Acta Cryst.(2001). E57, o707±o708 DOI: 10.1107/S1600536801010844 Yoshimitsu Moritaniet al. C5H12N+C7H4NO4ÿ
o707
organic papers
Acta Crystallographica Section E
Structure Reports
Online ISSN 1600-5368
Piperidinium
p
-nitrobenzoate
Yoshimitsu Moritani,a² Akihiko
Takedaband Setsuo Kashinob*
aGraduate School of Natural Science and
Technology, Okayama University, Tsushima, Okayama 700-8530, Japan, andbDepartment of
Chemistry, Faculty of Science, Okayama University, Tsushima, Okayama 700-8530, Japan
² On leave from Mitsui Chemical Analysis and Consulting Service Inc., Waki 6-1-2, Waki-cho, Kuga-gun, Yamaguchi 740-0061, Japan.
Correspondence e-mail: [email protected]
Key indicators Single-crystal X-ray study
T= 298 K
Mean(C±C) = 0.005 AÊ
Rfactor = 0.057
wRfactor = 0.205
Data-to-parameter ratio = 14.6
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2001 International Union of Crystallography Printed in Great Britain ± all rights reserved
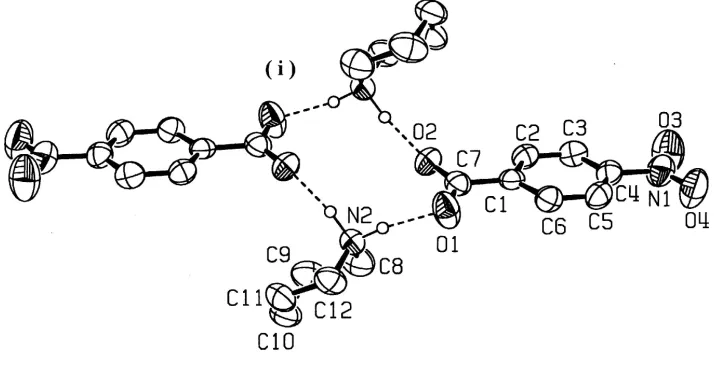
A new type of hydrogen-bond pattern for piperidinium p-substituted benzoates is reported; this is found in piper-idinium p-nitrobenzoate, C5H12N+C7H4NO4ÿ, (I). In the
crystal of (I), the cations and anions are linked by NÐ H O hydrogen bonds around a center of symmetry to form a cyclic dimer of the formula unit.
Comment
We report here a new type of hydrogen-bond pattern in the cyclic secondary amine±p-substituted benzoic acid (1/1) system. In piperidinium p-nitrobenzoate, (I), a centrosym-metric dimer of the formula unit is formed through NÐH O hydrogen bonds (Fig. 1 and Table 1).
In most crystalline salts formed between cyclic secondary amines andp-substituted benzoic acids, the cations and anions are arranged around a twofold screw axis to form NÐH O hydrogen bonds (Kashino et al., 1972, 1978, 1981; Kashino, 1973). The same type of hydrogen bonding is also found in both pyrrolidinium p-nitrobenzoate and hexamethyl-eneiminium p-nitrobenzoate (Takeda, 1992). In a few cases, the cations and anions are arranged along a glide plane to form NÐH O hydrogen bonds (Kashinoet al., 1973, 1981).
The possibility of the formation of a cyclic dimer through the NÐH O hydrogen bonds was deduced based on the result of a molecular weight measurement in benzene solution, combined with a geometrical consideration of the hydrogen-bonded system (Kashino, 1973). However, the formation of a cyclic dimer has been found only in one of the dimorphs of hexamethyleneiminiump-bromobenzoate, in which the dimer is formed around a twofold axis (Kashino et al., 1981). The present study established a centrosymmetric type as the fourth pattern of hydrogen bonding possible in salts of cyclic secondary amines andp-substituted benzoic acids.
Experimental
Stoichiometric amounts of piperidine andp-nitrobenzoic acid were dissolved in benzene at 340 K. After cooling the solution, crystals of (I) were grown by slow evaporation at room temperature.
Crystal data
C5H12N+C7H4NO4ÿ Mr= 252.27
Orthorhombic,Pccn a= 8.949 (2) AÊ
b= 22.451 (5) AÊ
c= 12.712 (4) AÊ
V= 2554.0 (11) AÊ3 Z= 8
Dx= 1.312 Mg mÿ3
MoKradiation Cell parameters from 25
re¯ections
= 10.6±11.4
= 0.10 mmÿ1 T= 298 K Prismatic, colorless 0.500.300.20 mm
Data collection
Rigaku AFC-5Rdiffractometer
!±2scans
Absorption correction: scan (Northet al., 1968)
Tmin= 0.952,Tmax= 0.980 2875 measured re¯ections 2509 independent re¯ections 1142 re¯ections withI> 2(I)
Rint= 0.022
max= 26.0 h= 0!11
k= 0!27
l= 0!15
3 standard re¯ections every 97 re¯ections intensity decay: 0.6%
Re®nement
Re®nement onF2 R[F2> 2(F2)] = 0.057 wR(F2) = 0.205 S= 0.91 2509 re¯ections 172 parameters
H atoms treated by a mixture of independent and constrained re®nement
w= 1/[2(F
o2) + (0.1P)2
+ 1.1042P]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.002 max= 0.13 e AÊÿ3 min=ÿ0.18 e AÊÿ3
Extinction correction:SHELXL97 Extinction coef®cient: 0.0059 (14)
Table 1
Selected geometric parameters (AÊ,).
O1ÐC7 1.253 (4) O2ÐC7 1.250 (4) O3ÐN1 1.219 (4) O4ÐN1 1.216 (4) N1ÐC4 1.476 (5) C1ÐC6 1.386 (5) C1ÐC2 1.387 (5) C1ÐC7 1.513 (5) C2ÐC3 1.378 (5)
C3ÐC4 1.377 (5) C4ÐC5 1.379 (5) C5ÐC6 1.378 (5) N2ÐC8 1.478 (5) N2ÐC12 1.489 (5) C8ÐC9 1.510 (6) C10ÐC11 1.502 (6) C11ÐC12 1.500 (6) O4ÐN1ÐO3 123.6 (4)
O4ÐN1ÐC4 118.5 (4) O3ÐN1ÐC4 117.9 (4) C6ÐC1ÐC2 119.4 (3) C6ÐC1ÐC7 120.5 (3) C2ÐC1ÐC7 120.1 (3) C3ÐC2ÐC1 120.3 (3) C4ÐC3ÐC2 119.1 (3) C3ÐC4ÐC5 121.8 (4) C3ÐC4ÐN1 119.2 (3) C5ÐC4ÐN1 119.0 (4)
C6ÐC5ÐC4 118.5 (3) C5ÐC6ÐC1 120.9 (3) O2ÐC7ÐO1 125.0 (4) O2ÐC7ÐC1 117.7 (3) O1ÐC7ÐC1 117.2 (3) C8ÐN2ÐC12 111.1 (3) N2ÐC8ÐC9 109.6 (4) C10ÐC9ÐC8 110.0 (4) C11ÐC10ÐC9 111.0 (3) C12ÐC11ÐC10 111.4 (4) N2ÐC12ÐC11 110.3 (3)
Table 2
Hydrogen-bonding geometry (AÊ,).
DÐH A DÐH H A D A DÐH A
N2ÐH5 O1 0.94 (4) 1.78 (4) 2.697 (5) 165 (4) N2ÐH6 O2i 1.02 (5) 1.71 (5) 2.731 (4) 171 (4)
Symmetry code: (i) 1ÿx;1ÿy;1ÿz.
All H atoms were found from a difference Fourier map. At the ®nal stage of the least-squares re®nement, all H atoms except those involved in hydrogen bonds were ®xed at the ideal positions, and their isotropic displacement parameters were ®xed to 1.2Ueqof the parent atoms.
Data collection: MSC/AFC Diffractometer Control Software
(Molecular Structure Corporation, 1994); cell re®nement:MSC/AFC Diffractometer Control Software; data reduction: TEXSAN for Windows(Molecular Structure Corporation, 1997±1999); program(s) used to solve structure: SIR92 (Altomareet al., 1993); program(s) used to re®ne structure: SHELXL97 (Sheldrick, 1997); molecular graphics: ORTEP-3 (Farrugia, 1997); software used to prepare material for publication:TEXSANfor Windows.
References
Altomare, A., Cascarano, G., Giacovazzo, C. & Guagliardi, A. (1993).J. Appl. Cryst.26, 343±350.
Farrugia, L. J. (1997).J. Appl. Cryst.30, 565.
Kashino, S., Sumida, Y. & Haisa, M. (1972).Acta Cryst.B28, 1374±1383. Kashino, S. (1973).Acta Cryst.B29, 1836±1842.
Kashino, S., Kataoka, S. & Haisa, M. (1978).Bull. Chem. Soc. Jpn,51, 1717± 1722.
Kashino, S., Sasahara, N., Kataoka, S. & Haisa, M. (1981).Bull. Chem. Soc. Jpn,54, 962±966.
Kashino, S., Sasaki, M. & Haisa, M. (1973).Bull. Chem. Soc. Jpn,46, 1375± 1379.
Molecular Structure Corporation (1994).MSC/AFC Diffractometer Control Software. MSC, 3200 Research Forest Drive, The Woodlands, TX 77381, USA.
Molecular Structure Corporation (1997±1999). TEXSAN for Windows. Version 1.06. MSC, 9009 New Trails Drive, The Woodlands, TX, USA. North, A. C. T., Phillips, D. C. & Mathews, F. S. (1968).Acta Cryst, A24, 351±
359.
Sheldrick, G. M. (1997).SHELXL97. University of GoÈttingen, Germany. Takeda, A. (1992). Master's thesis, Faculty of Science, Okayama University,
Japan.
Figure 1
supporting information
sup-1
Acta Cryst. (2001). E57, o707–o708supporting information
Acta Cryst. (2001). E57, o707–o708 [doi:10.1107/S1600536801010844]
Piperidinium
p
-nitrobenzoate
Yoshimitsu Moritani, Akihiko Takeda and Setsuo Kashino
S1. Comment
We report here a new type of hydrogen-bond pattern in cyclic secondary amine–p-substituted benzoic acid (1/1) system.
In piperidinium p-nitrobenzoate, (I), a centrosymmetric dimer of the formula unit is formed through N—H···O hydrogen
bonds (Fig. 1 and Table 1).
In most of cryslalline salts formed between cyclic secondary amines and p-substituted benzoic acids, the cations and
anions are arranged around a twofold screw axis to form N—H···O hydrogen bonds (Kashino et al., 1972, 1978, 1981;
Kashino, 1973). The same type of hydrogen bonding is also found in both pyrrolidinium p-nitrobenzoate and
hexamethyl-eneiminium p-nitrobenzoate (Takeda, 1992). In a few cases, the cations and anions are arranged along a glide plane to
form the N—H···O hydrogen bonds (Kashino et al., 1973, 1981).
The possibility of the formation of a cyclic dimer through the N—H···O hydrogen bonds was deduced based on a result
of a molecular weight measurement in a benzene solution, combined with a geometrical consideration of the
hydrogen-bonded system (Kashino, 1973). However, the formation of cyclic dimer has been found only in one of the dimorphs of
hexamethyleneiminium p-bromobenzoate, in which the dimer is formed around a twofold axis (Kashino et al., 1981). The
present study established a centrosymmetric type as the fourth pattern of hydrogen bonding possible in salts of cyclic
secondary amines and p-substituted benzoic acids.
S2. Experimental
Stoichiometric amounts of piperidine and p-nitrobenzoic acid were dissolved in benzene at 340 K. After cooling the
solution, crystals of (I) were grown by slow evaporation at room temperature.
S3. Refinement
All H atoms were found from a difference Fourier map. At the final stage of the least-squares refinement, all H atoms
except those involved in hydrogen bonds were fixed at the ideal positions, and their isotropic displacement parameters
Figure 1
A molecular view of (I) showing the hydrogen-bonding pattern and the atomic numbering scheme. Displacement
ellipsoids are drawn at the 50% probability level and H atoms involved in hydrogen bonds are shown as small spheres of
arbitrary radii. Hydrogen bonds are indicated by dashed lines. [Symmetry code: (i) 1 - x, 1 - y, 1 - z.]
Piperidinium p-nitrobenzoate
Crystal data
C5H12N+·C7H4NO4− Mr = 252.27
Orthorhombic, Pccn a = 8.949 (2) Å b = 22.451 (5) Å c = 12.712 (4) Å V = 2554.0 (11) Å3 Z = 8
F(000) = 1072
Dx = 1.312 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 25 reflections θ = 10.6–11.4°
µ = 0.10 mm−1 T = 298 K
Prismatic, colorless 0.50 × 0.30 × 0.20 mm
Data collection
Rigaku AFC5R diffractometer
Radiation source: Rigaku rotating anode Graphite monochromator
ω–2θ scans
Absorption correction: ψ (North et al., 1968) Tmin = 0.952, Tmax = 0.980 2875 measured reflections
2509 independent reflections 1142 reflections with I > 2σ(I) Rint = 0.022
θmax = 26.0°, θmin = 1.8° h = 0→11
k = 0→27 l = 0→15
3 standard reflections every 97 reflections intensity decay: 0.6%
Refinement
Refinement on F2 Least-squares matrix: full R[F2 > 2σ(F2)] = 0.057 wR(F2) = 0.205 S = 0.91 2509 reflections 172 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
supporting information
sup-3
Acta Cryst. (2001). E57, o707–o708w = 1/[σ2(F
o2) + (0.1P)2 + 1.1042P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.002 Δρmax = 0.13 e Å−3
Δρmin = −0.18 e Å−3
Extinction correction: SHELXL97, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 Extinction coefficient: 0.0059 (14)
Special details
Experimental. The scan width was (1.21 + 0.30tanθ)° with an ω scan speed of 4° per minute (up to 2 scans to achieve I/σ(I) > 10). Stationary background counts were recorded at each end of the scan, and the scan time:background time ratio was 2:1.
Ratio observed/unique reflections is too low. Although we tried to measure two set of data by using differnt sizes of crystals and scan times, the high ratio was not achieved. This is probably an effect of thermal puckering motions of the piperidinium ring and thermal libration of the nitro group.
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
The bond length N2—H6 1.02 (5) Å is longer than the normal. Such elongation is sometimes observed in the N—H bond involved in hydrogen bond, e.g. Kashino et al., (2001). Acta Cryst. C57, 627–631.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
The Ueq of N2 is low as compared to neighbors in the piperidinium ion. This is probably because the C atoms in the ring are effected by thermal puckering of the ring, while the thermal mortion of the N2 atom is suppressed by two hydrogen bonds in which the N2 atom is involved.
Precision (0.006 Å) on the C—C bond lengths is low in the piperidinium ring. This may be an effect of large thermal motion of the C atoms.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
H3 1.0134 0.2475 0.4928 0.069* H4 0.8483 0.3160 0.5672 0.063* H5 0.480 (5) 0.4309 (17) 0.605 (3) 0.069 (12)* H6 0.386 (5) 0.487 (2) 0.593 (3) 0.096 (15)* H7 0.3303 0.4154 0.4642 0.090* H8 0.2823 0.3695 0.5490 0.090* H9 0.1355 0.4773 0.5125 0.097* H10 0.0715 0.4155 0.4820 0.097* H11 −0.0330 0.4496 0.6393 0.090* H12 0.0503 0.3897 0.6559 0.090* H13 0.1620 0.5012 0.7092 0.085* H14 0.1176 0.4520 0.7890 0.085* H15 0.3138 0.3944 0.7367 0.077* H16 0.3755 0.4566 0.7670 0.077*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.075 (2) 0.111 (3) 0.0555 (18) 0.0372 (18) 0.0125 (15) 0.0146 (17) O2 0.0690 (18) 0.0521 (15) 0.0725 (18) 0.0103 (15) 0.0019 (15) 0.0147 (14) O3 0.127 (3) 0.116 (3) 0.075 (2) 0.038 (2) 0.001 (2) −0.031 (2) O4 0.119 (3) 0.076 (2) 0.102 (3) 0.042 (2) 0.005 (2) 0.005 (2) N1 0.079 (2) 0.057 (2) 0.076 (3) 0.006 (2) −0.003 (2) −0.013 (2) N2 0.047 (2) 0.055 (2) 0.0559 (19) 0.0030 (17) 0.0077 (16) 0.0026 (17) C1 0.0407 (19) 0.0418 (19) 0.051 (2) −0.0079 (17) −0.0002 (17) 0.0063 (17) C2 0.057 (2) 0.049 (2) 0.051 (2) 0.001 (2) −0.0024 (18) 0.0077 (19) C3 0.064 (2) 0.056 (2) 0.048 (2) −0.006 (2) −0.001 (2) −0.0006 (19) C4 0.050 (2) 0.046 (2) 0.059 (2) −0.0025 (18) −0.0033 (19) −0.0061 (19) C5 0.061 (2) 0.050 (2) 0.060 (3) 0.005 (2) −0.006 (2) 0.0085 (19) C6 0.053 (2) 0.056 (2) 0.048 (2) 0.0011 (19) −0.0036 (18) 0.0067 (18) C7 0.0414 (19) 0.057 (2) 0.060 (2) −0.0027 (18) 0.0002 (19) 0.007 (2) C8 0.078 (3) 0.086 (3) 0.062 (3) −0.013 (3) 0.010 (2) −0.014 (2) C9 0.061 (3) 0.105 (4) 0.076 (3) −0.020 (3) −0.012 (2) 0.010 (3) C10 0.049 (2) 0.079 (3) 0.096 (3) −0.009 (2) 0.009 (2) 0.022 (3) C11 0.063 (3) 0.083 (3) 0.067 (3) 0.002 (2) 0.022 (2) 0.006 (2) C12 0.062 (2) 0.080 (3) 0.050 (2) −0.003 (2) 0.004 (2) 0.014 (2)
Geometric parameters (Å, º)
supporting information
sup-5
Acta Cryst. (2001). E57, o707–o708C2—H1 0.95 C10—C11 1.502 (6) C3—C4 1.377 (5) C10—H11 0.95 C3—H2 0.95 C10—H12 0.95 C4—C5 1.379 (5) C11—C12 1.500 (6) C5—C6 1.378 (5) C11—H13 0.95 C5—H3 0.95 C11—H14 0.95 C6—H4 0.95 C12—H15 0.95 N2—C8 1.478 (5) C12—H16 0.95
O4—N1—O3 123.6 (4) N2—C8—C9 109.6 (4) O4—N1—C4 118.5 (4) N2—C8—H7 109 O3—N1—C4 117.9 (4) C9—C8—H7 109 C6—C1—C2 119.4 (3) N2—C8—H8 109 C6—C1—C7 120.5 (3) C9—C8—H8 109 C2—C1—C7 120.1 (3) H7—C8—H8 110 C3—C2—C1 120.3 (3) C10—C9—C8 110.0 (4) C3—C2—H1 120 C10—C9—H9 109 C1—C2—H1 120 C8—C9—H9 109 C4—C3—C2 119.1 (3) C10—C9—H10 109 C4—C3—H2 121 C8—C9—H10 109 C2—C3—H2 120 H9—C9—H10 110 C3—C4—C5 121.8 (4) C11—C10—C9 111.0 (3) C3—C4—N1 119.2 (3) C11—C10—H11 109 C5—C4—N1 119.0 (4) C9—C10—H11 109 C6—C5—C4 118.5 (3) C11—C10—H12 109 C6—C5—H3 121 C9—C10—H12 109 C4—C5—H3 121 H11—C10—H12 110 C5—C6—C1 120.9 (3) C12—C11—C10 111.4 (4) C5—C6—H4 120 C12—C11—H13 109 C1—C6—H4 120 C10—C11—H13 109 O2—C7—O1 125.0 (4) C12—C11—H14 109 O2—C7—C1 117.7 (3) C10—C11—H14 109 O1—C7—C1 117.2 (3) H13—C11—H14 110 C8—N2—C12 111.1 (3) N2—C12—C11 110.3 (3) C8—N2—H5 111 (2) N2—C12—H15 109 C12—N2—H5 114 (2) C11—C12—H15 109 C8—N2—H6 110 (2) N2—C12—H16 109 C12—N2—H6 109 (2) C11—C12—H16 109 H5—N2—H6 101 (4) H15—C12—H16 110
O3—N1—C4—C5 180.0 (4) C8—C9—C10—C11 55.7 (5) C3—C4—C5—C6 1.9 (6) C9—C10—C11—C12 −54.4 (5) N1—C4—C5—C6 −177.9 (3) C8—N2—C12—C11 −58.7 (5) C4—C5—C6—C1 −1.4 (6) C10—C11—C12—N2 55.2 (4)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N2—H5···O1 0.94 (4) 1.78 (4) 2.697 (5) 165 (4) N2—H6···O2i 1.02 (5) 1.71 (5) 2.731 (4) 171 (4)