); 230(A¡G)
T14 LN 1 del30 5.5⫹I China 1 213(C¡A); 214(G¡C); 230(A¡G)
T15 LN 1 wt 5.5 Alaskan 192(G¡C); 228(G¡C); 231(C¡G); 240(A¡G);
306(T¡C); 339(G¡A); 355(G¡A); 355(G¡C); 361(C¡A); 366(T¡A); 377(C¡A)
T16 LN 2 del30 5.5 China 1 189(A¡C); 192(G¡C); 208(A¡T); 220(A¡G);
225(A¡G)
T17 LN 1 del30 5.5⫹I China 1 213(C¡A); 214(G¡C); 230(A¡G)
T18 LN 1 wt 5.5 Med⫺ 212(G¡A); 330(A¡G); 349(A¡C)
T19 LN 1 del30 5.5⫹I China 1 213(C¡A); 214(G¡C); 230(A¡G)
T20 LN 1 wt 4.5⫹I B95.8 212(G¡A); 220(A¡G); 240(A¡G); 246(A¡C);
342(A¡T); 366(T¡A)
aLN, lymph node biopsy specimen; del30, del30 present; wt, del30 not present; No. of rep 33 units, number of rep33 units detected;⫹I, ins15 present. Accession numbers of the sequences in the table are JN820295 to JN820314.
on May 16, 2020 by guest
http://jcm.asm.org/
groups of patients were reconstructed and assessed for poly-morphism linkage (Table 4).
No complete linkage across the different genomic regions (EBNA1, EBNA3C, BZLF1, and LMP1) was found, but still the most frequent arrangement detected in this series (10 patients) was number 5 (Table 4). Despite the fact that arrangement 10 was detected in only 3 cases, it is worth mentioning because it com-bined two polymorphic variations which are predominant in our
geographic region. Briefly, the Zp-V3⫹49 variant, first described
by our group in this geographic region within the promoter region of BZLF1, was always detected in association with a V-ala variant of EBNA1 restricted to South America, along with the absence of del30 and ins15, and always with 3.5 rep33 units in the context of EBV1 (Table 4). Although no statistically significant complete genomic linkage was found, partial associations were disclosed. Zp-V3 variants in the promoter of BZLF1 significantly occurred in
association with del30 (P⫽0.046), a high copy number of rep33
(P⫽0.0363), and within the EBV2 genome (P⫽0.0001).
How-ever, EBNA1 variants did not show a consistent association with any other polymorphism within LMP1 or the promoter region of BZLF1 gene or with viral type.
DISCUSSION
Although LMP1 sequence variants have been deeply studied in a wide variety of malignancies in different geographic regions (6), only a few studies addressed this issue in pediatric patients (2, 5, 14), and to the best of our knowledge, only one of these included pediatric cases of IM (2). With the aim of disclosing current con-flicts arising from the different sample types used in other studies and/or particular health conditions, this work addressed sequence variation within the C-ter region of LMP1 in different compart-ments from children with IM during primary infection and its convalescence, and also from pediatric EBV-related lymphomas from Argentina.
As previously described by Edwards et al. (12), the phyloge-netic reconstruction of LMP1 variants proved that China 1, China 3, and Med strains were closely related and that China 2, NC, and Alaskan strains clustered together in a second related group. China 1 was the most common variant among pediatric patients, both with IM and with EBV-related lymphomas, included in this series. This led to the conclusion that China 1 is the predominant variant in our geographic region and is not related to any partic-ular pathological condition. Although to date no data using this classification system are available from South American patients, this observation is consistent with the literature, in which China 1 was previously reported to be the predominant LMP1 variant in other regions, such as North America and Asia (6). The same conclusion for China 1 regarding pathology association could be
made for the Med⫺variant, which was also evenly distributed
between disease statuses but with a lesser incidence. As far as could be concluded from the analysis of the sequence variants in this pediatric series, no variant was found to be statistically associated with lymphomas, not even the Alaskan variant, which was de-tected with low frequency but exclusively in malignant samples. Given that this variant has been described only in NPC samples from North America (6, 12) and that it has been reported to have
an increased capability of activating the NF-B signaling pathway,
it could be speculated that the association of this variant and lym-phomas would turn significant if the number of patients studied could be increased. However, considering the epidemiology of IM in a developing region, such as Argentina, and that only nearly 40% of pediatric B-cell lymphomas are EBV associated (unpub-lished data), the recruitment of 15 pediatric symptomatic IM pa-tients and 20 pediatric lymphoma cases makes this an interesting series. Despite these considerations, this is the first report to de-scribe the Alaskan variant in EBV-related lymphomas from South America.
[image:7.585.40.286.87.491.2]Several studies have revealed that both healthy and young
TABLE 3EBNA1 and BZLF1 polymorphism distribution in IM and lymphoma patientsa
Patient code
EBNA1 BZLF1
PBMC OS PBMC OS
IM1* P-thr⬙/P-ala P-thr⬙ Zp-P Zp-P
IM2 V-leu Ag V-leu Ag Zp-P Zp-P
IM3 P-thr= P-thr= Zp-P Zp-P
IM4 P-thr= P-thr= Zp-P Zp-P
IM5 V-leu Ag V-leu Ag Zp-P Zp-P
IM6* V-leu Ag/P-thr= V-leu Ag/P-thr= Zp-P Zp-P
IM7 V-leu V-leu Zp-V3 Zp-V3
IM8 V-leu Ag V-leu Ag Zp-P Zp-P
IM9* V-ala-i/P-ala V-ala-i Zp-P/Zp-V3 Zp-P
IM10 P-thr⬙ P-thr⬙ Zp-P Zp-P
IM11 P-thr= P-thr= Zp-P Zp-P
IM12 V-leu V-leu Zp-P Zp-P
IM13 P-thr⬙ P-thr⬙ Zp-P Zp-P
IM14 V-leu V-leu Zp-P Zp-P
IM15 V-ala-i V-ala-i Zp-V3⫹49 Zp-V3⫹49 EBNA1, lymph node
biopsy specimen
BZLF1, lymph node biopsy specimen
T1 V-leu Ag Zp-V3
T2 P-thr= Zp-V3
T3 V-ala-ii Zp-V3
T4 V-ala-iii Zp-V3⫹49
T5 V-leu-vii Zp-V3
T6 V-leu Zp-V3
T7 P-thr= Zp-P
T8 V-ala-iv Zp-V3⫹49
T9 V-leu Ag-i Zp-V3
T10 V-leu Ag-ii Zp-P
T11 P-thr= Zp-P
T12 V-leu Ag Zp-P
T13 V-leu Ag Zp-P
T14 V-leu Ag Zp-P
T15 V-ala-ii Zp-V3
T16 V-ala-v Zp-V3
T17 V-leu-viii Zp-P
T18 P-ala= Zp-V3
T19 P-thr= Zp-P
T20 V-leu Zp-P
aAn asterisk denotes coinfection at some point during follow-up. Coinfecting variants
are in boldface. The EBNA1 column describes variants in EBNA1 genes according to the variant signature amino acids alanine, threonine, and leucine in codon 487 as described previously (24). In EBNA1 variant nomenclature, P stands for prototype (i.e., being closely related to the B95.8 sequence). V stands for variant, as these sequences differ more from the prototype sequence. The terms ala, thr, and leu refer to the amino acid in codon 487, and the terms Ag, Ag-I, Ag-ii, thr=, thr⬙, ala=, and viii represent subvariants according to extra substitutions apart from the one in codon 487. The BZLF1 column represents variants in the promoter region of the BZLF1 gene according to substitutions in positions⫺141,⫺106,⫺100, and⫺49 with respect to the transcription start site as described previously (25). For BZLF1 variants, Zp is the abbreviation for the BZLF1 promoter region. V3 stands for variants with three additional substitutions, and⫹49 describes a V3 variant with an additional substitution at position⫺49.
on May 16, 2020 by guest
http://jcm.asm.org/
adults with IM were infected with multiple LMP1 variants, and their distribution fluctuated dynamically between OS and PBMC over time (8, 27, 35, 36); however, no coinfections with LMP1 variants were observed in the pediatric patients with IM studied here. On the other hand, coinfection with two EBV variants was detected in a low proportion (3/15) of IM patients in different compartments at some time point during the study when other EBV genes were assessed (Table 3) (24, 25). In certain samples, even though only one LMP1 variant was de-tected, two variants of EBNA1 or the promoter region of BZLF1 were identified. One explanation for this fact is viral coinfec-tion with two viral genomes containing different BZLF1 and EBNA1 variants but sharing a common LMP1 variant. An al-ternative reasoning was proposed by Gutierrez et al. when they described the presence of multiple EBNA1 variants in the ab-sence of more than one LMP1 variant in NK/T-cell lymphomas in adults. The authors suggested that EBNA1 variants could be
generated in vivo and not as a consequence of multivariant
infection (16). However, the low proportion of coinfection could be easily explained by the fact that children still were not exposed to reinfections during adolescence or early adulthood as adults have been. A similar observation was reported by Jin et al. in a Chinese pediatric population with IM, where only 15% of the cases showed the presence of multiple viral variants (21). On the other hand, no coinfection was detected for any gene in the lymphoma samples, supporting the idea of clonal expansion of the originally infected neoplastic cells as proposed for B-cell lymphomas.
Regarding the polymorphisms within the C-ter of LMP1, it has been suggested that variants containing del30 are more prevalent among tumors than in control samples from different geographic regions, excluding China, where del30 was evenly detected among both groups, but to date this augmented occurrence in tumors is controversial (reviewed in reference 6). Particularly in Argentina, the deleted LMP1 variant was previously preferentially detected in tumor samples rather than in samples from healthy individuals,
either adults (8, 9) or children (5). The predominance of the del30 variant within tumors also was described in the rest of South America and Mexico, and it is in this geographic region that this distinct distribution scheme is more clearly observed (6, 7, 11, 14, 18). In the present series, del30 variants were detected in tumor samples and in IM with a similar high incidence. Moreover, high numbers of rep33 also were predominant among IM and lym-phoma samples over variants with low numbers of rep33 copies, and this resulted in a statistical association between del30 and a high copy number of rep33. Furthermore, the association between ins15 and del30 was found to be significant. These results con-firmed previous observations in which an association between del30 and high rep33 numbers and between del30 and ins15 had been documented in HL samples from Argentina and Brazil (14). In contrast, in our region a very low incidence of del30 variants and a low number of rep33 copies was previously described among healthy individuals (4, 8). To the best of our knowledge, the 15-bp insertion encoding a JAK3 signaling motif has been described only within the third repeat of rep33 in B95.8-related sequences (14, 28, 40). This is the first report to describe ins15 within the fourth 33-bp tandem repeat, hence proving that the structural variation of the C-ter of LMP1 is greater than previously thought.
[image:8.585.38.545.80.284.2]The similarities found among LMP1 variants in benign and malignant EBV-related entities, which differ significantly from those described in healthy carriers, prompted us to suggest the existence of certain variants which are closely related to the development of EBV-positive pathologies in children and are not just linked to tumors. On the other hand, the fact that the onset of lymphoma in children younger than 10 years of age in our country (4, 5), together with EBV epidemiology showing primary infection during early childhood, are quite remark-able. Moreover, it has been shown that individuals with a his-tory of IM have a 3-fold elevated risk for EBV-positive HL and that this risk remains augmented for a lifetime (19, 20). Al-though the precise mechanism by which EBV contributes to the
TABLE 4EBV genomic arrangementsa
Arrangement
no. Frequency Patient code EBV gene polymorphism (EBNA3C, BZLF1, EBNA1, LMP1)
1 2 IM1*, T11 EBV1, Zp-P, P-thr, 5.5 rep33, ins15⫺, del30
2 1 IM2 EBV1, Zp-P, V-leu, 4.5 rep33, ins15⫺, wt
3 1 IM3 EBV1, Zp-P, P-thr, 4.5 rep33, ins15⫹, del30
4 2 IM4, T7 EBV1, Zp-P, V-leu, 5.5 rep33, ins15⫹, del30
5 10 IM5, IM6*, IM8, IM12, IM14, T10, T12,
T13, T14, T17
EBV1, Zp-P, V-leu, 5.5 rep33, ins15⫹, del30
6 4 IM7, T1, T5, T9 EBV2, Zp-V3, V-leu, 5.5 rep33, ins15⫹, del30
7 1 IM9* EBV1, Zp-P, V-ala, 4.5 rep33, ins15⫺, wt
8 1 IM10 EBV1, Zp-P, P-thr, 6.5 rep33, ins15⫺, wt
9 2 IM11, IM13 EBV1, Zp-P, P-thr, 5.5 rep33, ins15⫺, wt
10 3 IM15, T4, T8 EBV1, Zp-V3⫹49, V-ala, 3.5 rep33, ins15⫺, wt
11 1 T2 EBV2, Zp-V3, P-thr, 5.5 rep33, ins15⫺, del30
12 2 T3, T15 EBV1, Zp-V3, V-ala, 5.5 rep33, ins15⫺, wt
13 1 T6 EBV2, Zp-V3, V-leu, 6.5 rep33, ins15⫹, del30
14 1 T16 EBV2, Zp-V3, V-ala, 5.5 rep33, ins15⫺, del30
15 1 T18 EBV1, Zp-V3, P-ala, 5.5 rep33, ins15⫺, wt
16 1 T19 EBV1, Zp-P, P-thr, 5.5 rep33, ins15⫹, del30
17 1 T20 EBV1, Zp-P, V-leu, 4.5 rep33, ins15⫹, wt
aAn asterisk denotes patients with coinfection at some time point throughout the study, but only the most prevalent variant was included in this analysis. All gene polymorphisms
studied (EBNA3C, BZLF1, EBNA1, and LMP1) for each arrangement are listed under the EBV gene polymorphism column header.
on May 16, 2020 by guest
http://jcm.asm.org/
development of this malignancy is not fully understood, it is supposed that EBV rescues from apoptosis those B lympho-cytes whose differentiation is faulty, probably by means of pro-liferative and antiapoptotic signals (22, 29).
The resemblance described for LMP1 variants among EBV-related lymphomas and IM were made more evident when genomic arrangements, including EBNA3C, EBNA1, and BZLF1 along with LMP1 variants, were analyzed. The most frequent arrangement (Table 4, no. 5) was evenly distributed among the two groups.
Besides the strict associations between EBV2 and del30, EBV2 and a high number of rep33 units, and EBV2 and the Zp-V3 vari-ant, no complete linkage was found among polymorphisms from the different analyzed genes. Although not statistically significant, probably due to the low number of patients included in addition to its low frequency, arrangement no. 10 turned out to be very interesting, given that it combined a series of genetic variations which were preferentially or exclusively detected in our country (24, 25). In accordance with the present results, incomplete link-age has been previously reported for LMP1, EBNA1, and EBNA2 polymorphisms (15). These results reinforce the concept intro-duced by Chang et al. that EBV natural variation is far greater than previously described (6).
These observations highlight the importance of the follow-up of pediatric IM patients within a few years to moni-tor the possible involvement of EBV in lymphomagenesis and also to establish if del30, together with a high number of rep33 copies, in symptomatic acute infection truly represents an in-creased risk for EBV malignant conditions following IM in our geographical region.
ACKNOWLEDGMENTS
We thank G. Moscatelli and S. Moroni (Medical Staff at the Parasitology and Chagas Disease Laboratory, Ricardo Gutiérrez Children Hospital) for providing assistance in reviewing clinical records.
This study was supported in part by a grant from the National Agency for Science and Technology Promotion (PICT 2008 no. 0052 and PICT 2007 no. 1071). M.A.L. is supported by a fellowship from the National Research Coun-cil (CONICET), and M.G. is supported by a fellowship from the National Agency for Science and Technology Promotion (ANPCyT).
P.A.C. and M.V.P. are members of CONICET, Research Career gram. J.A. and E.D.M. are members of the CABA Research Career Pro-gram.
None of the authors has any competing interest.
REFERENCES
1.Bentz GL, Whitehurst C, Pagano JS.2011. Epstein-Barr virus latent membrane protein 1 (LMP1) C-terminal-activating region 3 contributes to LMP1-mediated cellular migration via its interaction with Ubc9. J. Virol.85:10144 –10153.
2.Berger C, et al.1997. The 30-bp deletion variant of Epstein-Barr virus-encoded latent membrane protein-1 prevails in acute infectious mononu-cleosis. J. Infect. Dis.176:1370 –1373.
3.Chabay P, Burna V, Moar A, Grinstein S. 1999. Prevalencia de la infección por el virus de Epstein-Barr en pacientes pediátricos. Rev. Hos-pital Ñiños Buenos Aires41:88 –91.
4.Chabay P, De Matteo E, Merediz A, Preciado MV. 2004. High frequency of Epstein Barr virus latent membrane protein-1 30 bp de-letion in a series of pediatric malignancies in Argentina. Arch. Virol. 149:1515–1526.
5.Chabay P, et al.2011. Epstein Barr virus in relation to apoptosis markers and patients’ outcome in pediatric B-cell non-Hodgkin lymphoma. Can-cer Lett.307:221–226.
6.Chang CM, Yu KJ, Mbulaiteye SM, Hildesheim A, Bhatia K.2009. The
extent of genetic diversity of Epstein-Barr virus and its geographic and disease patterns: a need for reappraisal. Virus Res.143:209 –221. 7.Chen WG, et al.1996. Genotyping of Epstein-Barr virus in Brazilian
Burkitt’s lymphoma and reactive lymphoid tissue. Type A with a high prevalence of deletions within the latent membrane protein gene. Am. J. Pathol.148:17–23.
8.Correa RM, et al.2004. Epstein-barr virus (EBV) in healthy carriers: distribution of genotypes and 30 bp deletion in latent membrane protein-1 (LMP-1) oncogene. J. Med. Virol.73:583–588.
9.Correa RM, et al.2007. Epstein Barr virus genotypes and LMP-1 variants in HIV-infected patients. J. Med. Virol.79:401– 407.
10. Delecluse HJ, Feederle R, O’Sullivan B, Taniere P.2007. Epstein Barr virus-associated tumours: an update for the attention of the working pa-thologist. J. Clin. Pathol.60:1358 –1364.
11. Dirnhofer S, et al.1999. High prevalence of a 30-base pair deletion in the Epstein-Barr virus (EBV) latent membrane protein 1 gene and of strain type B EBV in Mexican classical Hodgkin’s disease and reactive lymphoid tissue. Hum. Pathol.30:781–787.
12. Edwards RH, Seillier-Moiseiwitsch F, Raab-Traub N.1999. Signature amino acid changes in latent membrane protein 1 distinguish Epstein-Barr virus strains. Virology261:79 –95.
13. Edwards RH, Sitki-Green D, Moore DT, Raab-Traub N.2004. Potential selection of LMP1 variants in nasopharyngeal carcinoma. J. Virol.78:868 – 881.
14. Guiretti DM, et al.2007. Structural variability of the carboxy-terminus of Epstein-Barr virus encoded latent membrane protein 1 gene in Hodgkin’s lymphomas. J. Med. Virol.79:1730 –1732.
15. Gutierrez MI, et al.2000. Association of EBV strains, defined by multiple loci analyses, in non-Hodgkin lymphomas and reactive tissues from HIV positive and HIV negative patients. Leuk. Lymphoma37:425– 429. 16. Gutierrez MI, et al.1997. Sequence variations in EBNA-1 may dictate
restriction of tissue distribution of Epstein-Barr virus in normal and tu-mour cells. J. Gen. Virol.78:1663–1670.
17. Hall TA.1999. BioEdit: a user friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser.41:95–98.
18. Hayashi K, et al.1997. Deletion of Epstein-Barr virus latent membrane protein 1 gene in United States and Brazilian Hodgkin’s disease and reac-tive lymphoid tissue: high frequency of a 30-bp deletion. Hum. Pathol. 28:1408 –1414.
19. Hjalgrim H, Engels EA.2008. Infectious aetiology of Hodgkin and non-Hodgkin lymphomas: a review of the epidemiological evidence. J. Intern. Med.264:537–548.
20. Hjalgrim H, et al.2007. Infectious mononucleosis, childhood social en-vironment, and risk of Hodgkin lymphoma. Cancer Res.67:2382–2388. 21. Jin Y, Xie Z, Lu G, Yang S, Shen K.2010. Characterization of variants in
the promoter of BZLF1 gene of EBV in nonmalignant EBV-associated diseases in Chinese children. Virol. J.7:92.
22. Kapatai G, Murray P.2007. Contribution of the Epstein Barr virus to the molecular pathogenesis of Hodgkin lymphoma. J. Clin. Pathol.60:1342– 1349.
23. Librado P, Rozas J.2009. DnaSP v5: a software for comprehensive anal-ysis of DNA polymorphism data. Bioinformatics25:1451–1452. 24. Lorenzetti MA, et al.2010. EBNA1 sequences in Argentinean pediatric
acute and latent Epstein-Barr virus infection reflect circulation of novel South American variants. J. Med. Virol.82:1730 –1738.
25. Lorenzetti MA, et al.2009. Epstein-Barr virus BZLF1 gene promoter variants in pediatric patients with acute infectious mononucleosis: its comparison with pediatric lymphomas. J. Med. Virol.81:1912–1917. 26. Luzuriaga K, Sullivan JL.2010. Infectious mononucleosis. N. Engl. J.
Med.362:1993–2000.
27. Mainou BA, Raab-Traub N.2006. LMP1 strain variants: biological and molecular properties. J. Virol.80:6458 – 6468.
28. Miller WE, Edwards RH, Walling DM, Raab-Traub N.1994. Sequence variation in the Epstein-Barr virus latent membrane protein 1. J. Gen. Virol.75:2729 –2740.
29. Pinto A, et al.1998. Hodgkin’s disease: a disorder of dysregulated cellular cross-talk. Biotherapy10:309 –320.
30. Posada D, Crandall KA.1998. MODELTEST: testing the model of DNA substitution. Bioinformatics14:817– 818.
31. Rickinson AB, Kieff ED.2007. Epstein Barr virus, p. 2655–2700.InKnipe DM, et al. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
on May 16, 2020 by guest
http://jcm.asm.org/
32. Saechan V, Mori A, Mitarnun W, Settheetham-Ishida W, Ishida T. 2006. Analysis of LMP1 variants of EBV in southern Thailand: evidence for strain-associated T-cell tropism and pathogenicity. J. Clin. Virol.36: 119 –125.
33. Sample J, et al.1990. Epstein-Barr virus types 1 and 2 differ in their EBNA-3A, EBNA-3B, and EBNA-3C genes. J. Virol.64:4084 – 4092. 34. Sandvej K, et al.1997. Sequence analysis of the Epstein-Barr virus (EBV)
latent membrane protein-1 gene and promoter region: identification of four variants among wild-type EBV isolates. Blood90:323–330. 35. Sitki-Green D, Covington M, Raab-Traub N.2003.
Compartmentaliza-tion and transmission of multiple Epstein-Barr virus strains in asymptom-atic carriers. J. Virol.77:1840 –1847.
36. Sitki-Green DL, Edwards RH, Covington MM, Raab-Traub N.2004. Biology of Epstein-Barr virus during infectious mononucleosis. J. Infect. Dis.189:483– 492.
37. Swofford DL.2003. PAUP*. Phylogenetic analysis using parsimony (*and other methods), version 4. Sinauer Associates, Sunderland, MA. 38. Tamura K, et al.2011. MEGA5: molecular evolutionary genetics analysis
using maximum likelihood, evolutionary distance, and maximum parsi-mony methods. Mol. Biol. Evol.28:2731–2739.
39. Vetsika EK, Callan M.2004. Infectious mononucleosis and Epstein-Barr virus. Expert. Rev. Mol. Med.6:1–16.
40. Walling DM, et al.1999. The molecular epidemiology and evolution of Epstein-Barr virus: sequence variation and genetic recombination in the latent membrane protein-1 gene. J. Infect. Dis.179:763–774. 41. Wang D, Liebowitz D, Kieff E.1985. An EBV membrane protein
ex-pressed in immortalized lymphocytes transforms established rodent cells. Cell43:831– 840.
42. Young LS, Rickinson AB.2004. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer4:757–768.
on May 16, 2020 by guest
http://jcm.asm.org/
Distinctive Epstein-Barr Virus Variants Associated with Benign and
Malignant Pediatric Pathologies: LMP1 Sequence Characterization and
Linkage with Other Viral Gene Polymorphisms
Mario Alejandro Lorenzetti, Magdalena Gantuz, Jaime Altcheh, Elena De Matteo, Paola Andrea Chabay, and María Victoria Preciado Molecular Biology Laboratory, Pathology Division, Ricardo Gutiérrez Children Hospital, Gallo 1330 Buenos Aires, Argentina; Parasitology and Chagas Disease Laboratory, Ricardo Gutiérrez Children Hospital, Gallo 1330 Buenos Aires, Argentina; and Pathology Division, Ricardo Gutiérrez Children Hospital, Gallo 1330 Buenos Aires, Argentina
Volume 50, no. 3, p. 609 – 618, 2012. In Table 3 and throughout the text, an error was made in referring to a variant in the promoter
region of the BZLF1 gene described by our group and termed Zp-V3⫹49. After subsequent analysis of the sequence alignments, we
realized that the substitution G¡T described as being at position⫺49 was actually at position⫺51 with respect to the transcription start
site. This mistake has no consequence with regard to the data presented in this paper, but it does have a consequence with regard to the name of the variant. In order to correct the nomenclature of this BZLF1 promoter region variant, from this point forward this variant
with the G¡T substitution at position⫺51 should be referred to as Zp-V3⫹51.
Copyright © 2012, American Society for Microbiology. All Rights Reserved.