Scholarship at UWindsor
Scholarship at UWindsor
Electronic Theses and Dissertations Theses, Dissertations, and Major Papers
2010
Structure-Function Characterization of the Human Dual Specificity
Structure-Function Characterization of the Human Dual Specificity
Phosphatase hYVH1
Phosphatase hYVH1
Colleen Mailloux University of Windsor
Follow this and additional works at: https://scholar.uwindsor.ca/etd
Recommended Citation Recommended Citation
Mailloux, Colleen, "Structure-Function Characterization of the Human Dual Specificity Phosphatase hYVH1" (2010). Electronic Theses and Dissertations. 307.
https://scholar.uwindsor.ca/etd/307
This online database contains the full-text of PhD dissertations and Masters’ theses of University of Windsor students from 1954 forward. These documents are made available for personal study and research purposes only, in accordance with the Canadian Copyright Act and the Creative Commons license—CC BY-NC-ND (Attribution, Non-Commercial, No Derivative Works). Under this license, works must always be attributed to the copyright holder (original author), cannot be used for any commercial purposes, and may not be altered. Any other use would require the permission of the copyright holder. Students may inquire about withdrawing their dissertation and/or thesis from this database. For additional inquiries, please contact the repository administrator via email
Structure-Function Characterization of the Human Dual
Specificity Phosphatase hYVH1
by
Colleen Mailloux
A Thesis
Submitted to the Faculty of Graduate Studies through the Department of Chemistry and Biochemistry
in Partial Fulfillment of the Requirements for the Degree of Master of Science at the
University of Windsor
Windsor, Ontario, Canada 2010
iii
I hereby certify that I am the sole author of this thesis and that no part of this
thesis has been published or submitted for publication.
I certify that, to the best of my knowledge, my thesis does not infringe upon
anyone‟s copyright nor violate any proprietary rights and that any ideas, techniques,
quotations, or any other material from the work of other people included in my thesis,
published or otherwise, are fully acknowledged in accordance with the standard
referencing practices. Furthermore, to the extent that I have included copyrighted
material that surpasses the bounds of fair dealing within the meaning of the Canada
Copyright Act, I certify that I have obtained a written permission from the copyright
owner(s) to include such material(s) in my thesis and have included copies of such
copyright clearances to my appendix.
I declare that this is a true copy of my thesis, including any final revisions, as
approved by my thesis committee and the Graduate Studies office, and that this thesis has
iv
YVH1 is a highly conserved dual specificity phosphatase that possesses a novel
zinc-binding domain. Although studies implicate hYVH1 in cell survival and cell cycle
progression, it remains poorly characterized. In this study, association of hYVH1 with
the 60S subunit was demonstrated. Oxidative stress inhibits this association, with the
appearance of a 25kDa hYVH1 fragment. Domain deletion studies reveal that regions of
the catalytic and zinc-binding domains facilitate ribosomal binding. Collectively, our
results lead to a proposed mechanism whereby structural rearrangements in the
zinc-binding domain mediate dissociation of hYVH1 from the ribosome and exposure of a
proteolytic cleavage site.
We have also purified several hYVH1 variants for X-ray crystallography. To
date, we have obtained a low resolution solution structure of full length hYVH1
representing the first structure of any YVH1 orthologue. We anticipate that structural
analysis will offer invaluable insights concerning the regulation, mode of action, and
v
vi
After all the time and energy that has gone into this thesis, it is a pleasure to
thank those who made possible this feat. First and foremost I would like to express
immense gratitude to my supervisor Dr. Vacratsis. His enthusiasm for the sciences
influenced my enrolment into the Master‟s program, and his ceaseless guidance and
support throughout are greatly appreciated. He was an excellent mentor, and his
teachings were key in my scientific development.
I would like to thank my committee members, Dr. Boffa and Dr. Swan, for the
guidance they offered in our meetings. To the professors within the department: Dr.
Ananvoranich, Dr. Boffa, Dr. Lee, Dr. Mutus, and Dr. Pandey, I have spent a substantial
amount of time in your labs and to say „thank you for the use of your equipment‟ is most
definitely an understatement. The relaxed and open-door environment established
within the Biochemistry Department has definitely enhanced my experience as a
Master‟s candidate.
I am thankful to those who assisted me with the day to day affairs of a student
researcher: Beth, Marlene, Kerri, Kimberly, Kerri, and Michelle. From scholarship
applications to helplessly trying to solve the mystery of international dry ice shipment, a
helping hand was always available across the hall.
I am grateful to my labmates, both past and present. I must thank the hYVH1
pioneers of the Vacratsis lab: Khaled, Zareen, John, and Priya, for the knowledge they
vii
putting up with me in the months leading up to my defense. We definitely have some
unforgettable memories.
To my family and my husband. My personal support team. Thank you for
picking up the slack when I was overwhelmed with my studies, not only throughout my
time as a Master‟s candidate, but throughout my entire 21 years as a student. You are
viii
Author‟s Declaration of Originality...iii
Abstract...iv
Dedication...v
Acknowledgements...vi
List of Figures...xii
List of Abbreviations...xvi
CHAPTER 1: Introduction 1.1 Cellular Phosphorylation ...1
1.2 Protein Phosphatases...3
1.3 The Dual Specificity Phosphatase YVH1...6
1.4 The Human Dual Specificity Phosphatase hYVH1...8
1.5 Ribosome Biogenesis...11
1.5.1 Ribosome Biogenesis: From Nucleolus to Cytoplasm...13
1.5.2 Ribosome Biogenesis is Sensitive to Intra and Extracellular Environment……..16
1.5.3 Trans-Acting Factors Involved in Ribosome Biogenesis...18
1.5.4 Tools to Identify and Analyze Trans-acting Factors in Ribosome Biogenesis...19
1.5.5 YVH1 is a Trans-acting protein in Ribosome Biogenesis in Yeast...21
1.6 Methods to Explore Structural/Functional Features of Protein Tyrosine Phosphatases...23
ix
Protein Structure...26
1.7 Objectives...31
CHAPTER 2: Materials and Methods 2.1 Plasmids...32
2.2 Cell Culture and Transfections...35
2.3 Ribosome Profiling...36
2.4 Immunoprecipitation...38
2.5 Immunoblotting...38
2.6 Protein Expression and Purification...39
2.7 DiFMUP Assays………...42
2.8 pNPP Assays...43
2.9 Limited Proteolysis...44
CHAPTER 3: Results PART I) Characterization of the hYVH1:Ribosome Interaction 3.1 Human YVH1 Interacts With the 60S Ribosomal Subunit………45
3.2 Effect of Substrate Trap Mutants on Co-fractionation of hYVH1 With the Ribosome………...51
3.3 Effect of Phosphomimetic Mutants on Fractionation of hYVH1………...55
x
Interaction………..61
3.6 Investigation of Regions Required for Interaction of hYVH1 with
Ribosomal Subunits………...63
PART II) In Vitro Structure Function Analysis of hYVH1
3.7 Effect of Zinc-Binding Domain on Activity of hYVH1 Toward DiFMUP…………67
3.8 Effect of Mutating Conserved Residues on In Vitro Phosphatase Activity
of hYVH1………...70
3.9 Effect of DMSO and Glycerol on In Vitro Phosphatase Activity of hYVH1……….72
3.10 Preparation of Protein for Analysis by X-ray Crystallography………76
3.11 Small Angle X-ray Scattering Reveals Low Resolution Structure of hYVH1…….78
3.12 Limited Proteolysis Confirms Boundaries of Flexible N-terminal Region………..80
3.13 Design and Purification of Mutants Predicted by Surface Entropy
Reduction………...85
CHAPTER 4: Discussion
PART I) Characterization of the hYVH1: Ribosome Interaction
4.1 hYVH1 Interacts With Particles of the 60S Subunit...89
4.2 Overexpression of hYVH1 Does Not Affect Ribosome Profiles...91
4.3 Catalytic Mutants Do Not Affect hYVH1:60S Interaction...93
4.4 hYVH1:Ribosome Interaction is Not Regulated Through Phosphorylation
xi
the hYVH1: Ribosome Complex ...98
4.6 Ribosome Biogenesis and the Cell Survival Effect...106
4.7 Domain Deletion Studies Suggest Regions of Both Domains are Necessary for Complete Ribosome Binding...107
PART II) In Vitro Structure Function Analysis of hYVH1 4.8 Structural Effects on In Vitro Catalytic Activity of hYVH1………109
4.9 Elucidating the Structure of hYVH1……….113
4.10 Concluding Remarks………...117
References...123
xii CHAPTER 1: Introduction
Figure 1.1 Regulation of Cellular Phosphorylation Levels...2
Figure 1.2 General Mechanism of Dephosphorylation Employed by Protein Tyrosine Phosphatases...4
Figure 1.3 Classification of the Protein Tyrosine Phosphatases...5
Figure 1.4 Schematic of hYVH1 Protein Sequence...9
Figure 1.5 Summary of Effect of hYVH1 Mutants on Cell Cycle...12
Figure 1.6 Ribosome Biogenesis in Mammalian Cells...14
Figure 1.7 Proposed Role of YVH1 as a Trans-Acting Factor in Ribosome Biogenesis...24
Figure 1.8 Hydrolysis of the Artificial Phosphatase Substrates DiFMUP and pNPP...27
CHAPTER 3: Results PART I) Characterization of the hYVH1:Ribosome Interaction Figure 3.1 Schematic of Experimental Set-up for Ribosome Profiling...46
Figure 3.2 Fractionation of Endogenous hYVH1 with Respect to Ribosomal Subunits...47
Figure 3.3 Fractionation of Overexpressed hYVH1 with Respect to Ribosomal Subunits...49
xiii
Figure 3.6 Fractionation of Catalytically Significant Point Mutants...54
Figure 3.7 Fractionation of Phosphomimetic hYVH1 mutants...56
Figure 3.8 Ribosomal Profiles in Response to tert-Butyl Hydroperoxide
Treatment...58
Figure 3.9 Co-fractionation of Overexpressed hYVH1 in response to TBH
Treatment...59
Figure 3.10 Fractionation of Overexpressed hYVH1 in Response to TBH
Treatment...60
Figure 3.11 Fractionation of Catalytically Inactive C115S hYVH1 in
Response to TBH Treatment...62
Figure 3.12 hYVH1 Domain Deletion Constructs...64
Figure 3.13 Effect of Domain Deletion on Co-fractionation of hYVH1 with
Ribosomal Subunits...65
Figure 3.14 Effect of Partial Domain Deletion on Co-fractionation of hYVH1
with Ribosomal Subunits...66
PART II) In Vitro Structure Function Analysis of hYVH1
Figure 3.15 Purification of Recombinant hYVH1 and Zn∆hYVH1...68
Figure 3.16 Activity of Full Length and Zn∆hYVH1 Toward DiFMUP...69
Figure 3.17 Effect of Point Mutation of Conserved Residues on Catalytic
xiv
DiFMUP...74
Figure 3.19 Effect of DMSO on Catalytic Activity of hYVH1 Toward
DiFMUP...75
Figure 3.20 Effect of DMSO on Activity of hYVH1 toward pNPP...77
Figure 3.21 Low Resolution Structure Obtained using Small Angle X-Ray
Scattering...79
Figure 3.22 Limited Proteolysis of hYVH1...81
Figure 3.23 Mass Spectrometry of Peptide Fragments Obtained Using
Limited Proteolysis...82
Figure 3.24 Purification of Deletion Mutants as Determined Using Limited
Proteolysis...84
Figure 3.25 Surface Entropy Reduction Mutants Predicted In Silico for
Crystallization Trials...86
Figure 3.26 Purification and Preparation of SER mutants for X-ray
Crystallography...87
Figure 3.27 Activity of Purified and Concentrated SER mutants Toward
xv
PART I) Characterization of the hYVH1: Ribosome Interaction
Figure 4.1 Alignment of Zinc-Binding Domain of YVH1 Orthologues...99
Figure 4.2 Proposed Mechanism of hYVH1:Ribosome Dissociation
In Response To Oxidative Stress...103
Supplementary Figures
Supplementary Figure 1 Co-fractionation of Endogenous hYVH1 in
Response to TBH Treatment...119
Supplementary Figure 2 Approach to Elucidate Mechanism of Disruption of
hYVH1:Ribosome Interaction in Response to TBH...120
Supplementary Figure 3 Attempt at Elucidating Mechanism of TBH-
Induced hYVH1:Ribosome Dissociation...121
Supplementary Figure 4 RPL26 Does Not Co-Immunoprecipitate with
xvi ADP adenosine diphosphate
ATM ataxia telangiectasia mutated
ATP adenosine triphosphate
ATR ATM-related
BSA bovine serum albumin
Cal A calyculin A
CDC cell division cycle
CDK cyclin-dependent kinase
DAMMIN dummy atom model minimization
DEPC diethylpyrocarbonate
DiFMU- 6,8-difluoro-4-methylumbelliferyl
DiFMUP 6,8-difluoro-4-methylumbelliferyl phosphate
DMEM Dulbecco‟s Modified Eagle Medium
DMSO dimethyl sulfoxide
DNA deoxyribonucleic acid
DSP dual specificity phosphatase
DTT dithiothreitol
DUSP12 dual specificity phosphatase 12
EDTA ethylenediaminetetracetic acid
ERK extracellular signal-regulated kinase
FBS fetal bovine serum
xvii GST glutathione S-transferase
HEK 293 human embryonic kidney
HeLa Henrietta Lacks (cervical cancer cell line)
HPLC high performance liquid chromatography
IB immunoblot
IP immunoprecipitation
IPTG isopropyl β-D-1-thiogalactopyranoside
kDa kilodaltons
MALDI TOF matrix-assisted laser desorption ionization-time of flight
MAPK mitogen-activated protein kinase
MKP mitogen-activated protein kinase phosphatase
mRNA messenger ribonucleic acid
m/z mass to charge ratio
OD optical density
Pi inorganic phosphate
PBS phosphate buffered saline
PCR polymerase chain reaction
PEI polyethyleneimine
PMSF phenylmethylsulphonyl fluoride
pNP- para-nitrophenyl
pNPP para-nitrophenyl phosphate
xviii
rDNA ribosomal deoxyribonucleic acid
RNA ribonucleic acid
RNase ribonuclease
RPL26 ribosomal protein large subunit 26
rRNA ribosomal ribonucleic acid
SDS PAGE sodium dodecyl sulfate polyacrylamide gel electrophoresis
SER surface entropy reduction
siRNA small interfering ribonucleic acid
snoRNA small nucleolar ribonucleic acid
TAP tandem affinity purification
TBH tert-butyl hydroperoxide
TBST Tris-buffered saline-Tween 20
UV ultraviolet
1
Introduction
1.1 Cellular Phosphorylation
Protein phosphorylation is a highly relevant and ubiquitous post-translational
modification which serves as a key regulatory mechanism of several biological pathways
including cell cycle, cellular differentiation, growth, apoptosis and metabolism [1, 2]. It
is estimated that more than 30% of eukaryotic proteins are phosphorylated; with these
phosphorylation events most commonly occurring on the hydroxyl group of serine,
threonine, and tyrosine residues [3]. Introduction of a negatively charged phosphate
group onto one or several of these residues generally elicits a conformational change
which can alter protein activity, stability, localization, affinity for binding partners, or
allow it to adopt an entirely different function [4, 2]. Phosphorylation levels throughout
the cell are tightly regulated through the antagonistic action of protein kinases and protein
phosphatases [1]. As shown in Figure 1.1, protein kinases phosphorylate proteins by
mediating the transfer of the gamma phosphate from ATP, while protein phosphatases
catalyze the hydrolysis of a phosphate group from phosphotyrosine, phosphothreonine, or
phosphoserine residues, releasing inorganic phosphate as a biproduct of the
dephosphorylation reaction [2].
Specific phosphorylation and dephosphorylation events can be triggered by
numerous intracellular or extracellular stimuli, initiating signal transduction pathways,
and ultimately producing a specialized response. As phosphorylation participates in the
regulation of various cellular activities, aberrant phosphorylation has implications in
2
3
Protein phosphatases are a diverse family of proteins that remove a phosphate
group from a phosphotyrosine, phosphothreonine, or phosphoserine residue on its
substrate protein. Protein phosphatases are classified by their mode of action, by their
dependence on metals, and by their substrate specificity. The serine/threonine
phosphatases function as metalloproteins and dephosphorylate phosphoserine and/or
phosphothreonine residues [7]. A second group of phosphatases, the protein tyrosine
phosphatases (PTPs), or cysteine dependent PTPs, act through the formation of a
thiol-phosphate enzyme intermediate in the removal of a thiol-phosphate. All PTPs possess the
consensus sequence HC(X)5RS/T (also termed P-loop), as well as an aspartic acid on one
of the protein‟s outer loops which acts as a general acid/base in the phosphatase
mechanism. The conservation of this characteristic P-loop suggests that these
phosphatases proceed through a similar catalytic mechanism as outlined in Figure 1.2 [8].
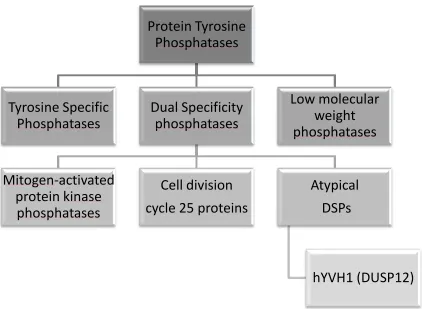
The PTP subfamily (summarized in Figure 1.3) consists of the tyrosine specific
phosphatases, the dual specificity phosphatases (DSPs), and the low molecular weight
phosphatases [7]. While the tyrosine specific phosphatases dephosphorylate exclusively
phosphotyrosine residues, the dual specificity phosphatases can dephosphorylate both
phosphotyrosine and phosphothreonine/phosphoserine residues. Although the DSPs
show limited sequence homology with other PTPs, they do share the consensus
D…HC(X)5RS/T catalytic cysteine sequence, and proceed through a parallel catalytic
mechanism [9]. It is postulated that the broader substrate specificity of these
phosphatases is derived from the depth of their catalytic cleft as well as the presence of
4
5
Figure 1.3: Classification of the Protein Tyrosine Phosphatases. The protein tyrosine phosphatases can be classified into three major groups: the tyrosine specific phosphatases, the low molecular weight phosphatases, and the dual specificity phosphatases (DSPs). One group of DSPs, the atypical DSPs, consists of a variety of poorly characterized enzymes which are thought to target substrates other than MAPKs or CDKs. Among these is the dual specificity phosphatase hYVH1, or DUSP12.
Protein Tyrosine
Phosphatases
Tyrosine Specific
Phosphatases
Dual Specificity
phosphatases
Mitogen-activated
protein kinase
phosphatases
Cell division
cycle 25 proteins
Atypical
DSPs
hYVH1 (DUSP12)
Low molecular
6
Among the dual specificity phosphatases are the mitogen-activated protein kinase
phosphatases (MKP), the cell division cycle (CDC) phosphatases, and the atypical DSPs
[11]. The MKPs act to dephosphorylate the mitogen-activated protein kinases (MAPK),
thereby deactivating them. The MAPK signalling pathways regulate several cellular
functions including cell proliferation, differentiation, and stress response [2]. In addition
to their catalytic domain, MKPs possess an N-terminal substrate-binding domain. Only
upon binding to a substrate MAPK molecule is the MKP catalytic domain active, hence
enhancing substrate specificity [2].
The atypical, or VH1-like DSPs are a poorly characterized group of phosphatases.
These phosphatases lack the MAPK recognition motifs, and hence dephosphorylate
substrates other than the MAPKs [11]. In addition to their VH1-like catalytic domain, the
atypical DSPs often possess additional domains or short sequences that participate in
substrate identification, localization, and protein-protein interactions [10]. While this
group of DSPs consists primarily of protein phosphatases, one of them (DUSP11) has
been shown to dephosphorylate mRNA [12].
1.3 The Dual Specificity Phosphatase YVH1
The first discovered eukaryotic atypical dual specificity phosphates was YVH1 in
yeast, which displays high evolutionary conservation with orthologues present in species
ranging from yeast to human [13]. YVH1 contains the P-loop consensus sequence,
characteristic of all PTPs, in its N-terminal catalytic domain. However, YVH1 is
7
conserved C-terminal zinc-binding domain in addition to its N-terminal catalytic domain
[7]. This cysteine-rich domain is able to coordinate two moles of zinc per mole of
protein. Through the use of truncated mutant variants, it has been found that the zinc
finger domain is essential for in vivo function of YVH1 [7,14,15]. This novel
zinc-binding domain is highly conserved throughout evolution amongst YVH1 orthologues,
suggesting that the domain is critical for proper protein function [7].
In yeast, it has been found that transcription of yvh1 is induced by nitrogen
starvation and low temperature [16]. Yeast with yvh1 gene knocked out display a slow
growth phenotype, sporulation defects and defective glycogen accumulation (all
independent of catalytic activity) [17].
To date, no substrates have been identified, however a study conducted by
Sakumoto et. al in 2001 showed interaction between YVH1 and YPH1 by the yeast
two-hybrid method. YPH1 is a dynamic protein that is critical for DNA replication, cell cycle
control, and biogenesis of the 60S ribosomal subunit [18]. Multicopy yph1 was able to
rescue both the slow growth defect and recover transcript levels of sporulation-specific
genes associated with yvh1 disruption mutants. Additionally, deletion studies showed
that the catalytic domain of YVH1 was sufficient for interaction with YPH1. It was
therefore suggested that YPH1 could be a candidate substrate; however, no further
evidence has been presented to support this hypothesis [13]. Interestingly, YVH1 has
recently been found to play a role in ribosome biogenesis in yeast, specifically, in the
8
1.4 The Human Dual Specificity Phosphatase hYVH1
The human orthologue, hYVH1 (also termed DUSP12), shares approximately
30% sequence identity with YVH1 and possesses the characteristic DSP domain and
C-terminal zinc-binding domain (Figure 1.4). hYVH1 is located on the chromosomal
region 1q21-q22, which is amplified in human liposarcomas and a variety of other solid
tumours, including ovarian cancer and hepatocellular carcinomas [7,20]. It has been
found that the human orthologue possesses the ability to rescue the slow growth defect
caused by yeast yvh1 disruption mutants [7]. The Vacratsis lab has identified HSP70 as a
binding partner of hYVH1. Further, our lab has recently shown that hYVH1 acts as a cell
survival phosphatase: its overexpression protects cells from apoptosis induced by heat
shock, oxidative stress and Fas receptor activation [21]. The notion of hYVH1 as a cell
survival phosphatase was preceded in a study conducted by MacKeigan et al., in which
systematic knockdown of phosphatases by transfection with siRNAs identified hYVH1 as
potential anti-apoptotic protein [22]. Notably, this was the first physiological role of
hYVH1 shown to necessitate its catalytic activity: substitution of the catalytic cysteine
residue with serine was unable to protect cells [21].
Interestingly, the catalytic activity of hYVH1 toward artificial substrates in vitro
at high temperature or in non-reducing conditions was affected to a lesser extent
compared to other PTPs, suggesting that hYVH1 may be resistant to inactivation under
these conditions [21]. This hypothesis was further investigated by Bonham et al. who
showed that the zinc-coordinating cysteines in the zinc-binding domain were able to
protect the active-site cysteine of hYVH1 from inactivation by oxidation [23]. Under
9
10
the coordinating cysteine residues in the zinc-binding domain are oxidized, triggering the
release of zinc. When the levels of oxidative stress exceed the redox buffering range of
the coordinating cysteines, the active site cysteine forms intramolecular disulfide bonds
with nearby cysteine residues. Upon return to normal cellular redox conditions, this zinc
ejection is readily reversed, and the active site returned to its active, reduced state. This
supports a mechanism in which the active site cysteine is capable of eluding irreversible
oxidation through the formation of intramolecular disulfide bonds [23].
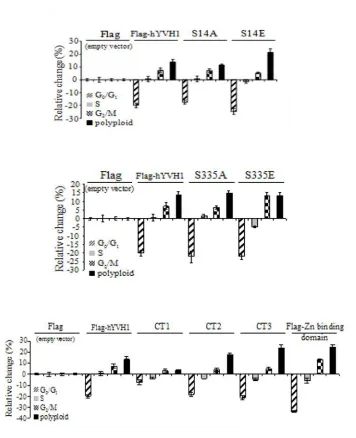
It has been shown that hYVH1 is itself regulated by phosphorylation. Through
the use of phosphatase inhibitors to boost cellular phosphorylation levels, followed by the
subsequent analysis of hYVH1 peptides by mass spectrometry, it has been found that
hYVH1 can be phosphorylated at three sites: serine 14, threonine 252 and serine 335,
located near the N-terminus, within the zinc-binding domain, and near the C-terminus,
respectively [24]. When mutating these residues to alanine or glutamic acid, hence
mimicking the non-phosphorylated and phosphorylated forms, respectively, it was
observed that phosphorylation may affect subcellular localization of hYVH1. Typically,
hYVH1 exhibits a nuclear, perinuclear, and cytoplasmic localization. However,
overexpressed S14A or S335A mutants in HeLa cells displayed nuclear localization,
while overexpressed S14E or S335E mutants resided mainly in the cytoplasm. This
distinction in localization was accompanied by changes in cell cycle profiles as analyzed
by flow cytometry.
When comparing HeLa cells overexpressing wild type hYVH1 to cells expressing
normal levels of endogenous hYVH1, a marked decrease in cells in G0/G1, coupled to an
11
S14A mutant produced cell cycle profiles comparable to that of cells overexpressing wild
type hYVH1, the overexpression of S14E had an augmented response with an even
greater decrease in G0/G1 cells and an increased number of polyploid cells when
compared to wild type. Similarly, overexpression of S335E resulted in an increased
number of cells in G2/M. This study, briefly summarized in Figure 1.5, showed that
human YVH1 may be involved in the control of cell cycle progression, and that this
function may be regulated through phosphorylation.
When investigating the effect of domain-deletion constructs on the cell cycle
profiles, it was found that the catalytic domain alone was unable to significantly affect
the cell cycle profile in comparison to untransfected cells. When expressing constructs
consisting of the catalytic domain and segments of the zinc-binding domain, increasing
amounts of cells in G2/M and decreasing amounts of cells in G0/G1 were observed,
culminating with the zinc-binding domain alone, which had the greatest amount of cells
in G2/M and least amount in G0/G1, as compared to wild type. And finally,
overexpression of a catalytically dead variant of hYVH1 did not have an effect on cell
cycle compared to overexpression of wild type hYVH1. This study suggests that the
zinc-binding domain alone is likely sufficient to elicit changes in cell cycle [24].
1.5 Ribosome Biogenesis
As mentioned previously, YVH1 in yeast has recently been identified as a
trans-acting factor in the biogenesis of the 60S ribosomal subunit, and it was suggested that
12
13
ribosomes consist of four ribosomal RNAs (25S/28S, 5.8S and 5S in the 60S subunit, and
18S in the 40S subunit) and approximately 80 ribosomal proteins [25]. Biogenesis of the
ribosomal 60S and 40S subunits (outlined in Figure 1.6) requires a considerable amount
of coordination between RNA polI for synthesis of 25S/28S, 5.8S and 18S rRNA, RNA
polII for synthesis of mRNA of the various ribosomal proteins, and RNA polIII for the
synthesis of 5S rRNA [26]. Additionally, ribosome biogenesis demands strict temporal
and spatial coordination of a vast range of trans-acting factors that facilitate the process,
which in yeast results in the synthesis of approximately 40 ribosomes per second [27].
These trans-acting factors include greater than 150 non-ribosomal proteins, and
approximately 70 small nucleolar RNAs (snoRNAs), which guide modifying factors and
ribonucleases via complementary base-pairing with rRNA [26,27]. It is therefore logical
that such a complex and dynamic process would consume a significant amount of the
cells resources, and hence require meticulous regulation [28]. Mutations in this intricate
process are often associated with human disease such as bone marrow failure syndromes
and tumorigenesis [29,30].
1.5.1 Ribosome Biogenesis: From Nucleolus to Cytoplasm
The first step in ribosome biogenesis is the transcription of ribosomal RNA and
the transcription and translation of ribosomal proteins. The nucleolus is the central
location for transcription of ribosomal genes. Here, ribosomal genes are organized in
tandem arrays, termed nuclear organizer regions, along chromosomes [26,31]. The
25S/28S, 5.8S and 18S rRNAs are transcribed as a single precursor transcript in the
14
15
transcription [29]. A high degree of regulation is observed at the transcriptional level,
where chromosomal modifications can render genes of essential ribosomal components
inactive, thereby inhibiting the entire biogenesis pathway [28].
Once transcribed, rRNA transcripts must be modified, cleaved by exonucleases
and endonucleases, folded, and assembled with ribosomal proteins [31]. Several of the
ribosomal proteins are imported from the cytoplasm into the nucleolus where they are
able to associate with rRNA. The earliest reported pre-ribosomal complex is the 90S
particle. This complex includes the uncleaved 35S/47S precursor rRNA with various
associated ribosomal and non-ribosomal proteins [32]. Cleavage of the rRNA then yields
two separate complexes: the 43S particle and the 66S particle, which are precursors to the
40S and 60S ribosomal subunits, respectively [27,29,33]. The 43S subunit is almost
immediately exported to the cytoplasm, where the few final stages of maturation occur,
including a final rRNA cleavage event. The pre-66S subunit however, has a considerably
higher ratio of protein:RNA than the mature 60S particle, and must reside in the nucleus
for an extended period of time. During this time, the pre-60S particle must undergo
several processing steps in which association and dissociation with various trans-acting
factors must occur prior to becoming competent for nuclear export [27,33]. While the
majority of ribosomal proteins are assembled onto the 60S subunit in the early steps of
maturation, there are a few that are presumed to associate with the complex following
16
1.5.2 Ribosome Biogenesis is Sensitive to Intra and Extracellular Environment
Because of the taxing demand imposed on cellular resources by ribosome
biogenesis, it is essential that this process be highly regulated. As mentioned previously,
a significant amount of regulation is observed at the level of transcription. Ribosomal
genes can be completely silenced by structural rearrangement of genes into
transcriptionally inactive heterochromatin [31]. This inactivation of ribosomal genes is
poorly understood and remains relatively constant within a cell. Alternatively,
transcription levels can be adjusted through the modulation of the transcriptional
machinery to provide a more immediate and transient response to cell cycle and growth,
as well as cellular environment. For example, rRNA production in metabolically active,
proliferating cells is quite high compared to the significantly reduced levels of rRNA
synthesis that can be observed in fully differentiated cells. Additionally, ribosome
biogenesis is closely coordinated with the cell cycle, with highest levels of rRNA
transcription occurring in S and G2 phases of the cell cycle (in mammalian cells) and
suppressed during mitosis [31]. The mitotic repression is due to phosphorylation of the
transcription factors SL1 and TTF-1 by cdk1/cyclin B, consequently inhibiting RNA
polymerase I activity and therefore hindering rRNA synthesis. Similarly, a drastic
reduction of rRNA production is observed in response to cellular insults such as deficient
nutrients and drug treatment [31]. In both yeast and mammalian cells, transcription of
ribosomal genes is closely coupled to cell cycle and growth in response to environmental
conditions via the TOR signalling pathway [34]. Further, the RAS-cAMP protein kinase
17
Although regulation of ribosome biogenesis is well characterized at the
transcriptional level, little is known concerning the various aspects of regulation
throughout the process of maturation. For one, it is clear that the regulation of ribosome
biogenesis by TOR extends beyond transcriptional control. TOR is responsible for the
activation of P70 S6 kinase. Phosphorylation of ribosomal protein S6 by S6 kinase is
required for recognition of ribosome protein mRNA by the translational machinery,
hence allowing for the synthesis of relevant ribosomal proteins [34]. Additionally, TOR
participates in the later stages of ribosome biogenesis in the nucleoplasm, as inhibition of
TOR in yeast causes defects in processing of the 35S rRNA precursor [34,35].
Furthermore, it is thought that there are several proteins that act as a surveillance
mechanism throughout ribosome biogenesis. These “quality control” proteins ensure that
pre-ribosomal subunits do not continue in the biogenesis pathway if improper assembly
has occurred. Because ribosome synthesis is a spatially ordered event, a common
mechanism of control is thought to be the inhibition of transportation of maturing
ribosomal particles through the requirement of proteins that function either as active
transport signals or to overcome retention signals [27]. When triggered, these
surveillance mechanisms may also result in the degradation of non-functional ribosomes
in response to improperly assembly, or damage due to insult by UV radiation or oxidative
stress [29, 36]. Studies in yeast have presented evidence of crosstalk between ribosome
biogenesis and other major synthetic pathways throughout the cell, such as the secretory
pathway [28]. While it is speculated that the plethora of trans-acting factors that mediate
18
pathway and allow for its coordination with intra- and extracellular environmental cues,
the specifics concerning these mechanisms have yet to be unveiled [27,28].
1.5.3 Trans-Acting Factors Involved in Ribosome Biogenesis
The maturation of ribosomal subunits is a highly ordered event, enabled by
several trans-acting factors (non-ribosomal proteins and snoRNPs), which transiently
associate with various pre-ribosomal complexes along the pathway from nucleolus to
cytoplasm, but are not a part of the final, mature 60S or 40S subunits. The trans-acting
factors may fulfill roles such as acting as endo/exonucleases, participating in ribosomal
transportation, mediating the association and dissociation of ribosomal/and
non-ribosomal proteins, recycling of export proteins or functioning in signal transduction
pathways to communicate with other cellular processes [29,36]. Several of these
enzymes are energy consuming GTPases and ATPases that, upon NTP hydrolysis,
undergo conformational changes that can trigger the desired response [27,29,36]. While
in many cases their role may seem trivial, their concerted action is essential for normal
cellular function, and even the smallest defect may alter biogenesis or translational
efficiency. For example, a defective ATPase involved in the release of certain
trans-acting factors from the ribosome may result in the inhibition of 40S and 60S subunit
association, and hence inhibit translation [29]. Although a significant number of these
trans-acting factors have been identified, in many cases their function and means of
19
1.5.4 Tools to Identify and Analyze Trans-acting Factors in Ribosome Biogenesis
Despite our comprehensive understanding of the translational mechanism
exercised by the mature ribosomal machinery, the process of ribosomal biogenesis and
the role of several of the involved non-ribosomal proteins in this very dynamic process
are poorly understood. Most of the studies of eukaryotic ribosomal biogenesis to date
were performed in yeast, and mammalian studies are still very premature [26,27].
Nonetheless, the yeast studies performed offer valuable insight into the mechanism of
ribosomal biogenesis in higher eukaryotes, as several of the ribosomal proteins and the
identified trans-acting factors are conserved between yeast and higher eukaryotes [31].
There have been various approaches taken in the investigation of the numerous
premature complexes associated with ribosome biogenesis. One such method involves
the use of tandem affinity purification (TAP) tagging of proteins known for their
involvement in the maturation pathway to isolate various intermediate, pre-ribosomal
complexes [27,29,36,37]. Once isolated, proteins within the complex are identified via
the use of mass spectrometry. In conjunction with TAP tagging, reverse tagging of the
identified proteins, assessment of rRNA content within the complex, and the use of GFP
fusion proteins to observe the subcellular localization of these intermediates allows for
the establishment of a timeline of sequential maturation events [27,29,36,37]. Because
ribosome biogenesis is spatially and temporally regulated, it can be concluded that
pre-ribosomal complexes in close proximity to the nucleolus are more premature than those
found within the nucleoplasm, which are more premature than the late complexes which
20
Another method used to investigate the role of non-ribosomal proteins in
ribosome biogenesis is ribosome profiling. Because ribosome particles sediment
differently, ribosomes can be fractionated on a density gradient, and levels of subunits
observed through the detection of rRNA. Therefore, alteration of expression of a protein
involved in the maturation pathway may consequently affect the levels of mature 60S,
40S, 80S (complexed 60S and 40S), polysomes (several 80S ribosomes complexed to a
single mRNA transcript), or halfmers (several 40S subunits and associated translational
machinery bound to a single mRNA transcript). These observations can lend insight into
the function of the protein under study as it pertains to ribosome synthesis [14,15,19,26].
In many cases, yeast and mammal protein orthologues involved in ribosome
biogenesis fulfill very similar roles in their respective organisms [27]. Despite the
numerous similarities, there are marked discrepancies in the function of these
orthologues, which stress the importance of studying this process in mammals and higher
eukaryotes in conjunction with the yeast model. The eukaryotic initiation factor (eIF6),
shares 77% sequence identity between its orthologues in yeast and humans [32]. This
protein has been shown to be involved in ribosome biogenesis in yeast, presumably in
stabilization of the 60S subunit. Although it is not characterized as well in humans,
studies have also shown eIF6 to be implicated in maturation of the 60S subunit in
humans, presumably fulfilling a similar role as in yeast, considering human eIF6 is able
to recover normal biogenesis in eIF6 deletion mutants in yeast. However, in contrast to
yeast, mammalian eIF6 has been shown to have an additional role in anti-association of
the 60S and 40S subunits, preventing the formation of the 80S particle in the absence of
21
disengage [32]. In addition to illuminating the potential differences between proteins in
this process in yeast and higher eukaryotes, these studies highlight the dual role that may
be fulfilled by several proteins involved in ribosome biogenesis, which could provide
means for crosstalk with other cellular processes.
1.5.5 YVH1 is a Trans-acting protein in Ribosome Biogenesis in Yeast
Recently in yeast, YVH1 has been found to play an important role in maturation
of the 60S ribosomal subunit. Although yeast two hybrid studies have shown YVH1 to
interact with YPH1/ Nop7, a known factor in ribosome biogenesis, the first strong
indication that YVH1 may be a trans-acting factor in ribosome assembly was in a study
that showed that yvh1 deletion strains containing a misfolded mutant form of the
membrane protein pma1-10 were still able to grow [19,38]. Upon further investigation
into the properties of yvh1 that were responsible for suppressing this mutant pma1-10, it
was found that HA-tagged YVH1 co-fractionated with the 60S subunit, and deletion of
the yvh1 gene caused defects in 60S subunit biogenesis [19]. Interestingly, this
phenotype was paralleled in yeast containing deletions of large (60S) subunit proteins
RPL19 and RPL35. Ribosome profiling revealed decreased levels of free 60S and 80S
ribosomal subunits and an increase in free 40S subunits as well as an accumulation of
halfmer polysomes (43S particles which consist of the 40S subunit, and associated
translation initiation factors stalled at the start codon) [14,15]. Complementation studies
using a YVH1 truncated mutant, containing the zinc-binding domain alone, were able to
22
which further investigation into the function of YVH1 in ribosome biogenesis not only
revealed that ribosome profiles change in response to yvh1 deletion, but also used
Northern blotting to detect an accumulation of early pre-60S rRNA (i.e. 27S rRNA) and a
reduction in late 60S rRNA (i.e. 25S rRNA) in response to yvh1 deletion, further
supporting its involvement in maturation of the 60S subunit. A reduction in 60S export
was also observed in these studies [14,15,19]. Interestingly, the zinc-binding domain
alone was found to be capable of restoring normal ribosome profiles, suggesting that the
catalytic domain is not required for ribosome biogenesis. Through this observation, the
slow growth phenotype described in yvh1 deletion yeast was attributed to the observed
defect in ribosome biogenesis. Co-purification of YVH1 with late pre-60S particles
using the TAP method further validates the implication of YVH1 in ribosome biogenesis
[14].
The ribosome stalk is an important structure of the ribosome, which is required
for the recruitment/association of several translation factors. P0 is a protein that forms
the base of the stalk and associates with the 60S subunit only in the cytoplasm. A highly
homologous protein, Mrt4, associates with the ribosome only localized in the nucleolus
and nucleoplasm. Because the association of Mrt4 and P0 with the 60S subunit is
mutually exclusive, it has been suggested that Mrt4 and P0 bind the same site on the 60S
ribosome, hence the association of P0 with the 60S subunit requires that Mrt4 and the
pre-60S subunit have dissociated from each other. However, upon deletion of yvh1, GFP
tagged Mrt4 is specifically mislocalized to the cytoplasm and co-fractionated with the
60S subunit on a sucrose gradient, suggesting its inability to dissociate from the 60S
zinc-23
binding domain of YVH1, but not the catalytic domain alone [14,15]. The human
orthologue hYVH1 was able to complement the yvh1 deletion in yeast, and knockdown
of hYVH1 in both HeLa and HEK 293 cells caused aberrant localization of human
MRTO4, suggesting a similar role in humans [15]. The proposed role for YVH1 in yeast
ribosome biogenesis is shown in Figure 1.7. Despite these observations, further
investigation of the role of hYVH1 in ribosomal biogenesis in human cells is
required with respect to evidence of 60S subunit association and characterization of
the interaction between hYVH1 and the ribosome, and is a major objective of the
proposed research.
1.6 Methods to Explore Structural/Functional Features of Protein Tyrosine
Phosphatases
1.6.1 In Vitro Phosphatase Assays Exploit Artificial Substrates
The first discovered dual specificity phosphatase is VH1 from the vaccinia virus
[9]. Its human orthologue, VHR, shares 30% sequence homology with the N-terminal
catalytic domain of human phosphatase hYVH1. It was therefore estimated that VHR
would serve as a good model for the catalytic activity of hYVH1. However, our lab has
found that the in vitro phosphatase activity exhibited by GST-VHR fusion protein toward
artificial substrates is 453 times greater than that exhibited by GST-hYVH1 fusion
protein [40]. This observation evokes interest as to the structural features responsible for
such a marked decrease in activity and to the functional significance of these
24
25
In vitro phosphatase assays are useful tools that can lend insight into the function
or regulation of protein phosphatases, and assess the consequence of effector molecules
and protein modifications on phosphatase activity [41]. Because of the challenges
associated with phosphatase substrate identification, artificial substrates are often
employed in these assays. There has been much emphasis placed on the design of
artificial substrates, and a researcher may choose from an assortment of substrates to best
suit the specific phosphatase. These substrates include peptides or single amino acids
that can accurately mimic natural substrates. In such cases, hydrolysis of the
phosphoester bond can be observed through the use of radioactive assays or
non-radioactive assays in which released phosphates are detected either through the use of
phospho-specific antibodies or phosphomolybdate colorimetry. Alternatively,
spectrophotometric substrates can be used, which absorb light or fluoresce upon
hydrolysis. These substrates are advantageous as their hydrolysis can be directly and
immediately detected, and although they are not phosphorylated amino acids, their
structures generally mimic that of a phosphorylated amino acid residue, facilitating
recognition by the protein phosphatase.
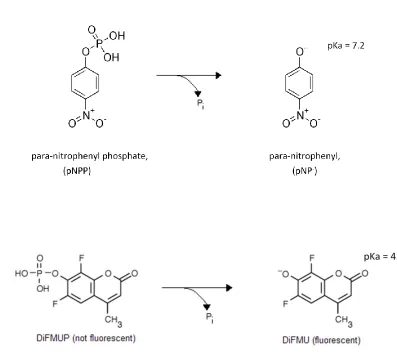
Para-nitrophenyl phosphate (shown in Figure 1.8) is an artificial substrate that
consists of a phenyl ring linked to a phosphate through a phosphoester bond, accurately
mimicking a phosphotyrosine residue. Upon hydrolysis of the phosphor-ester bond,
para-nitrophenyl absorbs light at a wavelength of 450 nm. The p-para-nitrophenyl product has a
pKa of 7.2, and therefore produces a more intense signal at alkaline pH. If the
26
used in a discontinuous assay: quenching the reaction with a base can allow for detection
of para-nitrophenyl [41].
Another spectrophotometric substrate is 6,8-difluoro-4-methylumbelliferyl
phosphate (DiFMUP) (shown in Figure 1.8). Upon incubation with DiFMUP, the
phosphatase under study catalyzes the hydrolysis of DiFMUP to its hydrolysis product
6,8-difluoro-4-methylumbelliferyl (DiFMU). The generation of the fluorogenic product
DiFMU, allows for the rate of formation of DiFMU, and hence the rate of catalysis, to be
monitored [42]. DiFMUP presents advantages over other spectrophotometric substrates
as it has a high fluorescent quantum yield, which allows for greater sensitivity than can
be achieved with alternative artificial substrates (such as OMFP and pNPP). Further,
DiFMU has a lower pKa than do the hydrolysis products of other artificial substrates such
as pNPP, making it a better candidate for continuous phosphatase reactions at a reduced
pH [41,43].
1.6.2 X-ray Crystallography and Supporting Techniques to Elucidate Protein
Structure
X-ray crystallography is a powerful technique that allows for the acquisition of a
protein structure by obtaining resolution at the atomic level [44]. Proteins suitable for
analysis by this method must be able to form highly ordered crystals. The formation of
suitable crystals has been a major obstacle faced by researchers. Various techniques have
27
28
a screening process is established in which various buffers are tested in order to
determine optimal conditions for crystallization of the protein in question. The
conditions tested may include variations in types of buffer and pH, temperature and
presence of ligands (which often enhances protein stability) [45,46].
In addition to these screening methods, crystal engineering is becoming a popular
field of study. Crystal engineering involves the modification of proteins to increase
propensity to form crystal contacts. This may be done through the fusion of the proteins
of interest to a readily crystallized carrier protein or through construct optimization in
which flexible regions/loops are excluded from the protein. Another emerging technique
is surface entropy reduction (SER), which involves site-directed mutagenesis of certain
high entropic residues (such as arginine, glutamine, glutamic acid, lysine) to residues of
lower entropy (such as alanine). This is based on the fact that crystallization is driven by
very small changes in Gibb‟s free energy and that reduction in the thermodynamic cost of
packing high entropy side chains into crystals increases propensity for crystallization
[47]. Therefore, it has been found that these regions of lower entropy are more suitable
to mediate crystal contacts [47,48]. Surface entropy reduction has been experimentally
validated as these mutations are often found to be involved in crystal interfaces [48].
Crystallographic analysis has proven to be important in determining the structural
features of PTPs that mediate kinetic properties of the phosphatase. In particular, through
the determination of the crystallographic structure, the reason for the low activity of some
phosphatases has been discovered by aligning the elucidated structure (of the active site)
of the low activity phosphatase (such as Prl1) with that of DSPs with a higher activity
29
crystallography has proven to be important in studying the substrate-induced activation
mechanism practiced by this class of phosphatases [50,51]. It is predicted that analysis of
hYVH1 via crystallography could reveal fundamental information as to the structural
relationship between the catalytic domain and the novel zinc-binding domain, as well as
provide structural insights regarding the regulation of this enigmatic phosphatase and
clues concerning substrate identification.
In addition to X-ray crystallography, there are other techniques available that can
be used to obtain protein structure, or complement that observed through X-ray
crystallography. One such technique is protein NMR. This technique has the added
advantage of allowing for the determination of protein structure in solution, as crystal
structures, while generally accurate, occasionally prove to be unfaithful to the protein‟s
native form. However, in the structure determination of large proteins, NMR data
analysis can become extremely complex, and in such cases NMR is more effectively used
for the structural determination of individual domains rather than full length proteins
[52].
Yet another technique that allows for the observation of protein structure in
solution is small angle x-ray scattering (SAXS). While this method reveals a very low
resolution image of the protein structure (protein envelope), it does not require isotopic
labelling and is often sufficient in the validation of protein structure as determined by
X-ray crystallography [53,54]. SAXS is of particular use as this technique does not have
molecular weight limitations as do other techniques, such as NMR, and may lend
structural insight into regions that are too flexible to be resolved using X-ray
30
three-dimensional protein shape and domain arrangement under a variety of conditions,
ranging from near physiological to denaturing [55]. This allows for the exploration of
structural changes in response to external conditions, affording the observation of global
changes in protein folding from one state to another [56,57]. Additionally, SAXS can be
used for the visualization of global conformational changes in the presence of effector
molecules or ligands, as well as the study of oligomeric complexes in solution. [55,57,58]
In most cases, structural information obtained from SAXS and X-ray
crystallography have been highly concordant [54]. However, while most crystal
structures are validated against those structures determined by SAXS, there are several
cases, particularly in the determination of quaternary structure of protein complexes, that
revealed large discrepencies in the crystallographic models, highlighting the importance
31 1.7 Objectives
This study focuses on elucidating novel structural features of the human dual
specificity phosphatase hYVH1. Particular emphasis is placed on the relationship
between the catalytic domain and novel zinc-binding domain as well as their relevance in
the proposed function of hYVH1 as a trans-acting factor in ribosome biogenesis.
The specific aims are as follows:
1) Confirm the interaction of hYVH1 with the ribosomal subunits and, in the event that
hYVH1 does interact with the ribosome, further characterize this interaction by:
a. Investigating the domains or regions of hYVH1 required for this
interaction;
b. Evaluating the effects of phosphorylation and oxidation of hYVH1 on
ribosomal binding.
2) Prepare samples for structural analysis of hYVH1 by X-ray crystallography as well as
explore conditions in vitro that may enhance activity of hYVH1 toward artificial
substrates with the common goal of elucidating information concerning modes of
32
CHAPTER 2
Materials and Methods
2.1 Plasmids
The synthesis of pGEX-4T1 vector containing wild type hYVH1 has been
previously described [40]. This plasmid was harvested from a 5mL overnight culture as
described by the manufacturer using GenElute Plasmid Miniprep Kit (Sigma-Aldrich,
Inc.). To generate pGEX-4T1 Zn∆hYVH1, PCR-based site directed mutagenesis was
performed on purified pGEX-4T1 plasmid containing the wild type hYVH1 insert.
Oligonucleotide primers (synthesized by Invitrogen Corp.) were designed to carry out the
mutation Y191Stop. The forward primer used is 5‟-GGTTACAGAGAAGTAGCCA
GAATTGC–3‟, while the reverse primer is 5‟-GCAATTCTGGCTACTTCTCTGTAA
CC–3‟. Following PCR, DpnI (Invitrogen Corp.) digestion was performed for 1hr at
37oC. Competent DH5a cells were then transformed with PCR products (that were
subjected to DpnI digestion) as described below. Plasmids were harvested using
GeneElute Plasmid Miniprep kit and plasmids verified by automated sequencing (ACGT
Corp.).
The truncated hYVH1 derivative lacking the first 28 N-terminal amino acid
residues, E29 hYVH1, and that lacking the first 45 N-terminal amino acid residues, D46
hYVH1, were obtained by PCR using wild type pGEX-4T1 hYVH1 as a template. The
forward primer used to generate E29hYVH1 is: 5‟-GCGAATTCGAAGTGCAGCCAG
33
GAATTCGATCACCTGAGGGAAGCGGGC-3‟, both of which contain an Eco-RI
restriction site. The reverse primer used for both mutants was: 5‟-CGATGCGGCCGCT
CTAGAACTAGTGG-5‟, which contains a NotI restriction site. PCR products were then
subjected to DpnI digestion to degrade template DNA, verified on a 1% agarose gel, and
purified using QIAquick® PCR Purification Kit (Qiagen, Inc.), according to
manufacturer‟s instructions. Subcloning of the purified inserts into the pGEX-4T1 vector
was achieved using the restriction enzymes EcoRI and NotI, and the ligated plasmids
transformed into highly competent DH5a cells. Plasmids were harvested using Sigma
miniprep kit, and the sequence verified by automated sequencing (ACGT Corp.).
Surface entropy reduction (SER) mutants were predicted by submission of
primary sequence of hYVH1 to http://nihserver.mbi.ucla.edu/SER/ and the top 5 clusters
of mutations were generated by site directed mutagenesis, using pGEX 4T1 containing
wild type hYVH1 as a template. SER1, which consists of the mutations of the glutamic
acid residues at positions 62 and 63 to alanines, was generated using a single set of
primers: sense 5‟-CAGTGGACTCGGCGGCGCCCAGCTTC-3‟, and antisense 5‟-GA
AGCTGGGCGCCGCCGAGTCCACTG-3‟. All other SER mutants were generated
through two (for SER3, SER4, SER5 variants) or three (for SER2 variant) separate
rounds of site directed mutagenesis, each round employing a new set of primers to
introduce a new mutation. The primers used to construct SER2 are as follows: SER2,
round 1 (E151A): Sense, 5‟-CCAGATTCTCAAACCAGCGGCTAAGATGAATGAG-
3‟ Antisense, 5‟-CTCATTCATCTTAGCCGCTGGTTTGAGAATCTGG-3‟; SER2
round 2 (E151A, K152A) Sense,
34
3(K149A, E151A, K152A) Sense, 5‟-GCTCCAGATTCTCGCACCAGCGGCTGCG-3‟,
Antisense, 5‟-CGCAGCCGCTGGTGCGAGAATCTGGAGC-3‟. The primers used to
generate SER3 are as follows: SER3 round 1 (E189A) Sense, 5‟-CAAAAGGTTA
CAGCGAAGTATCCAGAATTGCAG-3‟, Antisense 5‟-CTGCAATTCTGGATACTTC
GCTGTAACCTTTTG-3‟; SER3 round 2 (E189A, K190A) Sense, 5‟-CAAAAGGT
TACAGCGGCGTATCCAGAATTGCAG-3‟, Antisense, 5‟-CTGCAATTCTGGATAC
GCCGCTGTAACCTTTTG-3‟. Primers used to generate SER4 are as follows: SER4
round 1 (E305A) Sense, 5‟-CTGGTATGGTGCACAGTGCTCTTGTGGTAGGTG-3‟,
Antisense 5‟-CACCTACCACAAGAGCACTGTGCACCATACCAG-3‟; SER4 round 2
(E305A, Q306A) Sense, 5‟-CAACTGGTATGGTGCAGCGTGCTCTTGTGGTAGGT
GG-3‟, Antisense, 5‟-CCACCTACCACAAGAGCACGCTGCACCATACCAGTTG-3‟.
Primers used to generate SER5 are as follows: SER5 round 1 (E200A) Sense,
5‟-GCAGAATTTACCTCAAGCACTCTTTGCTGTTGACCC–3‟, Antisense 5‟-GGGTCA
ACAGCAAAGAGTGCTTGAGGTAAATTCTGC-3‟; SER5 round 2 (Q199A, E200A)
Sense, 5‟-GCAGAATTTACCTGCAGCACTCTTTGCTGTTGACCCAAC-3‟ Antisense,
5‟-GTTGGGTCACCAGCAAAGAGTGCTGCAGGTAAATTCTGC-3‟.
Following each round of site directed mutagenesis, the PCR product was
subjected to DpnI digestion, followed by transformation into highly competent DH5a
cells. The generated plasmids were then harvested using GenElute™ Plasmid Miniprep
Kit (Sigma-Aldrich, Inc.), verified by agarose gel electrophoresis and automated
sequencing (ACGT Corp.), and used as a template for the following round of site directed
35
pCMV2-flag DThYVH1 was generated by site directed mutagenesis using the
singly mutated pCMV2-flag D84AhYVH1 as a template. Sense and antisense primers
were designed to flank glutamine at amino acid position 161 and mutate this residue to an
alanine. Sense: 5‟-GGGGTTTGAGTGGGCACTGAAATTATACC-3‟, and Antisense:
5‟-GGT ATAATTTCAGTGCCCACTCAAACCCC-3‟. PCR products were subjected to
DpnI digestion and plasmids transformed, harvested, and verified as described above.
Construction of mammalian DNA plasmids used (pCMV2-flag
hYVH1,pCMV2-flag C115 S, pCMV2-hYVH1,pCMV2-flag CT1 hYVH1, pCMV2-hYVH1,pCMV2-flag CT2 hYVH1, pCMV2-hYVH1,pCMV2-flag CT3
hYVH1, pCMV2-flag S14A hYVH1, pCMV2-flag S14E hYVH1, pCMV2-flag S335E
hYVH1) have been previously described [24,40,60].
2.2 Cell Culture and Transfections
HEK 293 cells were obtained from American Type Tissue Culture Collection.
HEK 293 cells were cultured in Dulbecco‟s Modified Eagle‟s Medium/Nutrient F-12
HAM supplemented with 8% fetal bovine serum, 2mM glutamine, 100U/mL penicillin
and 100µg/mL streptomycin at 37ºC, 5% CO2. Approximately 2x106 cells were seeded
into 100mm cell culture plates 24 hours prior to transfections. Cells were grown to
approximately 70-80% confluency and subsequently transfected using polyethyleneimine
(PEI)-mediated transfection. Fresh media was replaced 4 hours prior to transfection. PEI
transfection proceeded as follows: 50µL of a 150mM solution of NaCl was mixed and
incubated with 5 to 20µg of DNA, while simultaneously 50µL of 150mM NaaCl was
36
minutes at room temperature. Following incubation, solutions containing DNA and PEI
were mixed and incubated at room temperature for an additional 10 minutes. DNA/PEI
transfection mixtures were then added dropwise to tissue culture plates. Plates were
gently mixed, and subsequently incubated at 37ºC, 5% CO2 for 24 to 36 hours. For
tert-butyl hydroperoxide treatments, cells were treated 24hrs after transfection with
concentrations of 200µM, 500µM, or 2mM tert-butyl hydroperoxide and incubated for 4
hours at 37ºC, 5% CO2.
2.3 Ribosome Profiling
All solutions used for ribosome profiling were made using DEPC treated water.
DEPC treated water was prepared by adding DEPC (Sigma-Aldrich, Inc.) to millipore
water to a final concentration of 0.1% (vol/vol). The DEPC solution was mixed
thoroughly, incubated at 37ºC for at least 18hours and subsequently autoclaved. All
sucrose solutions had a buffer composition of 80mM NaCl, 5mM MgCl2, 20mM
Tris-HCl pH 7.4, 1mM DTT. Sucrose gradients were made by carefully layering 2mL of a 5%
sucrose buffer on top of 2mL of a 60% sucrose buffer in a Beckman Coulter
Ultracentrifuge tube (Beckman Coulter, Inc.) and incubating the tube on its side for 2.5 to
3hrs at room temperature.
Lysates were prepared for fractionation according to a modified version of the
protocol described by Idol et al. [26]. Approximately 1x107 cells were washed in
phosphate buffered saline and were incubated in trypsin for 1 minute to facilitate
37
and collected by centrifugation at 1100xg for 7.5min at 4ºC. Following centrifugation,
cell pellets were gently resuspended in ice cold PBS containing 100µg/mL cycloheximide
and incubated for 10min on ice. Following a second centrifugation step, cell pellets were
resuspended in 100µL cold hypotonic buffer (1.5mM KCl, 2.5mM MgCl2, 5mM
Tris-HCl pH 7.4) supplemented with 4µL RNaseOUTTM Ribonuclease Inhibitor (Invitrogen
Corp.) before adding 100µL cold hypotonic lysis buffer (1.5mM KCl, 2.5mM MgCl2,
5mM Tris-HCl pH 7.4, 2% sodium deoxycholate, 2% Triton X-100, 2.5mM DTT). Cells
were lysed by 40 strokes in a pre-chilled Dounce homogenizer (Kontes) and centrifuged
at 8000xg for 10min at 4ºC to remove cellular debris. Total protein concentration was
determined by the Bradford method, and lysates were supplemented with 1.7mg/mL
heparin (Sigma-Aldrich, Inc.). Lysate volumes corresponding to equal amounts of total
protein (approximately 1.5mg) were loaded onto the sucrose gradient and fractionated by
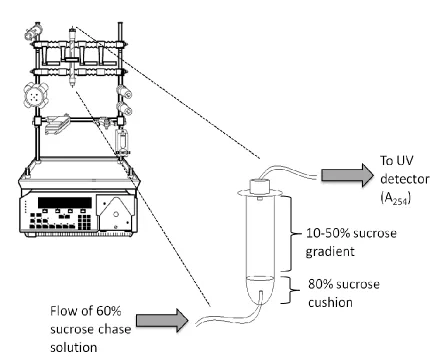
ultracentrifugation (Beckman Coulter Optima MaxE, Beckman Coulter, Inc.) at 245
000xg for 3hours at 4ºC on a swinging bucket rotor (MLS-50).
Following centrifugation, a 50% sucrose cushion was carefully layered on top of
the gradient and an 80% sucrose cushion was injected through the base of the
polyallomer centrifuge tube. The tube was capped and plumbed in line with a UV
detector with filter set to 254nm. A 60% sucrose chase solution was then continuously
pumped into the centrifuge tube at a flow rate of 1mL/min using a BioLogic LP
Chromatography System (Bio-Rad Laboratories). Fractions corresponding to
cytoplasmic RNA, intermediate fractions, 40S, 60S, 80S and polysomes were collected