www.ijicse.com 55
PREDICTION OF PROTEIN STRUCTURAL CLASSES BY PSEUDO AMINO ACID COMPOSITION USING IMPROVED HARMONY SEARCH RELATIVE REDUCT
FEATURE SELECTION AND ROUGH SET CLASSIFICATION ALGORITHMS
MRS. M. BAGYAMATHI DR. H. HANNAH INBARANI
Assistant Professor & Head Assistant Professor Dept. of Computer Science & Applications Department of Computer Science Gonzaga College of Arts and Science for Women Periyar University Krishnagiri, Tamil Nadu, India. Salem, Tamil Nadu, India. Email: [email protected] Email: [email protected] Abstract
The paper proposes classification of protein sequences using rough set based feature
selection and classification algorithm. Sequence-derived structural and physiochemical features
have been frequently used for analyzing and predicting structural, functional, expression and
interaction profiles of proteins. The pseudo amino acid (PseAA) composition can represent a
protein sequence in a discrete model without completely losing its sequence-order information,
and hence has been widely applied for improving the prediction quality for various protein
attributes. The general form pseudo-amino acid composition (PseAAC) has been widely used to
represent protein sequences in predicting protein structural and functional attributes. A two
major complexity is the large number of feature vectors and the accurate classification of its
structure. The proposed framework includes feature selection mechanism embedded with
classification technique for the efficient classification of the structure of the protein. In this
study, we employed rough set improved harmony search relative reduct algorithm for selecting
the optimum number of features, which is fed as an input to the rough set classification algorithm
to classify the protein sequence to its class. The performance analysis of the proposed framework
is compared with the Rough Set Improved Harmony Search Quick Reduct framework. The
experimental results revealed high accuracy of 90% from the proposed framework.
Keywords: Protein Structure Classification, Feature Selection, Improved Harmony Search, Relative Reduct, Rough Set Classification.
Introduction
Bioinformatics involves the
development and application of novel
informatics techniques in the genomic
sciences. Proteins play a fundamental role in
all living organisms and are involved in a
variety of molecular functions and
biological processes. Proteins are the most
essential and versatile macromolecules of
life, and the knowledge of their functions is
an essential link in the development of new
www.ijicse.com 56 development of synthetic biochemical such
as biofuels [23]. Traditionally,
computational prediction methods use
features that are derived from protein
sequence, protein structure or protein
interaction networks predict function [24].
Proteins are composed of one or more
chains of amino acids and show several
levels of structure [17]. In fact, according to
their chain folding pattern, proteins are
usually folded into four structural classes
such as all α, all β, all α + β and all α / β
[16].
Pseudo amino acid composition
(PseAAC) is an algorithm that could convert
a protein sequence into a digital vector that
could be processed by data mining
algorithms. The design of PseAAC
incorporated the sequence order information
to improve the conventional amino acid
compositions. The application of pseudo
amino acid composition is very common,
including almost every branch of
computational proteomics [21]. In this
study, the features are extracted from protein
primary sequence, based on amino acid
composition and K-mer patterns, or K-grams
or K-tuples [15]. Different classification
techniques have been used to classify the
protein sequence into its particular classes.
In the analysis of protein sequences and
structures based on their critical feature set.
High dimensional dataset degrades the
classification quality. The optimum number
of features helps to classify the sequence
into its family.
Rough set theory [14], provides a
mathematical tool which is used to deal with
vague and imprecise concept. A rough set
uses the concepts of lower and upper
approximations to classify the protein
sequences into disjoint categories. It can
also be used for feature selection and
knowledge discovery. It helps us to find out
the minimal attribute sets called „reducts‟ to
classify objects without deterioration of
classification quality.
In this study, both the feature selection
and classification technique is applied as a
single framework to predict the class of the
protein. The Rough Set Improved Harmony
Search Relative Reduct algorithm is
employed to reveal the optimum number of
features [13] and the Rough Set
Classification [12] algorithm is used to
classify the sequences precisely to its family
of classes.
The rest of the paper is structured as
Sections 2 to 5. Section 2 describes a
Review of the Literature; Section 3 explains
the Research Motivation; Section 4
www.ijicse.com 57 covers dataset description, feature selection
and classification algorithms and the
experimental analysis with the results and
discussion were described in Section 5 and
the chapter concludes with a discussion on
the interpretation and highlights the
possibility of future work in this area.
Review of Literature
In the past, many computational
intelligence techniques have been proposed
in the literature to provide deep insights into
protein sequence data. In this section, we
provide a concise overview of existing
feature selection and classification methods
involving different representations for the
purpose of function prediction and
classification. Thus, there is a need of a
highly accurate and efficient system that can
classify the newly discovered protein
sequences into existing super families in a
very short period of time [10].
References Technique Employed Operational Principle
Iqbal, MJ, et.al [18]
Decision tree, Naïve Bayes,
Neural Network, Random Forest
and SVM
Classification
Saha and Chaki [9] Neural Network, Fuzzy
ARTMAP, Rough Set Classifier Classification
Datta et al.[4] Principal Component Analysis
and Nearest Neighbor Classifier
Feature Selection and
Classification
Ahmad et al.[6] AAC and Growing
Self-Organizing Map
Feature Extraction and
Clustering
Bagyamathi and Inbarani
[17 ]
Rough Set Based Improved
Harmony Search algorithm
Feature Selection and
classification
Inbarani et al. [22] Rough Set based Particle Swarm
Optimization Algorithm
Feature Selection and
Classification
Inbarani et. al [28]
Hybrid Rough Set Improved
Harmony Search Quick Reduct
algorithm
Feature Selection and
classification
Pedergnana M et al. [25] Genetic Algorithm and Random Forest Classifier
Feature Extraction, Feature
www.ijicse.com 58
Jeong et.al [27] Random Forest Classification
Mansoori et al. [29] Fuzzy Rule based Classifier Classification
Bagyamathi and Inbarani
[26]
Rough Set Based Improved
Harmony Search Relative
Reduct
Feature Selection and
classification
Azar et al. [30] Unsupervised Particle Swarm
Optimization (PSO) based
Relative Reduct.
Feature Selection
Bagyamathi and Inbarani
[34 ]
Rough Set Based Improved
Harmony Search Quick Reduct
and Rough Set Classification
framework
Feature Selection and
classification
Research Motivation
Due to the growth of molecular
biology technologies and techniques, more
large-scale biological data sets are becoming
available. Identifying biologically useful
information from these data sets has become
a significant challenge. Computational
biology aims to address this challenge [31].
Many problems in computational biology,
e.g., protein function prediction, sub cellular
localization prediction, protein-protein
interaction, protein secondary structure
prediction, etc., can be formulated as
sequence classification tasks [32], where the
amino acid sequence of a protein is used to
classify the protein in functional and
localization classes.
In this study, the application of
feature selection techniques is focussed,
which do not alter the original representation
of the variables, but merely select a subset
of them. Thus, they preserve the original
semantics of variables, hence offering the
advantage of interpretability by a domain
expert [11]. While the feature selection can
be applied to both supervised and
unsupervised learning, this chapter
concentrates on the problem of supervised
learning (classification), where the class
labels are known in advance. In recent
decade, Population-based
algorithms have been attracting an increased
attention due to their powerful search
capabilities.
From the comparative analysis of the
www.ijicse.com 59 for only a particular kind of data set, but
may not perform well on a high dimensional
dataset of protein sequences. In protein
analysis, especially in protein sequence
classification, selection of features is most
important. Our main motivation in this study
is to design a classification model that uses a
novel feature selection and classification
algorithms in a single framework in order to
improve the classification accuracy.
Methods and Materials
The framework shown in Figure 1 has
been proposed for an efficient and highly
accurate classification of protein sequences
into super families. The framework is
divided into two phases: Feature Selection
and Classification. Both the techniques are
hybridized with rough set theory to deal the
problem of high dimensionality and
uncertainty in classifying the protein
sequences [34].
In the first phase of this study, the
protein primary sequences are collected
from Protein Data Bank (PDB) in fasta
format [16]. The fasta sequence file is used
as input data to the PseAAC-builder [5], that
generates the protein feature space using
amino acid composition and amino acid K-
tuples or K- mer patterns. The real valued
data generated by the PseAAC server to be
discretized, since the rough set theory best in
dealing with discrete values [17]. The
discretized dataset is the feature vector
extracted from the PseAAC server. In the
next step, the feature vector is fed as an
input to the Rough Set Improved Harmony
Search Relative Reduct (RSIHSRR) feature
selection algorithm. The RSIHSQR
algorithm is used to select the feature subset.
In the second phase, the feature subset
revealed by the RSIHSRR algorithm are
divided into training and testing based on
10-fold cross validation. The training set is
subjected to classification process by Rough
Set Classification (RSC) algorithm to
generate the decision rules. In the next step,
the generated rules are evaluated with the
test data to validate the RSC algorithm. The
various classification measures are applied
to evaluate the performance of the proposed
algorithm and also compared with various
classification algorithms for the accurate
prediction of protein structure.
Data Source
In this work, the protein primary
sequence dataset is derived from PDB
(http://www.rcsb.org/pdb) on SCOP
classification. The Structural Classification
of Proteins (SCOP) database is a largely
manual classification of protein structural
domains based on similarities of their
www.ijicse.com 60 The data set consists of sequences with 7623
of all α, 10672 of all β, 11048 of all α + β
and 11961 of all α / β [16]. Among one
thousand sequences with combinations of all
α , all β, all α + β and all α / β, each 250
sequences are taken for this study [17].
Feature Extraction
Protein sequences are consecutive
amino acid residues, and it is represented
with an alphabet A of size |A| = 20. Many
feature extraction methods have been
developed in the past several years.
Typically, these methods can be classified
into two categories. One is based solely on
amino acid composition. The other one is an
extension of the atomic length from only
one amino acid to a K amino acid tuple,
where K is an integer and larger than one,
which can also be referred as „Ktuple‟, such
as 2-tuple in [33].
In the first step of feature generation,
20 amino acid features are adopted as our
initial representative features, which is
simple and effective. Along with the 20
features, K-tuple features are introduced.
The high computational cost of K-tuple
prediction caused by large feature amount,
the Rough Set based feature selection
methods are introduced to reveal most
relevant K-tuples and eliminating the
irrelevant ones. In this study, the supervised
rough set based feature selection algorithms
such as Quick Reduct, PSO Quick Reduct,
and Improved Harmony Search Quick
Reduct were applied and the protein
sequence feature vectors are classified to
their respective structural classes [17].
Many researchers used one method
of feature extraction among the following
two different strategies. (a) Without
dimension reduction: the prediction can be
based on full K-tuple space without any
dimension reduction, and the maximum
value of K is set to 5. As a result, at most
205 = 3.2×106 features are extracted. (b)
With the dimension reduction: when the
proteins are represented in a high
dimensional space, the occurrences of many
K-tuples will be very scarce. Some K-tuples
just occur only once or even never occur in
the dataset. Thus, lots of them must be
irrelevant to the protein classification
process since they are too sparse. Motivated
by this phenomenon, in this chapter, the
feature selection techniques are adopted to
filter the K-tuple feature set. Therefore, only
the relevant tuples are selected as best subset
in order to reveal the better classification
result [17].
Feature Selection
An Improved HS algorithm was
www.ijicse.com 61 proposed algorithm, HMCR, PAR, bw
parameters are considered, but PAR and bw
are very important parameters in fine-tuning
of optimized solution vectors. The
traditional HS algorithm uses a fixed value
for both PAR and bw. In the HS method,
PAR and bw values are adjusted and cannot
be changed during new generations [2]. The
main drawback of the HS method is that the
numbers of iterations increases to find an
optimal solution. To improve the
performance of the HS algorithm and to
eliminate the drawbacks that lies with fixed
values of PAR and bw, IHS algorithm uses
variables PAR and bw in improvisation step
(Step 3) [3]. The pitch adjustment rate
parameter causes a musician to select a
value neighboring to its current choice. To
achieve better results, we define lower limit
and upper limit of the PAR and bw. PAR
and bw change dynamically with generation
number and expressed as follows:
PAR(gn) = PARmin + -
* gn (12)
Where, PAR(gn)is the Pitch
Adjusting Rate for each generation,
PARminand PARmaxis the minimum and
maximum Pitch Adjusting Rate respectively,
NIis considered as Number of
Improvisations and gnis the Generation
Number.
bw(gn) = bwmax* exp(c *gn); c = ln
[(bwmin / bwmax)] / NI (13)
Where, bw(gn)is considered as the
bandwidth for each generation bwminand
bwmaxare minimum and maximum
bandwidthrepectively.
The improved harmony search
reveals best features which yield high
classification accuracy when hybridized
with rough set quick reduct algorithm. The
Pseudocode of the RSIHSRR algorithm is
given in [7].
Classification
Classification is to find the best class
that is closest to the classified pattern. The
goal of classifying the selected features is, to
map every pattern of readings into an output
class, and the percent of correct mapping
forms the accuracy of the classifier.
We adopted rough set based
classification to predict the structure of the
protein sequences. The main advantages for
employing rough set theory in this study are
the following: RST does not require any
primary or additional information about the
dataset; RST can deliver a valuable analysis,
even with missing values; and RST can
www.ijicse.com 62 both quantitative and qualitative data [19].
The rough set classifier generates decision
rule based on equivalence relations of the
attribute set.
Rule generation is a critical task in
any learning system .In this approach, the
lower and upper approximations of the
training data set based on decision class are
constructed, based on the equivalence
relation [1]. In the next step, deterministic
rules are generated based on lower
approximation and non-deterministic rules
are generated based on upper approximation
[12]. The rules framed by the RSC
algorithm are validated with the test data set.
Experimental Analysis
For experimental studies, 1000
sequences were extracted from the SCOP
classification of Protein Data Bank. A
sequence of experiments was conducted to
show the efficacy of the proposed feature
selection algorithm. The proposed Improved
Harmony Search hybridized with Rough Set
Improved Harmony Search Relative Reduct
feature selection algorithm is implemented
and compared with the existing Rough Set
Improved Harmony Search Quick Reduct
algorithm, which produced best and minimal
feature set for the classification process [17].
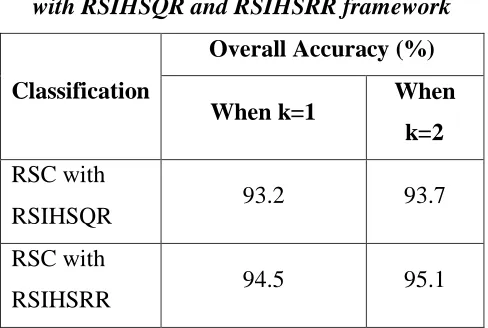
Table 2. Classification results of the RSC
with RSIHSQR and RSIHSRR framework
Classification
Overall Accuracy (%)
When k=1 When
k=2 RSC with
RSIHSQR 93.2 93.7
RSC with
RSIHSRR 94.5 95.1
Results and Discussion
In this study, the feature subset
selected by the RSIHSRR algorithm is
classified with Rough Set Classification
(RSC) algorithm. Accurate classification of
protein classes is the most critical part of the
sequence analysis. The result in Table 2
depicts the overall classification accuracy of
the existing and proposed framework. The
accuracy obtained from the proposed RSC
with RSIHSRR algorithm is compared with
RSC with RSIHSQR algorithm. In the Fig.
1, the overall accuracy of proposed
framework discloses the better classification
result when it is applied to the feature subset
revealed by the RSIHSRR algorithm. And
www.ijicse.com 63 procedure and the accomplishment rates of
test results, the proposed method can perk
up the prediction excellence of protein
structure class, and can afford as a
functional harmonizing framework.
Fig 1. Classification accuracy of RSC with
RSIHSQR and RSIHSRR algorithm (when
K=1, K=2)
Conclusion
The proposed framework well
predicts protein sequences to its family of
class or sub class based on its structure,
which is an effective combination of feature
extraction, feature selection and
classification methods. Feature extraction
from the chain of amino acid sequences
produces a huge number of feature vectors,
so the performance of the classification
model could decrease; hence, feature
selection method is essential. In this paper,
we applied RSIHSRR feature selection
algorithm to select or eliminate a minimal
subset of relevant and non-redundant
features. The proposed classification
approach is based on rough set is a novel
way to achieve protein sequence
classification. A comparison between the
obtained results of rough set with the
existing RSIHSQR and Rough Set classifier
algorithm has been employed. The Rough
set classification showed a higher overall
accuracy rates and it generates more
compact rules, when it is applied to the
reduct set from RSIHSRR feature selection
algorithm. Hence the analysis section clearly
proved the effectiveness of rough set based
approaches for Protein Sequence
classification.
References
[1] Inbarani, H.H., Kumar, S.S., Azar, A.T. and Hassanien, A.E., (2014), November. Soft rough sets for heart valve disease diagnosis. In International
Conference on Advanced Machine Learning
Technologies and Applications(pp. 347-356).
Springer, Cham.
[2] Al-Betar M, Khader A, and Liao I, (2010), “A harmony search with multi-pitch adjusting rate for the university course timetabling”, In: Geem, Z.W.
(ed.) Recent Advances in Harmony Search
Algorithm. SCI, vol. 270, pp. 147–161. Springer, Heidelberg.
[3] Chakraborty P, Roy GG, Das S, Jain D and Abraham A, (2009), “An improved harmony search algorithm with differential mutation operator”, Fundamenta Informaticae, Vol. 95, No. 4, pp. 1–26. [4] Datta A, Talukdar V, Konar A, and Jain LC, (2009), "A neural network based approach for protein structural class prediction," Journal of Intelligent and Fuzzy Systems, vol. 20, pp. 61-71.
90 92 94 96
RSIHSQR RSIHRRR
1-tuple
www.ijicse.com 64
[5] Du P, Wang X, Xu C, and Gao Y, (2012), “PseAAC-Builder: A cross-platform stand-alone program for generating various special Chou‟s
pseudo-amino acid compositions”, Analytical
Biochemistry Vol. 425, No. 2, pp. 117–119.
[6] Ahmad N, Alahakoon D and Chau R, (2008), "Classification of protein sequences using the growing Self-Organizing map," Colombo, pp. 167-172.
[7] Bagyamathi M and Inbarani HH, (2015), “Feature Selection using Relative Reduct hybridized with Improved Harmony Search for Protein Sequence Classification”, International Journal of Trend in Research and Development (IJTRD), ISSN:2394-9333, Special Issue | PCIT-15.
[8] Mahdavi M, Fesanghary M and Damangir E, (2007), “An improved harmony search algorithm for
solving optimization problems”, Applied
Mathematics and Computation, Vol. 188, No. 2, pp. 1567– 1579.
[9] Saha S and Chaki R, (2013), “A brief review of data mining application involving protein sequence classification”, International Journal of Database Management Systems, Vol. 4, pp. 469–477.
[10] Iqbal M.J, Faye E, Samir B.B and Md Said A, (2014) “Efficient feature selection and classification of protein sequence data in bioinformatics”, The Scientific World Journal, pp. 1–12.
[11] Saeys, Y., Inza, I.N., Larrañaga, P., (2007), A
review of feature selection techniques in
bioinformatics. Bioinformatics 23(19), 507–2517. [12]Hassanien AE, Jafar M.H. ALI (2004), “Rough Set Approach for Generation of Classification Rules of Breast Cancer Data”, Informatica, Vol. 15, No. 1, pp. 23-28.
[13] Inbarani HH, Bagyamathi M and Azar AT (2015), “A novel hybrid feature selection method based on rough set and improved harmony search”, Neural Computing and Applications, Springer London, pp. 1-22.
[14] Pawlak, Z., (1993), Rough Sets: Present State and The Future. Foundations of Computing and Decision Sciences 18(3-4), 157–166.
[15] Chandran, C.P., (2008), Feature Selection from Protein Primary Sequence Database using Enhanced Quick Reduct Fuzzy-Rough Set. In: Proceedings of International Conference on Granular Computing, GrC 2008, Hangzhou, China, August 26-28, pp. 111– 114, doi:10.1109/GRC.2008.4664758.
[16] Cao, Y., Liu, S., Zhang, L., Qin, J., Wang, J., Tang, K, (2006), Prediction of protein structural class with Rough Sets. BMC Bioinformatics 7(1), doi:10.1186/1471-2105-7-20.
[17] M. Bagyamathi and H. Hannah Inbarani, (2015), “A Novel Hybridized Rough Set and Improved Harmony Search based Feature Selection for Protein
Sequence Classification”, Big Data in Complex Systems: Challenges and Opportunities, Studies in Big Data series, Vol. 9, 2015, pp. 173-204. Springer-Verlag International, Switzerland.
[18] Iqbal M.J, Faye E and Samir B.B, (2014) “Data mining of protein sequences with amino acid position-based feature encoding technique”, Kuala Lumpur. Lecture Notes in Electrical Engineering, Vol. 285, pp. 119–126.
[19] Udhaya kumar S and Inbarani HH, (2016), “PSO-based feature selection and neighborhood rough set-based classification for BCI multiclass motor imagery task”, Neural Computing and Applications, Springer, DOI 10.1007/s00521-016-2236-5.
[20] Chinnasamy A, Sung W.K and Mittal A, (2005), “Protein Structure and Fold Prediction Using
Tree-Augmented Bayesian Classifier”, Journal of
Bioinformatics and Computational Biology, Vol. 3, No. 4, pp. 803–819.
[21] Du, P., Wang, X., Xu, C., Gao, Y, (2012), “PseAAC-Builder: A cross-platform stand-alone program for generating various special Chou‟s
pseudo-amino acid compositions”, Analytical
Biochemistry 425(2), 117–119.
[22] Inbarani, H.H., Azar, A.T., Jothi, G, (2014), “Supervised hybrid feature selection based on PSO and rough sets for medical diagnosis”, Computer Methods and Programs in Biomedicine 113(1), 175– 185.
[23] Nemati, S., Basiri, M.E., Ghasem-Aghaee, N., Aghdam, M.H, (2009), “A novel ACO–GA hybrid algorithm for feature selection in protein function prediction”, Expert Systems with Applications 36(10), 12086–12094.
[24] Rentzsch, R., Orengo, C, (2009), “Protein function prediction-the power of multiplicity”, Trends in Biotechnology 27(4), 210–219.
[25] Pedergnana M, Marpu PR, Mura MD, Benediktsson JA and Bruzzone L (2012). “A Novel supervised feature selection technique based on Genetic Algorithms”, In Proceedings of IEEE International Geoscience and Remote Sensing Symposium, 60-63.
[26] M. Bagyamathi and H. Hannah Inbarani, (2015), “Feature Selection using Improved Harmony Search Hybridized with Relative Reduct for Medical Data Classification”, International Journal of Applied Engineering Research (IJAER), 10(20): 19476-19480.
www.ijicse.com 65
[28] Inbarani HH, Bagyamathi M and Azar AT, (2015), “A Novel Hybrid Feature Selection Method based on Rough Set and Improved Harmony Search”, Neural Computing and Applications, Springer–
Verlag International, Switzerland, doi:
10.1007/s00521-015-1840-0.
[29] Mansoori, E. G., M. J. ZolghadrI, and S. D. Katebi. (2009), “Protein superfamily classification using fuzzy rule-based classifier”. IEEE Transactions on Nanobioscience, 8(1): 92–99.
[30] Azar, A.T., Banu, P.K.N., Inbarani, H.H, (2013), “PSORR - An Unsupervised Feature Selection Technique for Fetal Heart Rate”. In: 5th International Conference on Modelling, Identification and Control (ICMIC 2013), Egypt, August 31-September 1-2, pp. 60–65.
[31] Wei, X.(2010), “Computational approaches for biological data analysis. Doctoral Dissertation”, Tufts Uiversity Medford, MA, USA, ISBN: 978-1-124-21198-5
[32] Wong, A., Shatkay H, (2013), “Protein Function Prediction using Text-based Features extracted from the Biomedical Literature: The CAFA Challenge”. BMC Bioinformatics 14(3), S14, doi:10.1186/1471-2105-14-S3-S14.
[33] Park, K.J., Kanehisa, M, (2003) “Prediction of protein subcellular locations by support vector machines using compositions of amino acids and amino acid pairs”, Bioinformatics 19(13), 1656– 1663.