The Regulatory Regions Required for
B
ⴕ
Paramutation and Expression Are
Located Far Upstream of the Maize
b1
Transcribed Sequences
Maike Stam,*
,1Christiane Belele,* Wusirika Ramakrishna,
†Jane E. Dorweiler,*
Jeffrey L. Bennetzen
†and Vicki L. Chandler*
,2*Plant Sciences Department, University of Arizona, Tucson, Arizona 85721 and†Department of Biological Sciences, Purdue University, West Lafayette, Indiana 47907
Manuscript received May 23, 2002 Accepted for publication July 16, 2002
ABSTRACT
Paramutation is an interaction between alleles that leads to a heritable change in the expression of one allele. InB⬘/B-Iplants,B-I(high transcription) always changes toB⬘(low transcription). The newB⬘allele retains the low expression state in the next generation and paramutatesB-Iat a frequency of 100%. Comparisons of the structure and expression of B⬘ with that of a closely related allele that does not participate in paramutation demonstrated that transcription from the same promoter-proximal sequences is not sufficient for paramutation. Fine-structure recombination mapping localized sequences required forB⬘expression and paramutation. The entire 110 kb upstream of theB⬘transcription start site was cloned and sequenced and the recombination breakpoints were determined for 12 recombinant alleles. Sequences required for expression and paramutation mapped to distinct regions, 8.5–49 kb and 93–106 kb upstream of theB⬘ transcription start site, respectively. Sequencing and DNA blot analyses indicate that theB⬘region required for paramutation is mostly unique or low copy in the maize genome. These results represent the first example of long-distance regulatory elements in plants and demonstrate that paramutation is mediated by long-distancecisandtransinteractions.
P
ARAMUTATION was first described at r1(Brink reduction in transcription causes a dramatic decrease 1956) and b1 (Coe1959) in maize. Subsequently, in pigment in all plant tissues where B-I is normally paramutation was observed at two other maize loci,pl1 expressed.B⬘and heterozygousB⬘/B-Iplants are lightly (Hollicket al.1995) andp1(SidorenkoandPeterson pigmented whereas B-I plants are darkly pigmented 2001). All of these genes encode regulatory proteins (Coe1966;Pattersonet al.1993). The change ofB-Irequired for flavonoid pigment synthesis. Paramutation toB⬘is heritable as only B⬘ is transmitted to progeny. andtrans-interactions have also been described for both These newB⬘alleles are equivalent to theB⬘parent; they endogenous genes and transgenes in other plant species retain the reduced transcription state and induce para-(reviewed inBrink1973;Chandleret al.2000) and in mutation ofB-Iat a frequency of 100%. The paramuta-fungi (Colot et al. 1996; van West et al. 1999). Al- genic (pg)B⬘state is extremely stable; spontaneous changes though generally not meiotically heritable, allele inter- from B⬘ to B-I have not been observed in ⵑ100,000 actions are involved in trans silencing in animals (re- plants (Coe1966;Pattersonet al.1995), while theB-I
viewed inHenikoff and Comai 1998) and have been state is very unstable; it spontaneously changes to B⬘
speculated to be involved in diabetes in humans (Ben- at frequencies of 1–10% (Coe1966; Patterson et al.
nettet al.1997). Allele interactions also appear to be 1995).
involved in a transposon excision-repair mechanism in Comparison ofB-IandB⬘DNA sequences and geno-Petunia (van Houwelingenet al. 1999). The mecha- mic restriction maps generated with DNA-methylation-nism is not understood for any system, although a herita- sensitive and -insensitive restriction enzymes revealed no ble change in chromatin structure is a favored model differences in DNA sequences, DNA rearrangements, for paramutation (reviewed inHollicket al.1997;Chan- or DNA methylation patterns within 10 kb spanning the
dleret al.2000). transcribed region (Pattersonet al. 1993, 1995).
How-In plants heterozygous forB-IandB⬘, the transcription ever, the different transcription states are associated of theB-Iallele is always downregulated to aB⬘transcrip- with a difference in chromatin structure. A DNaseI hy-tion level (Patterson et al. 1993). The 10- to 20-fold persensitive site was observed near the transcription start site that is more hypersensitive inB-Ithan inB⬘( Chan-dler et al. 2000). Recombination mapping showed that
1Present address:Department of Developmental Genetics, Vrije
Uni-the sequences required for paramutation are tightly versiteit, De Boelelaan 1087, 1081 HV Amsterdam, The Netherlands.
linked toB⬘andB-I (ⱕ0.1 cM) and located upstream
2Corresponding author:Plant Sciences Department, University of
Ari-zona, Tucson, AZ 85721. E-mail: [email protected] of the transcribed region (Patterson et al.1995).
fragments ofⵑ685 bp (tmp-I) inB⬘andB-Iorⵑ614 bp (tmp-P)
We report here results from two approaches to identify
inB-Peru(MS30, CACCATGCCTGGGCTGACC; Mtl-tmp, CCG
where the sequences required for paramutation are
lo-GCAGCATCTCGAAGC). PCR conditions were the following:
cated. First, we characterized ab1allele,B-615, that does 1⫻PCR buffer (Sigma, product no. P2192), 0.1 mmdNTPs, not participate in paramutation, but unlike all other 0.8mof each primer, 1 unit of Taq DNA polymerase (Sigma),
reaction volume of 25l; 2 min at 94⬚, 30 cycles (1 min at
knownb1alleles, it shares upstream sequences in
com-94⬚, 1 min at 62⬚, 3 min 30 sec at 72⬚), 10 min at 72⬚, 1 hr at 4⬚.
mon withB⬘andB-I. Second, we used fine-structure
recom-Plants carryinggl2 tmp-I B⬘andGl2 tmp-P B-Peruwere crossed
bination mapping to localize theB⬘sequences required
and the resulting F1crossed withgl2 tmp-I B-Iplants. To isolate for paramutation. In addition, we identified a distinct recombinantB⬘alleles, colorless seeds (B⬘/B-Igenotype) were region required forB⬘expression. planted in trays and 10-day-old seedlings scored for theirglossy2
phenotype (wild typevs.mutant have differences in wax con-tent, easily visualized in seedlings). The recombinant seedlings (Gl2) were transplanted to the field and organized in grids MATERIALS AND METHODS
containing 16 or 18 rows of 20 plants each. Leaf pieces were pooled for all plants in each row and in each column. DNA
Plant stocks: In maize nomenclature (http://www.agron.
was isolated as described (Dellaportaet al.1983), and PCR missouri.edu/maize_nomenclature.html), a gene is designated
analyses were done on all pooled samples to identify candidate with lowercase italics (b1). Specific alleles are indicated with
recombinants carrying thetmpallele linked toB-Peru. an allele designation separated from the gene designation
To isolate recombinantB-Perualleles, purple seeds (B-Peru/ with a hyphen, dominant alleles are uppercase (B-I), and
B-I) were planted in trays. Recombinant seedlings (gl2) were recessive alleles are lowercase (b-K55). Throughout the
manu-transplanted to the field and vegetative pigment scored at script, a single allele listing indicates homozygosity, whereas
maturity. Plants with lightly pigmented sheaths were carrying heterozygous individuals are indicated with allele designations
a paramutagenicB-Peruallele or the parentalB-Iallele under-separated by a slash (/). All plant stocks contained dominant
went spontaneous paramutation. To identify recombinants functional alleles for all of the anthocyanin biosynthetic genes
every light plant was tested for loss oftmp-P. The nomenclature required in vegetative plant and seed tissues. Stocks containing
used for recombinants is: n, neutral; pg, paramutagenic; , variousb1alleles have been maintained in the Chandler
labo-recombinant allele with the parental allele contributing se-ratory. They were originally obtained from various sources: E. H.
quences upstream of the breakpoint indicated to the left of the Coe, Jr. (University of Missouri, Columbia) providedB-I(inbred
vertical bars and the parental allele contributing the coding W23 background), B⬘ (inbred K55 background), andgl2 b
region and promoter-proximal region indicated on the right. wt (inbred K55 background); M. G. Neuffer (University of
To follow recombinants in crosses, a PCR assay was used to Missouri, Columbia) provided B-Peru (inbred W22
back-specifically amplify a 427-bp fragment oftmp-P(primers: MS32, ground); and S. Evola (Ciba-Geigy, now Syngenta) provided
CTGTTGCTAACTAGTTTCACTGTTAA and MS33, GGCACA B-615. Thegl2(glossy2) andwt(white tip) loci are
morphologi-AATCGGTGAGAGTGAGAG). Primers amplifying thenpi402 cal markers that flankb1. TheB-615allele was in a proprietary
marker were used as an internal control (MS22, ACACGGCA line (Selingeret al.1998). We were using this line as a
recipi-CCTACGTATAAGG and MS23, CTGCACTCTCCGATAGAA ent for transformation and noticed that it carried ab1allele
with the same restriction map asB⬘, which was unusual. Be- TGG; 591-bp fragment that maps 1.1 cM upstream of b1; http://www.agron.missouri.edu/maps.html). PCR conditions cause theb1andr1loci encode functionally duplicate genes,
our stocks carriedr1alleles (r-gorr-r) that are not expressed were the following: 1⫻PCR buffer (Sigma, product no. P2192), 1l MgCl2 [for PCR, QIAGEN (Chatsworth, CA)], 0.2 mm
in the seed or in the same juvenile and mature plant tissues
asB-I,B⬘, andB-Peru. dNTPs, 0.8mof MS32 and MS33, 0.2mof MS22 and MS23, 1 unit of Taq DNA polymerase (Sigma), reaction volume of
Analyses of RNA levels and transcription rates:RNA was
isolated and RNA levels assayed by RNase protection experi- 25l; 2 min at 94⬚, 35 cycles (1 min at 94⬚, 1 min at 62⬚, 1 min 20 sec at 72⬚), 10 min at 72⬚and 4⬚.
ments as previously described (Dorweileret al.2000).
Tran-scription rates were assayed using nuclear run-on analyses as Isolation of pBACBⴕ1 clone:To clone theⵑ100-kb genomic MluI fragment, the pBeloBAC11 vector (Kimet al.1996) was previously described (Dorweileret al.2000).
DNA blot analyses:Standard blot analyses were done with modified to contain a uniqueMluI site by cutting withBamHI
andHindIII and ligating with a phosphorylatedBamHI-Mlu
I-ⵑ4g genomic DNA from leaves (Dellaportaet al.1983)
digested with several enzymes and enzyme combinations ac- AatII-HindIII linker (GATCCACGCGTGACGTCAAGCT), cre-ating the pBeloBAC-Mlu vector. This vector was purified, cut cording to manufacturers’ specifications and size fractionated
by electrophoresis in 0.5⫻TBE agarose gels followed by DNA withMluI, and dephosphorylated as inBirrenet al.(1999). To remove the background of nondephosphorylated ends, blot analyses (Stamet al. 1997). For blots with
large-molecular-weight DNA, high-molecular-large-molecular-weight DNA was isolated (Kaszas the DNA was ligated in bulk (Osoegawa et al. 1998) and separated on a pulsed-field agarose gel, and linear DNA was andBirchler1998), the resulting plugs were incubated with
the desired restriction endonuclease(s), and DNA was sepa- isolated from SeaPlaque low-melting-point agarose using wiz-ard PCR preps (Promega, Madison, WI).
rated by pulsed-field gel electrophoresis (PFGE) using a
CHEF-DRIII apparatus (Bio-Rad, Richmond, CA), in 0.5⫻ Nuclei were isolated fromB⬘ plants according to Kaszas
andBirchler(1998). Small DNA was removed from agarose TBE, 1% agarose gels [Sigma (St. Louis) A2929].
Recombinant screen: To isolate recombinant alleles be- plugs with PFGE [6 V cm⫺1, 10-sec pulse (constant), 120⬚
angle, 14⬚, 1% agarose, 0.5⫻ TBE for 3 hr] using a CHEF-tweenB⬘andB-Peru, we used the phenotypic markergl2, located
19 cM upstream (http://www.agron.missouri.edu/maps.html), DRIII apparatus (Bio-Rad). The plugs (0.1 ml, containing
ⵑ10–20g of DNA) were incubated with the desired restric-and the tightly linked gene tmp (transmembrane protein),
located 0.18 cM upstream of theb1transcription start (Stam tion endonuclease and PFGE was performed essentially as described (Osoegawa et al. 1998). Fragments smaller than et al.2000). Thetmpalleles of thegl2 B⬘andgl2 B-Istocks were
identical, but polymorphic relative to theGl2 B-Peru tmpallele ⵑ50 kb were removed by running the DNA toward the top of the gel [5 V cm⫺1, 15-sec pulse (constant), 120⬚angle, 14⬚,
in the third intron of tmp. Primers that amplified the third
conditions. The remaining DNA was separated at 6 V cm⫺1, digested genomic maizeB⬘ DNA. The 275 clones that gave
no or a very weak hybridization signal were rearrayed onto 2.7- to 20-sec pulse, 120⬚angle, 14⬚, 0.5⫻TBE for 16 hr. DNA
(ⵑ100 kb) was concentrated by running it into 4% agarose five sets of three Hybond N⫹filters. These filters were hybrid-ized with all the previously identified single copy fragments followed by electroelution for 41 hr at 4⬚ as described by
(Osoegawaet al.1998). from the regions 0–8 kb (Pattersonet al. 1995; Figure 2)
and 94–106.6 kb (Figure 7) upstream of theB⬘coding region. The dephosphorylated vector and genomic DNA were
li-gated overnight (room temperature) in a 1:5 molar ratio (Bir- They were also hybridized with digested genomic maizeB⬘ DNA a second time and with labeled pBeloBAC-Mlu DNA or
renet al.1999) followed by dialysis against sterile H2O and
the DNA was concentrated by incubation in 30% PEG8000, DraI-cut genomicE. coliDNA. The empty vector clones were identified by restriction digestion and gel electrophoresis. To 0.5⫻TE buffer.Escherichia coliDH10B cells were transformed
according toBirrenet al.(1999) using the electro cell manipu- estimate the actual repetitiveness in the maize genome of the remaining 95 nonhybridizing clones, inserts were PCR lator ECM 600 (BTX). A total of 10,584 colonies were arrayed
using the Biomek 2000 laboratory automation workstation amplified, radioactively labeled, and hybridized to filters con-tainingBglII andEcoRI,SalI double-digested genomicB⬘DNA. (Beckman, Fullerton, CA) essentially as described (Birrenet
al.1999). The resulting filters were hybridized with aB⬘pro- To position the fragments onto the pBACB⬘1, the plasmid DNAs of the 95 clones were sequenced by the Arizona GATC moter probe (SpeI/SalI, 1486–522 bp upstream of the start
site) radioactively labeled using random hexamer priming facility and aligned relative to the pBACB⬘1 sequence using BLAST 2 sequences (http://www.ncbi.nlm.nih.gov/gorf/bl2. (FeinbergandVogelstein1983). The results were visualized
using a Storm 860 phosphorimager and ImageQuant software html) and FAKtory, a sequence assembly program (http://bcf. arl.arizona.edu/faktory/).
(Molecular Dynamics, Sunnyvale, CA). One positive colony,
pBACB⬘1, was identified. DNA of this colony was prepared by Sequencing promoter-proximal region ofB-615:The pro-moter-proximal region ofB-615was amplified with the prim-alkaline lysis (Birrenet al.1999) and verified to be the desired
fragment by direct end sequencing. ers B-I1086ds, CGGCGAATTCGTGAGATGGTAGATTGGTT GCGAC (anneals 1063 bp 5⬘of transcription start) and BPSac-Multiple restriction digestions using single or multiple
en-zymes were done to identify the approximate number of re- US, GGAGGAGGAGCTCACCTCCGCT (anneals 93 bp 5⬘of transcription start). The PCR product was sequenced by the striction sites present and their positions relative to each other.
To order restriction fragments within the pBACB⬘1 clone, the Arizona GATC facility using the primers B-I1086ds, BPSac-US, TMDDE1 (GCCGAATTCGACTAACCTTAGGCAAAGTG), and insert was excised from the vector withNotI, followed by partial
restriction with a dilution series of a second enzyme. DNA B-I Ddeus (CGGCGGATCCTGTCACACTTTGCCTAAGGTTA GTC). The accession number for this sequence is AF205792. blots were hybridized with bacterial artificial chromosome (BAC)
end probes. Sequencing of pBACBⴕ1 clone:DNA from pBACB⬘1 was
ex-tracted using the large construct kit (QIAGEN) and sheared
Inverse polymerase chain reaction:AdditionalB⬘sequence,
upstream of the most 5⬘ MluI site in the pBACB⬘1 clone, was with a hydroshear device (Genemachines) to two different average sizes, 2 and 9 kb. The ends of the sheared fragments isolated by inverse polymerase chain reaction (IPCR). Genomic
B⬘DNA was cut withHinfI orBstYI followed by ligation (Snow- were made blunt with mung-bean exonuclease, extracted with a mixture of phenol and chloroform followed by chloroform
den and Napoli 1998). IPCR was performed, in turn, on
200 ng of theHinfI orBstYI ligation mixtures. The resulting extraction, and precipitated with isopropanol. The DNA was dephosphorylated with shrimp alkaline phosphatase, and “A” fragments were cloned into pGEM-T Easy according to the
manufacturer’s recommendation (Promega) and sequenced tails were added by incubation withTaqpolymerase, run on a gel, and eluted using the gel elution kit (QIAGEN). Inserts by the Arizona GATC facility. Primer sets used were VC28:
GAGCACCGAGCGGGGGTAGTACGTCG and VC29: AGTC were then ligated into pCR4-TOPO using the TA cloning system (Invitrogen, San Diego) and electroporated intoE. coliDH10B GCAGCGCGCCATTGGAAGAGTC; VC41: GTCAAATGGAC
GGTCGAACAAACATTCAG and VC42: CGAGCGTGAATAC electrocompetent cells (GIBCO BRL, Gaithersburg, MD). Sub-clones were sequenced from both directions using big dye TTTTTCTGATATCTAG; VC61: GATCTTGAAGTTAGCCTA
AACAAGATC and VC62: CAAGTTTGAACTATATCTTTACC terminator chemistry and run on an ABI3700 capillary se-quencer. Base calling and quality assessment were done using TTCACTC; VC89: GAAGCAACTTTACTGGATCGAGGG and
VC90: GGATATTCATGCATAACCATTGCTTC. A total of 3.9 kb PHRED (EwingandGreen1998), the sequences were assem-bled by PHRAP, and the resulting contigs edited with CON-of additional upstreamB⬘sequences was obtained (accession
no. AF475145). SED (Gordonet al.1998). To close gaps between the contigs and to go through GC-rich regions, a combination of different
Shotgun library construction for low-copy probe isolation:
The pBluescript SKII⫹vector was cut withEcoRV, dephosphor- approaches was used: (1) sequencing using different chemis-tries, (2) sequencing using thermofidelase enzyme, (3) PCR ylated, self-ligated, and subjected to agarose gel
electrophore-sis, and the linear vector fragment was excised, isolated from amplification with primers flanking a gap, (4) shotgun se-quencing of transposon-inserted subclones flanking a gap, agarose by phenol extraction, and resuspended in TE. pBACB⬘1
DNA was isolated according toBirrenet al.(1999), purified and (5) direct sequencing of the BAC template. When gaps were associated with repetitive regions, subclones that start by CsCl gradient ultracentrifugation, and sheared by
nebuliza-tion for 2.5 min at 10 psi essentially as described (Wilson and end in unique regions but otherwise consist of repetitive DNA were assembled separately and inserted in the main andMardis1997). Fragment ends were repaired by treating
with T4 DNA polymerase, Klenow, and polynucleotide kinase. assembly. The final error rate was estimated using CONSED. The accession number for the BAC sequence is AY078063. DNA fragments ofⵑ800 bp were gel purified (Osoegawaet
al.1998) and 6 ng were ligated to 5 ng of vector DNA, dialyzed, Assembly programs suggested three tandem repeats in the upstream region, but restriction mapping indicated seven re-concentrated, and transformed into E. coli DH10B cells as
described above. peats. To obtain the complete sequence of this region, a 9-kb shotgun fragment spanning the repeats was cloned into pCR4-A total of 631 white colonies were picked, arrayed, and
repli-cated as described (Birrenet al.1999) onto Hybond N⫹filters TOPO (Invitrogen) and a series of deletion derivatives were generated. The plasmid DNA was digested withSdaI, which using the Biomek 2000 laboratory automation workstation
double-nuclease III, with aliquots removed at 2-min time points. The reactions were stopped by heat incubation, the DNA was treated with mung bean nuclease to create blunt ends, and the vector was self-ligated following the manufacturer’s in-structions (Invitrogen). The DNA was used to transformE. coli (strain DH10B) and the resulting plasmids were sized on agarose gels. Clones with increasing deletions were then sequenced and the information assembled as described above.
RESULTS
Characterization of theB-615allele suggests that
se-quences required for paramutation are ⬎13 kb
up-stream of the coding region: The vast majority of b1
alleles do not participate in paramutation, but they have 5⬘sequences that are distinct from those ofB-IandB⬘
(Radicellaet al.1992;SelingerandChandler1999). Initial DNA blot analyses suggested that theB-615allele was an exception, because digestion with several en-zymes revealed the same map asB⬘,B-I(the maps ofB⬘
andB-Iare identical) within at leastⵑ12 kb upstream of the coding region. This was intriguing becauseB-615
does not participate in paramutation; when B⬘/B-615
plants are crossed withB-I, dark (B-I/B-615) and light (B⬘/B-I) pigmented plants segregate 1:1, indicating that
B-615is insensitive (neutral) to paramutation (K.Kubo, M.Stamand V.Chandler, unpublished data). We hy-pothesized a more thorough investigation of its expres-sion and DNA sequence relative toB⬘might reveal differ-ences, which would be candidates for requirements for paramutation.
Because paramutation involves an alteration in tran-scription, one possibility was that transcription from the same promoter-proximal sequences might be required for paramutation. In the original inbred genetic
back-Figure1.—B-615andB⬘specify similar levels of
transcrip-ground, theB-615allele gave rise to green plants, poten- tion. (A) An RNase protection assay on husk RNA usingb1 tially because it is not transcribed. Alternatively, a very andactin1RNA probes. The samples containing a probe
with-out the addition of maize RNA show the size of the probes
weak or null pl1 allele could be present (functional
before digestion with RNaseT1. (B) The bar graph shows the
alleles ofpl1and eitherb1orr1are required to activate
b1transcript levels normalized toactin1transcript levels from
the anthocyanin biosynthetic pathway in plant tissues;
each of the lanes in A (indicated by micrograms of RNA). (C)
Coe1985). To investigate this,B-615was introgressed An example of a run-on transcription assay with nuclei isolated into a b-K55 r-r Pl-Rh background by backcrossing six from husk tissue showing the signals forb1,c2(an anthocyanin biosynthetic gene regulated by b1), and ubiquitin2 (ubi) in
times. Ther-rallele is expressed in the anthers, which
B-615, B-I, and B⬘ individuals. (D) Normalization of the b1 allows scoring of thepl1allele independently of B
func-andc2signals to theubiquitin2signal from two experiments.
tion by monitoring anthocyanin pigment levels in
an-Results were visualized and quantified using a Storm 860
phos-thers. Pl-Rh is a fully functionalpl1allele (Cone et al. phorimager and ImageQuant software (Molecular Dynamics).
1993), producing dark purple anthers in the presence of r-r. The resulting purple anthered plants, B-615/
b-K55 r-r Pl-Rh, had weakly pigmented sheaths, similar DNA, using multiple enzyme combinations (BamHI,
BclI,BglII, EcoRI,EcoRV,HindIII, KpnI, PacI, SacI,XbaI, to B⬘, suggesting thatB-615 might be transcribed at a
level similar to that ofB⬘. To test this hypothesis, mRNA BamHI/BglII,BamHI/EcoRI,EcoRI/BglII, PstI/BglII,XbaI/
EcoRI,XbaI/HindIII, andXbaI/BglII) and coding and up-(Figure 1, A and B) and transcription levels up-(Figure 1,
C and D) were compared in husk tissues fromB-615and stream sequences as probes, detected no differences between the two alleles withinⵑ13 kb upstream of the
B⬘. The promoter regions of these two alleles conferred
similar transcription levels. Thus, the different paramu- transcription start site (map in Figure 2). An 1154-bp promoter-proximal fragment fromB-615was PCR ampli-tation properties ofB-615andB⬘are not caused by
differ-ences in transcription level. fied and sequenced (accession no. AF205792). The
Figure2.—TheB⬘, B-Ialleles have the same restriction map asB-615up toⵑ13 kb upstream of the transcription start site. All indicated restriction sites were tested by DNA blot analyses and are located at the same position in theB-615allele as in the B⬘andB-Ialleles. The open, numbered boxes are exons. The restriction sites in the region indicated by the solid bar (including exons positioned on this bar) were verified by sequencing ofB⬘ and/orB-Icloned genomic DNA. The restriction sites in the region indicated by the shaded bar were determined by DNA blot analyses on genomicB-IandB-615DNA. The DNA fragments used as probes are indicated. The region indicated by the striped box is the promoter-proximalB-615region that was sequenced (accession no. AF205792). The sequence is exactly the same as theB⬘,B-Isequence (accession no. X70790). The restriction sites are B,BamHI; Bc,BclI; E,EcoRI; EV,EcoRV; G,BglII; H,HindIII; K,KpnI; P,PstI; S,SacI; X,XbaI.
B⬘andB-I. Thus, either very subtle differences between sequences. BecauseB⬘ always paramutates B-I, all B⬘/B-I
progeny should be lightly pigmented except when
re-B⬘,B-I, andB-615in the 13-kb promoter-proximal region
mediate paramutation or the sequences are located far- combination separatesB⬘paramutation sequences from theB⬘promoter-proximal region, creating an altered B⬘
ther upstream.
Isolation of recombination-derived neutralBⴕalleles: allele that can no longer paramutateB-I. Such an allele
would produce a dark plant when heterozygous withB-I. Previous studies had shown that the sequences required
for paramutation were upstream of the coding region In the initial test of this approach, ⵑ9000 colorless seeds were planted and the resultingⵑ6500 plants scored (Pattersonet al.1995). Our comparison ofB-615with
B⬘suggested that the sequences required for paramuta- for mature plant pigment. All but 2 plants were lightly pigmented. The 2 darkly pigmented plants were candi-tion might reside⬎13 kb upstream. To determine more
precisely where the sequences required for paramuta- dates for rare recombinants that had replaced the B⬘
sequences required for paramutation with the neutral tion mapped, we employed a fine-structure
recombin-ation mapping approach. To facilitate the isolrecombin-ation of sequences fromB-Peru. A third recombinant was isolated in a separate experiment in which 1006B⬘/B-I(colorless) recombinants we usedB-Peru, which is insensitive
(neu-tral) to paramutation and produces a nearly green plant seeds derived from a cross between B⬘/B-Peruand B-I/ B-Peruplants were planted in the field. Among theⵑ650 and purple seed. In contrast, the paramutagenicB⬘
al-lele produces a lightly colored plant and colorless seed. plants that survived to maturity, 1 dark plant was identi-fied. PCR analyses using a polymorphic molecular The immediate upstream 2.5-kb promoter-proximal
re-gion ofB-Perucontains a unique sequence that confers marker 0.18 cM upstream ofb1(Stamet al.2000;tmp) demonstrated that all 3 were recombinants, as they car-seed pigmentation (Radicellaet al.1992;Selingeret
al.1998). The seed pigment and unique 2.5 kb inB-Peru ried the upstream molecular marker linked to B-Peru
and the B⬘ promoter-proximal and coding sequences relative toB⬘provide visual and molecular markers to
distinguish the promoter-proximal region ofB⬘andB-Peru (data not shown).
The recombinants were outcrossed withB⬘andB-Ito in crosses.
We hypothesized that there are distinct upstream se- determine their ability to participate in paramutation and were self-pollinated and outcrossed to recessive b1
quences inB⬘required for paramutation and that these
sequences are either missing or mutated inB-Peru. We alleles to investigate their pigment phenotype. All three recombinant alleles were neutral, as all dark purple further hypothesized that exchange of upstreamB-Peru
and B⬘ sequences via recombination would generate a (B-I-like) plants (98) and no light purple (B⬘-like) plants (55) carried recombinant alleles; the three alleles were neutral B⬘ allele, which we could easily identify as
de-scribed below. Figure 3 shows the phenotypes and cross- designated BPB⬘-n1, BPB⬘-n2, and BPB⬘-n3, with n
symbolizing neutral. The indicates a recombination ing strategy used. B⬘/B-Peru plants were crossed with
plants carryingB-I, the allele sensitive to paramutation event, the allele on the right indicates the promoter-proximal and coding region, and the allele on the left (colorless seeds and dark plants); colorless and dark
seeds segregated 1:1. Planting colorless seeds selects indicates the sequences upstream of the recombination breakpoint.
Figure4.—Polymorphisms betweenB⬘,B-Peru,B-615, and three neutralBPB⬘alleles revealed byMluI digestion. High-molecular-weight DNA was cut withMluI, separated by PFGE, and blotted. The blot was hybridized with the S/G700 probe (see Figure 2). Figure 5 shows restriction maps for the different alleles.
sites. It was difficult to map restriction sites far upstream in theB-Peru parental allele for two reasons. First, the most upstream 5⬘G probe did not hybridize well toB-Peru
DNA, and second, a large insertion of ⵑ30 kb is just Figure3.—Diagram illustrating the crossing strategy to
iso-upstream of theB-Perupromoter (Pattersonet al.1995;
late recombinantB⬘alleles that had lost the ability to cause
paramutation.B⬘plants (light pigmented plant, colorless ker- Selingeret al. 1998), which contained sites for several
nels) were crossed with B-Peru plants (nearly green plant, of the restriction enzymes used for mapping. Thus, we purple kernels). The resulting seeds (B⬘/B-Peru; purple ker- primarily compared the recombinants relative to B⬘. nels) gave rise to B⬘ colored F1 plants. The F1 plants were
The recombination breakpoints were assumed to have
crossed toB-Iplants (dark pigmented plant, colorless kernels),
taken place between the most upstream conserved site
giving rise to an ear segregating colorless (B⬘/B-I) and purple
(B-Peru/B-I) kernels. When the colorless seeds were planted, and the first polymorphic site relative toB⬘. Restriction
the vast majority of the resulting plants showed a B⬘ plant maps representing the key sites delimiting these recom-phenotype (B⬘/B-I⬘ plants; the paramutation of B-Iin these
bination intervals are shown in Figure 5. Two neutral
plants is indicated asB⬘*). Two dark individuals were isolated
alleles retain the SwaI site 50 kb upstream of the B⬘ in which theB-Iallele was not paramutated. TheB⬘allele in
transcription start site, but instead of the ⵑ100-kb B⬘ these individuals is neutral for paramutation (B⬘-n).
MluI fragment, they have anⵑ85-kbMluI fragment (Fig-ures 4 and 5). This positions the recombination
break-Sequence polymorphisms between Bⴕ, B-Peru, BP points in BPB⬘-n2andBPB⬘-n3between 50 and 100
Bⴕ-nalleles, andB-615:As a first step toward determining kb upstream of the coding region. The recombination
where the recombination breakpoints occurred, sequence breakpoint in theBPB⬘-n1allele could be pinpointed polymorphisms were mapped between B⬘, B-Peru, and more accurately because it occurred close to the B⬘
the BPB⬘-n alleles. Genomic DNA was isolated from transcription start, between 8.4 and 8.9 kb upstream plants containing each allele, digested with different (Figure 5). TheB-615allele retained theB⬘restriction enzymes, and subjected to conventional gel electropho- map up to 22 kb upstream of the transcription start site, resis or PFGE and DNA blot analysis using different but lacked theSwaI site 50 kb upstream and gained a probes.B-615was included in the analysis to determine SnaBI polymorphism relative toB⬘. The observation that more precisely where differences betweenB⬘and B-615 BPB⬘-n2andBPB⬘-n3retained 50 kb ofB⬘sequences, begin. A blot withMluI-digested DNA fromB⬘,B-Peru, yet are neutral with respect to paramutation, indicates
B-615, and the threeB⬘-neutral recombinants illustrates that the sequences up to 50 kb upstream of theB⬘ tran-one of the polymorphisms observed (Figure 4). We sus- scription start site are not sufficient for paramutation. pect the doublet seen with the recombinants and B⬘ The results withB-615are also consistent with sequences reflect partial digestion caused by DNA methylation, as farther upstream being required for paramutation.
MluI is methylation sensitive.MluI cuts once in theB⬘ Isolation of recombinant alleles delimiting the
se-quences required for paramutation: Isolation of the
Figure 5.—Restriction maps of B⬘, B-Peru, three neutral BPB⬘ alleles, and B-615. The restriction maps were generated by restric-tion digesrestric-tion of genomic DNA using multiple differ-ent enzymes, followed by gel electrophoresis and DNA blot analyses using the three probes indicated at the top. The exon probe was a 962-bp SacI cDNA fragment span-ning exons 7 and 8. The pre-cise locations of the 5⬘G and S/G700 probes are shown in Figure 2. The solid box represents the transcribed region of b1. B-Peru has a large insertion ofⵑ30 kb rela-tive toB⬘, indicated by the tri-angle. Within the triangle, re-striction sites (not shown) precluded mapping the po-sition of additional sites far-ther upstream in B-Peru. The shaded ovals indicate the intervals in which re-combination took place or where polymorphisms were detected (B-615). The re-striction sites indicated are B,BamHI; G,BglII; H, Hin-dIII; L,SalI; M,MluI; P,PstI; Sn,SnaBI; Sw,SwaI; X,XbaI.
three neutral B⬘ alleles indicated that sequences re- interval II recombinants. However, a few light plants could result fromB-Ispontaneously paramutating toB⬘, quired for paramutation could be further defined by
isolating more recombinants. Additional mapping ex- which would obscure the presence of interval I recombi-nants. To test for paramutagenicity, each independent periments were performed withB⬘andB-Peruusing the
strategy outlined in Figure 6. Three recombination in- recombinant allele was either crossed directly to B-I
or first self-pollinated and the resulting homozygous tervals can be distinguished between gl2 and the b1
coding region. Recombination betweenB⬘ andB-Peru recombinants were crossed toB-I. All 30BPB⬘ recombi-nants that produced viable seed were paramutagenic in interval I, downstream of P (sequences required for
paramutation), should generate B⬘ neutral alleles (as (Table 1).
We used a slightly different strategy with the purple previously isolated) andB-Peru pg alleles.
Recombina-tion events in intervals II and III will have the parental seed. From 19,267 purple seed that germinated, 2850 recombinants were identified betweenb1andgl2.These paramutation phenotypes (colorless seeds will have
paramutagenic alleles, and purple seeds neutral alleles), were transplanted to the field and scored for dark pur-ple vs. light purple pigment in the vegetative part of but they can be distinguished from each other using
thetmpmolecular marker (materials and methods). the plant. The lightly pigmented individuals (168) ei-ther were paramutagenicB⬘BPalleles or were carrying A total of 21,027 colorless seeds were planted,
seed-lings were scored for recombination between gl2 and a neutral B⬘BP allele and were light because the B-I
allele with which they were heterozygous had
spontane-b1, and the resulting 3332 recombinants were assigned
to one of the three intervals using thetmp molecular ously changed toB⬘. Screening the light purple plants by PCR for the tmpmarker identified 3 recombinants marker and genetic tests for paramutation. The
molecu-lar markers classified 32 recombinants to intervals I or in intervals I or II, which were then tested for their paramutation properties by crossing withB-I. One allele, II and 3300 recombinants to interval III. These numbers
do not include the three previously isolated interval I B⬘BP-n1, was a neutral interval II recombinant, while the other two light B⬘BP alleles were paramutagenic
B⬘neutral alleles. All 32 candidates for intervals I or II
showed a B⬘ phenotype when heterozygous with B-I, interval I recombinants.
TABLE 1
Paramutation properties of recombinant alleles
No. of
Allele Paramutation plants Recombination name phenotypea testedb site (kb)
BPB⬘-n1 Neutral 52 ⵑ9
BPB⬘-n2 Neutral 8 50–94
BPB⬘-n3 Neutral 38 50–94
BPB⬘- pg1 Paramutagenic 46 107–110
BPB⬘- pg2 Paramutagenic 29 107–110
BPB⬘- pg3 Paramutagenic 48 107–110
BPB⬘- pg4 Paramutagenic 44 107–110
BPB⬘- pg5 Paramutagenic 28 ⬎110
BPB⬘- pg6 Paramutagenic 51 ⬎110
BPB⬘- pg7 Paramutagenic 39 ⬎110
BPB⬘- pg8 Paramutagenic 12 ⬎110
BPB⬘- pg9 Paramutagenic 20 ⬎110
BPB⬘- pg10 Paramutagenic 25 ⬎110
BPB⬘- pg11c Not tested 0 ⬎110
BPB⬘- pg12 Paramutagenic 29 ⬎110 BPB⬘- pg13 Paramutagenic 15 ⬎110 BPB⬘- pg14 Paramutagenic 21 ⬎110 BPB⬘- pg15 Paramutagenic 18 ⬎110 BPB⬘- pg16 Paramutagenic 26 ⬎110 BPB⬘- pg17 Paramutagenic 14 ⬎110
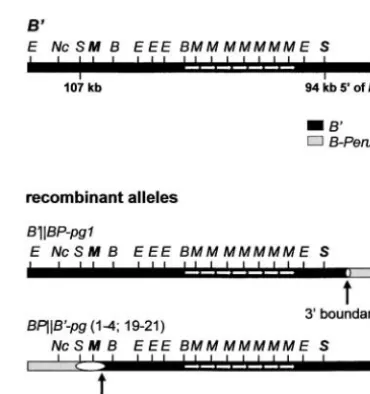
Figure6.—Recombination mapping strategy. To identify
BPB⬘- pg18 Paramutagenic 25 ⬎110 the 5⬘and 3⬘boundary of the region containing the
paramuta-BPB⬘- pg19 Paramutagenic 23 107–110 tion sequences, recombination experiments were performed
BPB⬘- pg20 Paramutagenic 19 107–110 using theB⬘andB-Perualleles. The solid box (P) represents
BPB⬘- pg21 Paramutagenic 26 107–110 the hypothetical sequences required for paramutation. The
BPB⬘- pg22 Paramutagenic 11 ⬎110 phenotypic markergl2, locatedⵑ19 cM upstream ofb1, and
the molecular markertmp(located 0.18 cM upstream ofb1) BPB⬘- pg23 Paramutagenic 34 ⬎110 were used to identify the recombinants. Both markers are BPB⬘- pg24 Paramutagenic 16 ⬎110 polymorphic betweenB⬘andB-Peru, which is indicated by the BPB⬘- pg25 Paramutagenic 18 ⬎110 differently shaped symbols. The three different recombination BPB⬘- pg26 Paramutagenic 31 ⬎110 intervals that can be distinguished between gl2 and the b1 BPB⬘- pg27c Not tested 0 ⬎110
coding region are indicated. TheB-Peru promoter-proximal BPB⬘- pg28 Paramutagenic 42 ⬎110 region contains sequences directing purple seed color (sc), BPB⬘- pg29 Paramutagenic 26 ⬎110 which are lacking at the B⬘ promoter-proximal region, re- BPB⬘- pg30 Paramutagenic 46 ⬎110 sulting in colorless seeds. These sequences provide a visual and BPB⬘- pg31 Paramutagenic 25 ⬎110 molecular marker to distinguish theB⬘andB-Perupromoter- BPB⬘
- pg32 Paramutagenic 24 ⬎110
proximal region.n, neutral; pg, paramutagenic; , recombi- B⬘
-BP- pg1d Paramutagenic 226 ⵑ10–94
nant allele with the parental allele contributing the upstream B⬘
-BP- pg2 Paramutagenic 146 ⵑ1 sequences indicated on the left side of the vertical bars and the
B⬘-BP-n1d Neutral 38 ⬎110
parental allele contributing the coding region and
promoter-proximal region indicated on the right side. aEach recombinant allele was tested for its paramutation
properties. The paramutagenic B⬘ allele was crossed with plants homozygous for each recombinant allele and the
re-transferred toB-Peru, this allele becomes paramutagenic; sulting F
1was crossed with the paramutableB-Iallele.B⬘/B-I it is fully capable of paramutatingB-I in trans. However, progeny are always lightly pigmented because of paramuta-the aleurone color remains dark in paramuta-the paramutagenic tion. Plants that were recombinants/B-Iwere identified with molecular markers: if they were light, the recombinant allele B-Perualleles. Thus, although theB⬘BP- pgalleles have
became paramutagenic; if they were dark, the recombinant
light plant pigment and paramutateB-I, causing
herita-allele did not become paramutagenic;i.e., it is neutral.
ble, reduced expression throughout the plant, the seed bThe number of plants with the recombinant allele that expression directed by theB-Peru promoter is not de- were tested in the genetic experiments.
tectably affected. cBPB⬘- pg11and-27were tentatively identified as
paramuta-genic alleles because when heterozygous withB-I, the plants
Cloning of an ⵑ100-kb fragment upstream of the Bⴕ
were lightly pigmented. However, the paramutagenic
geno-coding region: Additional upstream sequences were
type could not be confirmed in subsequent crosses because
needed to map the recombination breakpoints more
pre-no viable seeds were obtained.
cisely. PFGE analyses onB⬘DNA indicated that in geno- dBecause B-Peru has a large insertion downstream of the mic DNAMluI cuts in theB⬘coding region andⵑ100 kb recombination site, its position is defined relative to theSwaI site, set at 94 kb upstream of theB⬘transcription start site.
Figure7.—Sequence features of theⵑ107-kb region upstream of theB⬘transcription start site. The top map is an expanded version of the DNA regionⵑ100 kb upstream of theB⬘transcription start site. The tandem repeats shown with arrows are not repeated elsewhere in the maize genome. The individual boxes on this map are restriction fragments tested for repetitiveness using maize genomic DNA blot analyses. The large map shows the repetitiveness of various other regions within the BAC; the individual boxes are contigs of shotgun clones with a similar degree of repetitiveness in the maize genome (tested with DNA blot analyses) and are placed on the restriction map using sequence assembly programs. For both maps, open boxes represent unique or low-copy-number sequences in maize genomic DNA (a single or a few restriction fragments on DNA blots), lightly shaded boxes represent intermediate repetitive DNA (light smear or discrete, multiple fragments visible on blots), and darkly shaded boxes indicate highly repetitive DNA (results in black smear when used as probe on maize genomic DNA blots). The arrow at 0 kb represents theB⬘transcribed region and transcription start. The complete BAC sequence was used to search the National Center for Biotechnology Information database. Regions of similarity to transposable elements, aZ. maysroot EST, and two ORFs are indicated below the restriction map with stippled bars. ORF1 is similar to part of a hypothetical rice protein, and ORF2 is similar to part of a validated Arabidopsis phosphatidyl inositol 4-phosphate 5-kinase. Two small regions flanking the zeon element show similarity to part of the maize starch synthase I precursor (accession no. AF036891). Inverted and direct repeats are indicated with horizontal arrows below the map. The direct repeats (Dr1 and Dr2) areⵑ200 bp and each inverted repeat is 385 bp. The restriction sites shown are B,BamHI; E,EcoRI; M,MluI; S,SwaI.
as described inmaterials and methods. Direct end clones containing sequences that were either unique or low copy number in the maize genome were identified sequencing showed that one end of the BAC contained
theB⬘coding sequences upstream of theMluI site. Re- (materials and methods).
Shotgun sequencing of pBACBⴕ1 clone:To assist with
striction digestions with eight different enzymes
fol-lowed by PFGE showed the fragment sizes expected all subsequent analyses, the pBACB⬘1 clone was com-pletely sequenced. Shotgun library preparation, sequenc-from genomic DNA blot analyses and the previously
sequenced upstream B⬘ sequences (data not shown), ing, finishing the complete sequence, and sequence analysis of pBACB⬘1 were done as described (
Dubcov-indicating that the correct clone was isolated and that
it was not rearranged or chimeric. Several different ap- sky et al. 2001). Most of the sequences up to 80 kb upstream ofB⬘were related to transposons (Figure 7). proaches were used to generate a restriction map of the
pBACB⬘1 clone (materials and methods). This region contained five full-length long terminal re-peat (LTR) retrotransposons (class I elements);Ji,Opie, Identification of single- and low-copy-number regions
in the pBACBⴕ1 clone:To localize the breakpoints in the PREM-2,Zeon, andHuck.Ji,Opie, andPREM-2were
in-serted into a partialJielement and an additional, partial, recombinant alleles, additional probes from the pBACB⬘1
clone suitable for DNA blot analyses,i.e., fragments of Opiefragment (Opie-p) was inserted into the full-length
Opie element. The retrotransposons were flanked by low copy number in maize genomic DNA, were isolated.
Previous PFGE experiments (data not shown) showed sequences related to transposons that move via DNA intermediates (class II elements). These include a re-that the most upstream region of the 100-kb clone was
hypomethylated, suggesting that it might contain se- gion homologous to the transposase ofDoppia4 (acces-sion no. AF187822) and a transposon homologous to quences that were either genic or low copy in the maize
genome. The pBACB⬘1 clone was cut to completion the MuDR element of maize (D. Lisch, D. Selinger
and M.Stam, unpublished results). Two small regions withSwaI and religated, resulting in a clone containing
3.6 kb upstream of theMluI site in theB⬘coding region flanking theZeonelement were homologous to part of the maize starch synthase I precursor (accession no. and 12.6 kb downstream from the other end of the
clone. Subclones were isolated and sequenced, and sin- AF036891). The most upstream 20 kb of pBACB⬘1 con-tained a 108-bp region homologous to part of an Arabi-gle copy regions were identified using DNA blot analyses
(Figure 7). To identify additional single and low-copy dopsis phosphatidyl inositol 4-phosphate 5-kinase [T51821, 683 amino acids (aa); 68% identity], and a 530-bp re-regions throughout the pBACB⬘1 clone, a shotgun
(accession no. AC084763, 513 aa;ⵑ35% identity; Figure blots using probes from the shotgun library (Figure 7), in combination with the BAC sequence, showed that 7). Upstream of the two hypothetical open reading frames
(ORFs) was a region with seven tandem repeats, each B-615had the same map asB⬘up to 44.3 kb upstream, with the first detectable difference in restriction sites containing a singleMluI site. Farther upstream, a region
of 299 bp was 100% identical to an expressed sequence occurring atⵑ49 kb.
Pigment phenotype of recombinant alleles reveals an tag (EST) from a stressed root cDNA library fromZea
mays(accession no. AI649454). There was no evidence additional regulatory region: Examination of the pig-ment phenotypes of several recombinant alleles and for any class I or class II elements in this 20-kb region.
The previously isolated subclones, which had been B-615revealed the location of sequences required for
B⬘ expression (Table 2). The B⬘BP- pg2 allele with a tested for their repetitiveness in the maize genome, were
placed on the pBACB⬘1 restriction map by aligning their recombination breakpoint at ⵑ500 bp upstream gave rise to plants that looked likeB⬘, demonstrating that it sequences with the pBACB⬘1 sequence (shown
graphi-cally in Figure 7). Most of the retrotransposons were is possible to transfer sequences mediatingB⬘expression levels to the promoter-proximal region ofB-Peru. Plants not represented in these subclones, because they are
highly repeated in the genome (SanMiguelet al.1998) with B⬘BP-pg1 (B-Peru sequences to ⵑ93 kb, B⬘ se-quences upstream) looked like B-Peru, indicating that and were removed from the analyses by hybridization
with total genomic DNA. Six of the high-copy-number they did not gain the sequences required forB⬘ transcrip-tion. Plants containing the paramutagenicBPB⬘-pgalleles regions that made it through the screen correspond
to the Ji retrotransposon. The intermediate repetitive 1–4, 19–21, and the neutral BPB⬘-n2, -n3 alleles had the same pigment levels as B⬘ plants, indicating that regions between 0–10 kb and 30–40 kb correspond to
theMuDR-related element and the region between the they retained the sequences required forB⬘expression. Of these alleles, theBPB⬘-n2allele retained the least
Zeon and Huck elements, respectively. The
low-copy-number regions are just upstream of B⬘, at the other amount ofB⬘sequences (up to 92 kb), demonstrating that these sequences are sufficient forB⬘transcription. end of the BAC and between 40 and 50 kb (Figure 7).
Recombination breakpoint mapping:To localize the In contrast,BPB⬘-n1with a recombination breakpoint
at ⵑ8.5 kb was essentially green, demonstrating the breakpoints in the different recombinant alleles,
geno-mic DNA was isolated from plants containing the various 8.5 kb upstream ofB⬘is not sufficient forB⬘transcription levels. These data indicate that the regulatory sequences alleles, digested, and subjected to standard gel
electro-phoresis or PFGE, followed by DNA blot analyses using responsible forB⬘expression are located betweenⵑ8.5 kb (BPB⬘-n1) and ⵑ92 kb (BPB⬘-n2). Comparison ofB⬘
the unique or low-copy-number probes identified. These
data, summarized in Table 1, localized the region re- andB-615(same pigment levels and transcription rate) showed that they have essentially the same sequences quired for paramutation to betweenⵑ50 andⵑ110 kb
upstream of the B⬘ transcription start site, consistent untilⵑ49 kb upstream, suggesting that the sequences required forB⬘expression lie between 49 and 8.5 kb. with ourB-615results.
To define more precisely the breakpoints, the recom-bination regions were sequenced. For the seven BP
DISCUSSION B⬘-pgalleles with a breakpoint between 107 and 110 kb
upstream of theB⬘transcription start site, additionalB⬘ We used recombination mapping to localize the se-quences inB⬘required for paramutation to a 13-kb region sequences were needed as this was upstream of the BAC
clone. IPCR was used to amplify an additional 3.9 kb between 93 and 106 kb upstream of the transcription start site. Consistent with the recombination mapping of upstreamB⬘ sequence (materials and methods).
PCR primers were designed using theB⬘sequence and results, the neutral B-615allele shares very similar se-quences (possibly the same sese-quences) with B⬘ until DNA fragments were amplified from B-Peru and the
recombinant alleles in the regions surrounding the re- ⵑ49 kb upstream of the transcription start site. The fact that these two alleles are transcribed at the same rates combination breakpoints. All fragments were cloned
and sequenced and the recombination breakpoints as- demonstrates that transcription from the same promoter-proximal region is not sufficient for the ability to partici-signed to regions delimited by DNA sequence
polymor-phisms betweenB-PeruandB⬘(summarized in Table 2 pate in paramutation.
Experiments have been performed to identify the and Figure 8). These data revealed that the region of
B⬘between 93 and 106 kb upstream of theB⬘transcrip- sequences required for paramutation for two other genes,r1andp1. Ther1paramutagenic alleles are com-tion start site contains sequences required for
paramuta-tion. This 13-kb region in B⬘ is mostly unique or low plex, with three or four copies of ther1gene and flank-ing DNA located in large tandem arrays (Egglestonet
copy in the maize genome and it does not contain any
recognizable transposable elements, nor does it share al.1995;Panavaset al.1999). The sequences required for paramutagenic activity were mapped using unequal homology with the promoter-proximal region. The
TABLE 2
Pigment phenotype and recombination breakpoints of recombinants defining the key regulatory regions upstream ofBⴕ
Recombination Recombination Pigment
Alleles interval breakpoint (bp)a phenotype
BPB⬘-pg1 II 107,150–107,143 B⬘
BPB⬘-pg2 II 106,659–106,530 B⬘
BPB⬘-pg3 II 107,061–106,911 B⬘
BPB⬘-pg4 II 107,370–107,273 B⬘
BPB⬘-pg19 II 106,516–106,375 B⬘
BPB⬘-pg20 II 106,014–105,986 B⬘
BPB⬘-pg21 II 106,516–106,375 B⬘
BPB⬘-n1 I 8,901–8,429 Essentially green
BPB⬘-n2 I 92,190–91,924 B⬘
BPB⬘-n3 I 92,760–92,190 B⬘
B⬘BP-pg1 I 93,009–92,760 B-Peru
B⬘BP-pg2 I 885–411 B⬘
B-615(neutral) NA 49–44.3 kbb B⬘
aAll numbers are base pairs upstream of theB⬘transcription start site, set relative to theSwaI site at 94,021
bp. The recombination breakpoints were determined by DNA sequencing of PCR amplification products for all the alleles exceptBPB⬘-n1, which was determined by DNA blot analysis. The numbers define the interval in which the recombination breakpoint occurred as determined by DNA sequence polymorphisms. For BPB⬘-n1, the numbers are the location of the restriction site polymorphisms upstream and downstream of the recombination breakpoint.
bThese numbers represent the interval where theB-615restriction map diverges from theB⬘map.
the strength of paramutation correlated with the num- genome and shows no sequence or structure characteris-tic of transposons, but does contain seven tandem re-ber of repeats (Kermicle et al. 1995; Panavas et al.
1999). At thep1locus, a transgenic approach identified peats of a sequence not represented elsewhere in the maize genome. Intriguingly, DNA blot analyses indicate a 1.2-kb fragment normally located 5 kb upstream and
8 kb downstream of thep1transcription start site, which, that two neutral alleles,B-615andB-Peru, have approxi-when fused to GUS and reintroduced into maize, was
capable of paramutating the endogenous allele ( Sidor-enkoandPeterson2001). Once paramutated, the en-dogenous allele often is paramutagenic in the absence of the transgene, but the sequences required in the endogenous allele have not been defined. The paramu-tagenic transgene loci contained multiple copies of the transgene while the endogenousp1gene is flanked by 5.2-kb direct repeats containing ⵑ1.5 or 2 copies of the 1.2-kb region, consistent with the involvement of repeated sequences. However, no transgene loci with a single copy of the p1 transgene were obtained, pre-venting a direct test of the requirement for repeats. A correlation between gene silencing and the presence of repeated sequences has also been observed in many, but certainly not all, examples of transgene-induced gene silencing (Hobbset al.1990;Assaadet al.1993;Dorer
andHenikoff 1994; Stam et al. 1997; Muskens et al.
2000).
There is no sequence similarity between the 13-kb Figure8.—The paramutation sequences map between 93
and 106 kb upstream of theB⬘ transcription start site. The
region fromB⬘, the 1.2-kb region fromp1, or the
pub-restriction maps are shown for the recombinant alleles that
lishedr1 sequences. The 1.2-kb region ofp1 contains
have recombination breakpoints defining the 5⬘and 3⬘
bound-moderately repetitive DNA as well as sequences unique
ary of the paramutation sequences. The white ovals indicate
to thep1locus (Sidorenkoet al.1999); 78 bp is 100% the region in which recombination events took place. The identical to thep1transcript. AtB⬘, the region required restriction sites shown are B,BamHI; E,EcoRI; M,MluI; Nc,
NcoI; S,SwaI.
mately one copy of the sequence repeated in B⬘(data sons are arranged as nested insertions. Upstream ofB⬘,
PREM-2,Opie,Opie-p, and aJielement are all inserted not shown). Further analyses of the region required for
b1paramutation using additional recombination experi- into a partialJielement. In contrast, the region around thebz1 locus is relatively gene rich (Fuet al.2001). ments should enable further delineation of the key
se-quences and determine if the tandem repeats inB⬘are Our studies demonstrate that classical fine-structure recombination mapping is an excellent approach to involved in paramutation.
All of the recombination events that we isolated oc- identify the key sequences required for paramutation. It will be important to use a similar approach to deter-curred immediately upstream of theB⬘coding region,
ⵑ92–107 kb upstream, or even farther upstream. For the mine if the sequences required forB-Ito participate in paramutation map to the same region as inB⬘.The ability two recombinagenic regions sequenced, recombination
occurred in gene-like, unique, or low-copy-number se- to transfer the paramutation sequences to the neutral
B-Peruallele, causing it to become paramutagenic, also quences that had a low level of DNA methylation
com-pared to the sequences in between. For example, the suggests that a transgenic approach to further dissect the region required for paramutation should be feasi-methylation-sensitive enzymesMluI,SalI, andSnaBI cut
only genomic DNA in the sequences within and immedi- ble. Once the minimal sequences are defined in both alleles, it will be important to investigate whether there ately flanking theB⬘coding region and 92–107 kb
up-stream; the multiple sites located in between were not are sequence differences or differences in chromatin structure betweenB-IandB⬘.
cut and are thus presumably methylated. These results
are consistent with the recent findings from studies of Our recombination mapping experiments also identi-fied an additional regulatory region required forB⬘ expres-recombination around the bz1 locus in maize, which
indicate that recombination takes place in maize genes sion, located downstream of the sequences required for paramutation, but far upstream of the transcription start and not in the intergenic regions (Fuet al.2001, 2002),
and earlier work noting the high frequency of recombi- site. The observation thatB-615has the same transcrip-tion rate as B⬘ suggests that it retains these necessary nation within maize genes (Civardi et al. 1994;
Pat-tersonet al.1995;DoonerandMartinez-Ferez1997). regulatory sequences, which might be located within the region conserved withB⬘(betweenⵑ8.5 andⵑ49 One caveat is that most of the 80-kb region between the
two recombinagenic regions inB⬘consists of transposons. kb upstream of theB⬘ transcription start site). To our Because most of this region in B-Peru has not been knowledge this is the first example in plants of key cloned and sequenced, it is possibleB-PeruandB⬘do not regulatory sequences being located so far upstream of share the same transposon sequences in this intergenic a gene. There are hundreds of reports in the literature region, which would be expected to reduce recombina- of transgene analyses in many different plant species, tion dramatically. It is also possible that the two alleles all of which demonstrate that key regulatory sequences do share many of the same insertions, but recombina- controlling developmental and tissue-specific expres-tion is suppressed in the regions containing repetitive sion are usually near the transcription start site (for DNA (Fuet al.2002), potentially because of chromatin review, seeSingh1998 and references therein). While structure. Suppression of recombination between transpo- the number of regulatory regions characterized is lower sons would serve to decrease ectopic recombination and in maize than in plant species with more efficient trans-is likely to be a common feature of eukaryotic genomes. formation methodologies, the theme is the same (for In Drosophila and Arabidopsis the rate of meiotic re- example,RussellandFromm1997;Hueroset al.1999; combination is also reduced in transposon-rich regions Sidorenko et al. 2000). Additional studies on more of chromosomes (Charlesworthet al. 1994; CSHL/ maize genes are needed, but it is tempting to speculate WUGSC/PEB 2000). It will be necessary to clone and that our findings withB⬘are related to its unique para-sequence the comparable region fromB-Peruto address mutation properties, which we have speculated involve
this hypothesis rigorously. boundary elements gone awry (Chandleret al. 2000,
The sequences upstream ofB⬘consist of a large region 2002). In contrast to most plant promoters, it is not containing primarily retrotransposons, flanked by a few uncommon for regulatory sequences to be located far DNA transposons. A similar large block of retrotranspo- from transcriptional start sites in animals (for example, sons has been observed for the intergenic regions flank- Dillonet al.1997;Siposet al.1998;Belland Felsen-ing several other maize genes such asadh1(SanMiguel feld2000). The ability to do fine-structure
recombina-et al.1996) anda1(Chenet al.1997). As seen with the tion and large-scale mutagenesis experiments in maize
Adh1-F allele, the highly repetitive Ji, Opie, and Huck makesB⬘an excellent model system for further
investi-elements that constitute a substantial portion of the gations of the mechanisms underlying long-distancecis maize genome (SanMiguel et al.1996) are present in andtransinteractions that control transcription. the intergenic region upstream of B⬘. As observed in
We are indebted to Pascal Lennertz and Zahra Mobasher for their other regions of the maize genome (SanMiguelet al. expert technical assistance, which was crucial for identifying the