1
ABSTRACT
The term “epigenetics” refers to heritable changes in gene expression not associated with
alteration of DNA sequence. There is considerable research exploring how changes at the
epigenetic level influence tumor progression. Preservation of tissue samples with the formalin
fixed, paraffin embedded (FFPE) technique is commonly used to archive tumor biopsy samples.
There is currently no method for extracting chromatin from FFPE tissue that is compatible with
all downstream analysis techniques. We investigated if a cavitation enhancement reagent called
“nanodroplets” employed during acoustic sonication would improve chromatin recovery and
fragmentation from FFPE tissue. We assessed the quality of the chromatin extracted using
Formaldehyde Assisted Isolation of Regulatory Elements (FAIRE), which separates nucleosome
depleted or “open” chromatin from protein rich or “closed” chromatin. FAIRE requires
high-quality chromatin to achieve a significant open chromatin signal to background ratio by the
quantitative polymerase chain reaction (qPCR) or high throughput sequencing (HTS). Ewing
sarcoma cell lines and rodent xenograft tissue were used as a model system in our study because
they contain a known, disease-specific aberrantly open chromatin signature. We performed
FAIRE on rodent tumor xenograft tissue followed by qPCR and found that nanodroplets
improved data consistency and significantly increased the FAIRE signal to background ratio at
Ewing sarcoma-specific regions. Next, we will use HTS to determine if the genome-wide FFPE
FAIRE signature is comparable to that from fresh/frozen tissue samples. If successful across a
variety of tissue types, our protocol could be applied to chromatin signature-based cancer
2
ACKNOWLEDGMENTS
This research was partially supported by The Sarah Steele Danhoff Undergraduate
Research Award. I would like to thank my thesis advisor Samantha Pattenden for her wisdom
and expertise in overseeing my project. Her patience and editing throughout the span of this
project has been truly invaluable. Thank you to my PI Paul Dayton for his input and oversight of
my work. I would like to thank Ian Davis for providing his wealth of knowledge and mentoring.
Thank you to Daniel Mckay for volunteering his assistance as my Biology Faculty Mentor. I
would like to thank Sunny Kasoji for both mentoring me during the early stages of this project
and bringing such a positive attitude to the lab each day. I would also like to thank other
members of my lab Suud Ashur and Anna Kenan for their technical assistance and friendship.
3
Introduction
Epigenetic signatures such as DNA methylation and histone modification are important
regulators of gene expression. The term epigenetics refers to changes in gene expression
unrelated to modification of the actual gene sequence1. Chromatin consists of DNA wrapped around histone protein octamers (dimers of H3, H4, H2A and H2B), often described as being
similar to “beads on a string2”. The functional unit of chromatin is the nucleosome, which
consists of approximately 146 base pairs of DNA wound around the histone octamer and
separated by a linker DNA-histone H1 unit3. Nucleosomes are a significant barrier to transcription because they block the progression of RNA polymerase II4. Genome wide nucleosome occupancy studies have demonstrated that nucleosomes are depleted at promoters
associated with active transcription5. Cis acting DNA sequences and DNA binding proteins alter nucleosome stability6. Disruption of these mechanisms for nucleosome positioning can lead to the loss of gene regulation in vivo7. This change in nucleosome positioning can be referred to as “chromatin signature”. Tumor cells have altered gene expression and therefore key changes in
chromatin signature. Ewing sarcoma is an example of a cancer whose oncogenic effects result
from disrupted nucleosome positioning.
Chromatin accessibility
The propagation of regulatory elements along the genome is determined by “open”
nucleosome depleted sites8. There are 3 assays for isolating accessible regions of the genome9. DNase1 hypersensitivity utilizes enzymatic digestion to cut nucleosome free regions of DNA,
4
Elements (FAIRE) is a chemical-based method for looking at a genome wide analysis of
nucleosome depleted regions of open chromatin. Differences in the efficiency of crosslinking
chromatin across the genome allows for the isolation of noncoding regions such as promoters
that are accessible to RNA polymerase II, as first demonstrated in the yeast, Saccharomyces
cerevisiae12. FAIRE has been optimized for use on Eukaryotic tissue13 and can extensively map regulatory elements. We chose to utilize a modified version of FAIRE over the other techniques.
Because it is a chemical-based method, FAIRE does not degrade the enriched nucleosome
depleted active chromatin and is not affected by enzyme bias14. Conducting FAIRE followed by high throughput sequencing (FAIRE-seq) produces an overall view of active regulatory elements
and is protein target independent. Isolating global euchromatic regions associated with higher
levels of gene expression would enable the identification of disease specific loci, such as the
GGAA microsatellite repeat signature in Ewing sarcoma (see below)15.
Ewing sarcoma
Ewing sarcoma tumor cells are characterized by an oncoprotein that results from a
chromosomal translocation. EWS-FLI1 is the most common type of translocation making up
85% of Ewing sarcoma tumors, but there are other ETS fusions in Ewing sarcoma and other
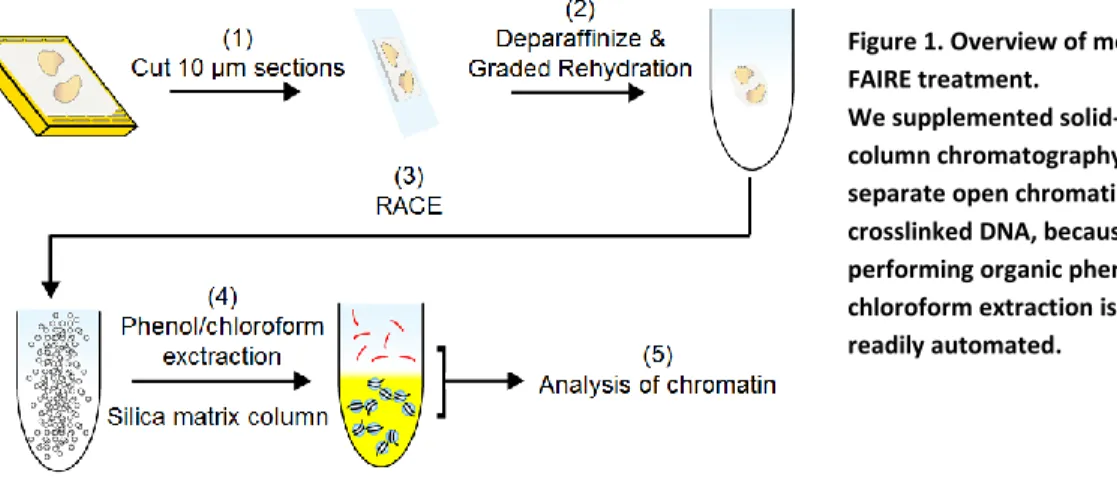
Figure 1. Overview of modified FAIRE treatment.
5
cancers16. The most common cancer causing EWS fusions are ERG, FLI1, and ETV117. The EWSR1 gene product contains a transcription activation domain and an RNA binding domain18, whereas the ETS proteins are DNA binding transcription factors. Following chromosomal
translocation, the activation domain of EWSR1 is fused to the DNA binding domain of the ETS
protein to create the EWS-ETS oncoprotein17.
EWS-FLI1 alters downstream gene expression by binding to GGAA microsatellite repeat
loci19,20. GGAA microsatellite binding subsequently increases expression of the EGR2
transcription factor in Ewing sarcoma compared to normal tissue15. EGR2 silencing results in the inhibition of cell proliferation and tumor growth when conducted in vivo on Ewing sarcoma
xenografts21. Ian Davis’ laboratory at UNC demonstrated that EWS-FLI1 binding results in nucleosome depletion at GGAA microsatellite repeats, while silencing EWS-FLI1 restores the
nucleosomes in these regions, and halts cell proliferation22. Disease specific changes in
nucleosome occupancy present an opportunity for both diagnostics and targeted therapy of this
cancer. Diagnosing Ewing sarcoma can be done using fluorescence in situ hybridization (FISH)
and RT-PCR on formalin fixed, paraffin embedded tissue. FISH uses DNA probes to test for the
presence of a breakpoint in the EWSR1 gene. These assays are limited as they do not distinguish
the fusion partner23 and require prior knowledge about the loci of interest.
FFPE and Diagnostics
The FAIRE protocol takes advantage of the molecular changes involved in crosslinking
DNA to histones using formaldehyde12. Formalin-Fixed, Paraffin Embedded (FFPE) tissue is a widely used method for preserving patient biopsy samples through formaldehyde crosslinking
6
quality of DNA may be reduced. The reduction in quality is a result of the DNA becoming
fragmented and inadequate purification methods during the process24. Ensuing molecular analysis techniques, such as the polymerase chain reaction (PCR), is affected by the limited
amount of purified DNA that can be obtained. Next generation sequencing is typically performed
on DNA isolated from fresh or frozen tissue, but these clinical samples are limited in that they
deteriorate quickly and are difficult to obtain26. We would like to utilize the availability and convenience of patient FFPE tissue samples for use in cancer diagnostics through the isolation
and subsequent identification of specific epigenetic signatures.
Fragmentation is essential to the FAIRE protocol for isolating regulatory elements.
Chromatin can be fragmented by mechanical disruption (sonication) or enzymatic digestion.
Enzymatic digestion often uses micrococcal nuclease, which degrades chromatin with enzymes
that digest the DNA between nucleosomes27. A major drawback to the use of enzymes is biased digestion that can lead to non-random fragmentation prior to sequencing28. Mechanical
fragmentation of chromatin generally produces more random DNA fragments29, but is often inconsistent and time-consuming. Current sonication methods, such as the Covaris, use high
intensity focused ultrasound in order to sheer samples in short bursts. A cavitation enhancing
agent called nanodroplets (developed in Paul Dayton’s lab at UNC) when added in solution with
DNA has been show to improve the consistency of a mechanical fragmentation in a low
frequency bench top waterbath30. Nanodroplets are comprised of a liquid core made up of a stable perfluorocarbon and a single layered lipid membrane31. Cavitation is the expansion and contraction of a nanodroplet when induced by acoustic pressure. The energy released by
cavitating nanodroplets when exposed to high frequency ultrasound waves will enhance DNA
7
We are interested in exploiting chromatin signature for diagnostics. Ewing sarcoma
served as our model system for chromatin. In order to achieve a broad spectrum analysis, we
also had the opportunity to test uncrosslinked genomic DNA in cells. The Ramsey lab provided
whole blood cells intended for a malaria diagnostic kit. These blood samples were used to
explore the application of nanodroplets to low frequency sonication of DNA from intact cells.
An optimized RACE protocol could then be applied to FFPE crosslinked chromatin that is more
challenging to break apart and requires high frequency sonication.
We can use cavitation enhancement to isolate high-quality chromatin from FFPE tissue,
which, for the first time, will make it possible to study changes in nucleosome occupancy in
Ewing sarcoma associated with cancer in archived patient samples. By comparing the chromatin
signatures through quantitative PCR (qPCR), we are able to test for the presence or absence of
nucleosome depleted sites with nanodroplet treatment during sonication. Our study will then
proceed to exploring different EWS-ETS fusions containing a chromosomal break other than
EWS-FLI. Different DNA binding domains fused to the EWS activation domain may result in
unique chromatin signatures whose presence can be identified utilizing FAIRE.
Methods
Low Frequency DNA Fragmentation. One hundred microliters (µL) of SDS/PAGE treated whole blood cells were sonicated in a Branson 2510 ultrasonic waterbath with 10 µl
perfluorobutane nanodroplets or buffer saline. The water bath was set at a height of 67.7mm,
temperature of 4°C, and centrally positioned in a floating plastic holder. DNA was extracted
8
determined using Qubit fluorometer. Twenty µL of each DNA sample was run on a 1.3% agarose gel.
Determining Cell Lysis Efficiency. EWS894 and EWS502 mouse xenografts were obtained by injecting approximately 106 cells subcutaneously into a nude mouse. The tumor was allowed to grow for six weeks. The tumor was sectioned and placed into 10% formaldehyde in order to
create an FFPE tissue block. MHH, SKES, REDES, and EWS502 FFPE patient derived cell
pellets were sectioned into 10 micron (μm) slices and one section was placed per slide. Slides
were deparaffinized in xylene and rehydrated in decreasingly concentrated (100%, 85%, 70%,
and 0%) ethanol solutions. Suspended tissue was sonicated in a Covaris Sonicator at 4°C (20%
duty cycle; intensity = 8; and 200 cycles per burst) for 12 min. Lysates were centrifuged and the
supernatant removed. DNA from both pellet and supernatant was extracted using Zymo Research
ChIP DNA clean and concentrate kit (#D5201) according to manufacturer’s instructions. DNA
concentration was determined using Qubit fluorometer.
Standard FAIRE and qPCR. Tissue was isolated fromEWS894 and EWS502 xenografts as described above and preserved by FFPE. FAIRE was performed after sonication. Solutions were
cold room centrifuged and the pellet discarded. We transferred 10% of the supernatant from each
tube to new 1.5mL Eppendorf tubes to serve as inputs. Input samples were digested with
proteinase K overnight through incubation at 55⁰C. The remaining supernatant was purified
using the Zymo Research ChIP DNA clean and concentrate kit, eluted as FAIRE DNA and
stored overnight at 0⁰C. Both input and FAIRE samples were digested with RNase and then
purified to a final volume of 25µl. Relative nucleosome occupancy was determined using qPCR
on FAIRE and input samples. AURKAIP1 and BC006361 primer sets served as positive and
9
μL of DNA sample was pipetted into the wells of a 384-well plate, and then 8 μL of qPCR
master mix [2.8µl de-ionized water, 0.1µl of Primer 1, 0.1µl Primer 2, 5µl of 2 X SYBR green
master mix (Roche)] was added. PCR was run on a ViiA7 Real-Time PCR system (Applied
Biosystems) and analyzed using the delta delta Ct method32. Results
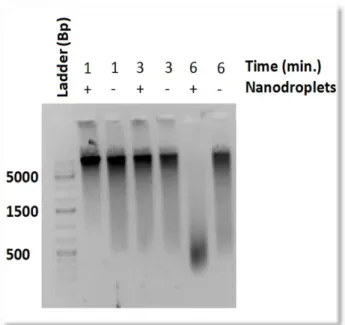
Validation of RACE on Genomic DNA. We began our analysis by first optimizing the use of RACE on blood cells. RACE employs nanodroplets in order to enhance the number of cavitation
events, in this case during low frequency
sonication, increasing consistency of
genomic DNA fragmentation for use in
sequencing30. While low frequency
sonication is suitable for blood cells, FFPE
tissue requires a high frequency sonicator in
order to disrupt crosslinking. Because
chromatin fragmentation is essential to the
FAIRE technique, we first evaluated the
performance of RACE on fresh whole blood
samples. A time titration was conducted
using a Branson low frequency ultrasonic water bath, ranging from 4 to 6 minutes of sonication
on intact blood cells. The whole blood cell solution contained either 10 µl of nanodroplets or
phosphate buffered saline (PBS). Fragmented DNA was isolated at approximately 500 base pairs
in length, the optimal range for use in high throughput sequencing, after six minutes of
10
sonicating with nanodroplets (Figure 2). There was minimal fragmentation without nanodroplets
at 6 minutes.
Application of FAIRE to FFPE Tissue. Having looked at genomic DNA, we then proceeded to determining the impact of nanodroplets on isolating chromatin from FFPE tissue. RACE was
adapted to our analysis of chromatin isolation by performing FAIRE and qPCR on EWS894
rodent xenograft FFPE tissue. FFPE xenografts derived from nude mice provide a uniform tissue
with known biomolecular markers, in this case originating from a Ewing sarcoma cell line. The
rehydration method for FFPE tissue was modified here to utilize slides rather than tissue scrolls,
which provided a reliable method for isolating tissue pellets. The modified protocol reduced the
risk of sample loss due to pipetting error and sped up the rehydration process. Predetermined
primers not specific to Ewing sarcomas were used correlating to open chromatin (AURKAIP1)
or closed chromatin (BC00361). P1 and P7 are Ewing sarcoma specific microsatellite repeat
regions that have aberrantly open chromatin signatures. RACE treatment during sonication of
FFPE pellets increased the percent input signal at determined by qPCR at 3 of the 4 regions
compared to RACE negative samples (Figure 3A). These samples are currently being sequenced
to compare nanodroplet-isolated FFPE FAIRE samples to those from fresh tissues.
We conducted the same FAIRE qPCR procedure on EWS502 xenograft cell lines (Figure
3B). The EWS502 tissue was more challenging for FAIRE as 0.06 ± 0.03 ng/µl of FAIRE DNA
was released per individual slide compared to the 12.8 ± 1.35 ng/µl FAIRE DNA released from
EWS894 tissue using RACE. The use of RACE on EWS502 did not have a significant difference
on the qPCR signal obtained, which is likely due to the low chromatin yields obtained from the
11 Figure 3. FAIRE-qPCR after RACE treatment improved signal over background noise of some rodent xenograft tissue.
AURKAIP1 is a region of chromatin that is always open, BC006361 is a region of chromatin that is always closed, ES1 and ES2 are Ewing sarcoma disease-specific regions of aberrantly open chromatin. The percent input is a ratio of the qPCR signal from DNA isolated from open chromatin to the total DNA. (A) Percent input is enriched at P1 and P7 for FFPE EWS894 rodent xenograft tissue. Error bars are the SD of 3 replicates. P values were calculated using a student’s T-test. (B) No enrichment after RACE treatment for FFPE EWS502 xenograft tissue; this is likely due to the difficulty of working with these xenografts as a result of low chromatin yields.
0.00 2.00 4.00 6.00 8.00 10.00 12.00 14.00 16.00
AURKAIP1 BC006361 P1 P7
Per ce n t In p u t qPCR Amplicon
A
RACE positiveRACE negative
p=0.024
p=0.016
p=0.069 p=0.023
0 0.02 0.04 0.06 0.08 0.1 0.12
AURKAIP1 BC006361 P1 P7
Per ce n t In p u t qPCR Amplicon
B
RACE positive12 0 0.5 1 1.5 2 2.5 3 3.5 4
MHH SKES RDES EWS502
D N A R e co ve ry R atio (Su p e rn atan t/ Pel le t) Pellet Type RACE positive RACE negative p=0.059 p= 0.046 p=0.172 p=0.016
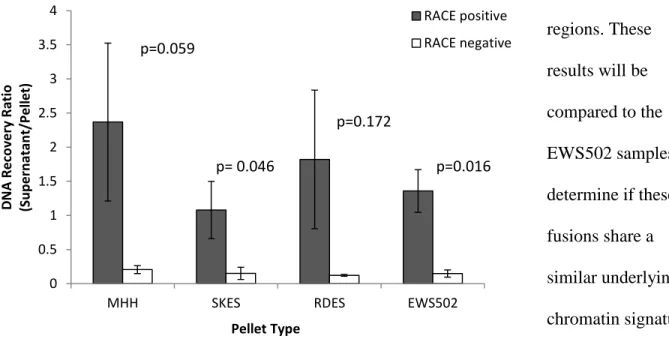
Our investigation into the presence of chromatin signatures in ETS family translocations
began with 4 samples containing similar EWS-FLI1 fusions: MHH, SKES, RDES, and EWS502.
Prior to performing FAIRE, we evaluated the percent yield of fragmented DNA that can be
isolated from these FFPE patient derived cell pellets. RACE was shown to significantly improve
the recovery of fragmented chromatin for SKES, and EWS502 cell pellets when sonicated for 12
minutes (Figure 4). For EWS502 cell pellets, RACE negative samples had 31.2 ± 5% of DNA
found in the supernatant relative to the cell pellet. RACE positive samples containing 10 µl of
nanodroplets prior to sonication were found to have more fragmented DNA in the supernatant
than pellet. We are in the process of conducting FAIRE and qPCR on these various EWS fusion
samples in order to identify the presence of aberrantly open chromatin at the P1 and P7 Ewing
sarcoma-specific
regions. These
results will be
compared to the
EWS502 samples to
determine if these
fusions share a
similar underlying
chromatin signature.
High throughput
sequencing of these
samples will ensure
13
that FAIRE does not disrupt open chromatin. Sequencing will also verify that molecular analysis
of FFPE tissue is comparable to fresh and frozen tissue. Human bladder tissue will serve as a
control as it lacks the Ewing sarcoma specific open chromatin.
Discussion
In contrast to utilizing fresh or frozen tissue in cancer based diagnostic screening, FAIRE
offers an alternative use of FFPE tissue through the isolation of regulatory elements associated
with gene regulation. The identification of a specific chromatin signature through qPCR
demonstrates the effectiveness of FAIRE at functioning as a screen for changes in chromosomal
accessibility. FAIRE does not depend on the enzymatic digestion step found in other chromatin
accessibility techniques, such as DNase I or ATAC, and subsequently eliminates the possibility
of enzyme bias. We will proceed with further sequencing of the FAIRE enriched FFPE EWS894
xenograft DNA to determine if FAIRE disrupts aberrant chromatin assembly.
The employment of biologically inert cavitation enhancing nanodroplets has the potential
to streamline the effectiveness of the FAIRE protocol by facilitating FFPE tissue dissociation. A
lack of nanodroplets during sonication generally resulted in a lower yield of FAIRE DNA, and
we showed several examples where RACE was able to overcome the difficulties associated with
obtaining high quality chromatin from FFPE tissue33. The ability to manufacture stable, size controlled nanodroplets for use in biological applications makes these agents potentially readily
accessible for research and clinical applications34. While RACE improved the percent input during qPCR for EWS894 xenografts, there was not a clear distinguishable FAIRE signal with
14
xenograft and combining multiple tissue slices prior to qPCR may improve the detection of
FAIRE signatures and reveal a difference in RACE samples.
Mutations in chromatin modifiers or their co-factors that disrupt either the nucleosome
contacts with DNA or nucleosome associated proteins are recognized events in a variety of
cancers3536. The EWS-FLI1 fusion in Ewing sarcoma is well studied, however it remains unknown if various other tumors containing different EWS fusion oncoproteins have similar or
different FAIRE signatures. Our modified FAIRE protocol has applications for any cellular
change that is associated with chromatin accessibility.
In conclusion, we have developed a novel protocol that isolates regions of open
chromatin that are accessible for gene expression from FFPE tissue. There have been challenges
associated with isolating high quality DNA when performing FAIRE on FFPE tissue.
Nanodroplet-mediated RACE improves the extraction of chromatin from FFPE tissue and
enables us to study nucleosome-depleted sites in cancer regulatory pathways. Ewing sarcoma
was used as a model system in our study, since it has known disease-specific regions of open
chromatin. We will use that model system to identify key regulatory elements in tumor tissue
15 Literature Cited
1. Deans, C. & Maggert, K. A. What do you mean, ???Epigenetic???? Genetics199, 887– 896 (2015).
2. Szerlong, H. J. & Hansen, J. C. Nucleosome distribution and linker DNA: connecting nuclear function to dynamic chromatin structure. Biochem. Cell Biol.89, 24–34 (2011).
3. Luger, K., Mäder, a W., Richmond, R. K., Sargent, D. F. & Richmond, T. J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature389, 251–260 (1997).
4. Durrin, L. K., Mann, R. K. & Grunstein, M. Nucleosome loss activates CUP1 and HIS3 promoters to fully induced levels in the yeast Saccharomyces cerevisiae. Mol Cell Biol12,
1621–1629 (1992).
5. Yuan, G.-C. et al. Genome-scale identification of nucleosome positions in S. cerevisiae. Science309, 626–30 (2005).
6. Gosalia, N. & Harris, A. Chromatin dynamics in the regulation of CFTR expression. Genes6, 543–558 (2015).
7. Gossett, A. J. & Lieb, J. D. In vivo effects of histone H3 depletion on nucleosome occupancy and position in Saccharomyces cerevisiae. PLoS Genet.8, (2012).
8. John, S. et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat. Genet.43, 264–8 (2011).
9. Tsompana, M. & Buck, M. J. Chromatin accessibility: a window into the genome. Epigenetics Chromatin7, 33 (2014).
10. He, H. H. et al. Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. Nat. Methods11, 73–8 (2014).
11. Goryshin, I. Y. & Reznikoff, W. S. Tn5 in vitro transposition. J. Biol. Chem.273, 7367– 7374 (1998).
16
13. Simon, J. M., Giresi, P. G., Davis, I. J. & Lieb, J. D. Using formaldehyde-assisted
isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nat. Protoc.7,
256–67 (2012).
14. Hörz, W. & Altenburger, W. Sequence specific cleavage of DNA by micrococcal nuclease. Nucleic Acids Res.9, 2643–2658 (1981).
15. Gomez, N. C. & Davis, I. J. Linking germline and somatic variation in Ewing sarcoma. Nat. Publ. Gr.47, 964–965 (2015).
16. Burchill, S. A. Ewing’s sarcoma: diagnostic, prognostic, and therapeutic implications of molecular abnormalities. J. Clin. Pathol.56, 96–102 (2003).
17. Feng, F. Y., Brenner, J. C., Hussain, M. & Chinnaiyan, A. M. Molecular Pathways: Targeting ETS Gene Fusions in Cancer. Clin. Cancer Res.20, 4442–4448 (2014).
18. Duggimpudi, S., Larsson, E., Nabhani, S., Borkhardt, A. & Hoell, J. I. The Cell Cycle Regulator CCDC6 Is a Key Target of RNA-Binding Protein EWS. PLoS One10,
e0119066 (2015).
19. Lessnick, S. L., Braun, B. S., Denny, C. T. & May, W. A. Multiple domains mediate transformation by the Ewing’s sarcoma EWS/FLI-1 fusion gene. Oncogene10, 423–431 (1995).
20. Gangwal, K. et al. Microsatellites as EWS/FLI response elements in Ewing’s sarcoma. Proc. Natl. Acad. Sci. U. S. A.105, 10149–54 (2008).
21. Grünewald, T. G. P. et al. Chimeric EWSR1-FLI1 regulates the Ewing sarcoma susceptibility gene EGR2 via a GGAA microsatellite. Nat. Genet.47, 1073–8 (2015).
22. Patel, M. et al. Tumor-specific retargeting of an oncogenic transcription factor chimera results in dysregulation of chromatin and transcription. Genome Res.22, 259–70 (2012).
23. Choi, E.-Y. K., Gardner, J. M., Lucas, D. R., McHugh, J. B. & Patel, R. M. Ewing sarcoma. Semin. Diagn. Pathol.31, 39–47 (2014).
17
25. Lou, J. J. et al. A review of room temperature storage of biospecimen tissue and nucleic acids for anatomic pathology laboratories and biorepositories. Clin. Biochem.47, 267–273 (2014).
26. Carrick, D. M. et al. Robustness of next generation sequencing on older formalin-fixed paraffin-embedded tissue. PLoS One10, (2015).
27. Skene, P. J. & Henikoff, S. A simple method for generating high-resolution maps of genome wide protein binding. Elife4, e09225 (2015).
28. Chung, H.-R. et al. The effect of micrococcal nuclease digestion on nucleosome positioning data. PLoS One5, e15754 (2010).
29. Schoppee Bortz, P. D. & Wamhoff, B. R. Chromatin immunoprecipitation (chip): Revisiting the efficacy of sample preparation, sonication, quantification of sheared dna, and analysis via pcr. PLoS One6, 1–10 (2011).
30. Kasoji, S. K. et al. Cavitation Enhancing Nanodroplets Mediate Efficient DNA
Fragmentation in a Bench Top Ultrasonic Water Bath. PLoS One10, e0133014 (2015).
31. Sheeran, P. S. & Dayton, P. a. Improving the performance of phase-change
perfluorocarbon droplets for medical ultrasonography: current progress, challenges, and prospects. Scientifica (Cairo).2014, 579684 (2014).
32. Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods25, 402–8 (2001).
33. Lin, J. et al. High-quality genomic DNA extraction from formalin-fixed and paraffin-embedded samples deparaffinized using mineral oil. Anal. Biochem.395, 265–267 (2009).
34. Martz, T. D., Bardin, D., Sheeran, P. S., Lee, A. P. & Dayton, P. A. Microfluidic generation of acoustically active nanodroplets. Small8, 1876–1879 (2012).
35. Lunning, M. A. & Green, M. R. Mutation of chromatin modifiers; an emerging hallmark of germinal center B-cell lymphomas. Blood Cancer J.5, e361 (2015).
36. Taberlay, P. C., Statham, A. L., Kelly, T. K., Clark, S. J. & Jones, P. A. Reconfiguration of nucleosome-depleted regions at distal regulatory elements accompanies DNA