Original Article
CODEN: IJPNL6
DEVELOPMENT AND VALIDATION OF A NEW RP-HPLC METHOD FOR THE
DETERMINATION OF DACLATASVIR DIHYDROCHLORIDE IN BULK AND
PHARMACEUTICAL DOSAGE FORMS
Abdul Kader
1, Milon Mondal
1*, Mohammad Asikur Rahman
1, Mukta Parvin
2,
Md.
Rafiquzzaman
1, Sukalyan Kumar Kundu
11
Department of Pharmacy, Jahangirnagar University, Savar, Daka-1342, Bangladesh
2Department of Pharmaceutical Science, North South University, Bashundhara, Dhaka-1229,
Bangladesh
*Corresponding author e-mail: [email protected]
Received on: 07-08-2017; Revised on: 11-09-2017; Accepted on: 15-09-2017
ABSTRACT
A simple, rapid reverse phase high-performance liquid chromatographic method has been developed and validated for the estimation of Daclatasvir Di-hydrochloride (DTDH) in bulk and in a pharmaceutical dosage form. Chromatography was carried out by Spherical Octyl Silane (C8 silica, column 250 x 4.6mm internal diameter was 5-µm), using mobile phase of composition of tri-ethylamine buffer (pH 5.00): acetonitrile (50:50 (v/v)). The flow rate was 1.0 mL min-1 and a peak was observed at about 6.13 minute as detected by a UV detector at 315 nm. The method was validated according to ICH guideline, checking the different analytical parameters such as linearity, precision, accuracy, specificity and robustness. The calibration curve was found to be linear (r2 =0.9997) for the analyte DTDH in the concentration range of 15-45µg/mL. The average recovery was found to be 98.42% to 100.64% for DTDH.
Keywords:Daclatasvir di-hydrochloride; Estimation; RP-HPLC; Pharmaceutical dosage form; Validation
INTRODUCTION
Hepatitis C virus (HCV) infection affects about 160 million people [4] around the globe and DTDH is used for the treatment of hepatitis C Virus (HCV). Chemical name of DTDH is Methyl [(2S)-1-{(2S)-2-[4-(4’-{2-[(2S)-1-{(2S )-2- [(methoxycarbonyl)amino]-3-methylbutanoyl}-2-pyrrolidinyl]-1H -imidazol-4-yl}-4-biphenylyl)-1H -imidazol-2-yl]-1-pyrrolidinyl}-3-methyl-1-oxo-2-butanyl]carbamate; dihydrochloride. It has an empirical formula of C40H50N8O6.2HCl and a
molecular weight of 811.8068. It inhibits the HCV nonstructural protein NS5A [1, 2]. Recent research suggests that it targets two steps of the viral replication process, enabling rapid decline of HCV RNA [3]. It is on list of essential medicine of
the World Health Organization and being marketed worldwide. Molecular formula of this drug is as below Figure 1.
The quantification of direct antiviral agents simeprevir, daclatasvir, ledipasvir, sofosbuvir/GS-331007, dasabuvir, ombitasvir and paritaprevir, together with ritonavir, in human plasma performed by UHPLC-MS/MS [7] and for the imultaneous quantitation of three novel hepatitis C antivirals, daclatasvir, asunaprevir, and beclabuvir in human plasma performed by Multiplexed LC-MS/MS method [8]. Another worked has been reported on validation and method development of assay and dissolution method for estimation and pharmaceuticals tablet dosage form by reverse phase HPLC [9] for the analysis of DTDH where
International Journal of Pharmacy
Mondal, et al. Int J Pharm 2017; 7(4):
7-13
ISSN 2249-1848
methanol in mobile phase was used 80%, which would be more costly.
The present research work has been carried out by using 50% of acetonitrile and tri-ethylamine buffer (pH 5.00), for buffer preparation only need about 2.4 mL tri-ethylamine and less percentage organic solvent used would be cost effective for analysis of DTDH. Column used of octyl silane (C8 silica, column 250 x 4.6mm internal diameter was 5-µm) which is more available and common use in most of pharmaceutical company. So this development work would be beneficiary for the analysis of DTDH in pharmaceuticals dosage form.
This validation study is defined as the process by which it is established, by laboratory studies, that the performance characteristics of the method meet requirements for the intended analytical application [6]. A method was developed and validated according to the guideline of ICH [5]; under the present study to achieve an analytical method with acceptable characteristics of suitability, reliability and feasibility etc.
MATERIALS AND METHODS
Equipment: The analysis of the drug was carried out by HPLC (model: Prominence, Shimadzu, Japan) which contained a quaternary low pressure gradient pump, UV Detector equipped with temperature controlled auto sampler and control column oven.
Chemicals: Daclatasvir Di-hydrochloride was a
gift sample and it was used without further purification, Acetonitrile (HPLC grade) was purchased from Marks, Germany and Tri-ethylamine and orthophosphoric acid 85% were from the university laboratory. HPLC grade demonized water was used in the present study. Tablets used for the development and validation collected from local market, savar, Dhaka in Bangladesh (60 mg). Excipients, lactose monohydrate, microcrystalline cellulose pH 102, croscarmellose sodium, colloidal silicon dioxide, magnesium stearate, opadry yellow were for placebo preparation in this study, which is collected from university laboratory.
Chromatographic Condition: The HPLC system
was of Prominence (Shimadzu). Analysis was carried out at 315 nm with a (C8 silica, column 250 x 4.6mm internal diameter was 5 µm) column using a mobile phase composition of Buffer of tri-ethylamine (pH 5.00): acetonitrile (50:50 v/v). To prepare the buffer solution, 2.38 mL of tri-ethylamine dissolved in about 500 mL of HPLC
grade water. Once dissolved, the pH was adjusted to 5.0 ± 0.05 with orthophosphoric acid. Both, the acetonitrile and the buffer solution were filtered through a 0.45 µ membrane filter.
The flow rate was set to 1.0 mL min-1 for the analysis, temperature for the analysis was set to
30°C, and injection volume used was 20µL. The wave length for detection was set at 315 nm, and the run time was for about 10 min.
Preparation of Standard and sample Solution:
Thirty milligram (30.0 mg) of DTDH (Working Standard) was weighed out and put into a 100 mL dry clean volumetric flask. Sixty milliliter (60 mL) of diluent was added and shook and executed sonication to dissolve DTDH. It was diluted to 100 mL with the diluent and mixed well. Five(s) milliliter of this solution was taken into a 50 mL volumetric flask and was dilute to 50 mL with diluent and that was mixed well. Sample solutions were prepared in the same way as that of the DTDH (WS) and accordingly 30 mg of sample was weighed and used for sample solution preparation.
Content of DTDH was calculated using the following formula:
Daclatasvir Di-hydrochloride content
= AsamAstd x 100Ws x 505 x Wsam100 x 505 x Pstd100 x 100 % Where,
Asam=Peak area of Daclatasvir Di-hydrochloride in Sample Solution chromatogram
Astd=Average Peak area of Daclatasvir Di-hydrochloride in Standard Solution chromatogram Ws =Weight of Daclatasvir Di-hydrochloride Working Standard in mg
Wsam=Weight of sample in mg
Pstd=Potency of Daclatasvir Di-hydrochloride Working Standard in percentage
Validation study: Validation study was carried out for checking the following parameters based on ICH guidelines: System suitability, Specificity, Linearity, Precision, Accuracy (recovery) and Robustness.
RESULTS AND DISCUSSION
Method development: DTDH is being marketed as
trial and error study it has been possible to develop a RP-HPLC method to identify and quantify DTDH. In this method a satisfactory chromatogram was found at RT=6.138 min (Fig. 2)
using the mobile phase of tri-ethylamine buffer (pH 5.00) and acetonitrile (50:50 v/v) with an octyl silan column (250 x 4.6 mm, 5-µm).
System suitability: System suitability test was carried out to verify whether the analytical system was working properly and whether it was able to give accurate and precise results. The system suitability was evaluated for the proposed method as follows. Data from five injections (30.0µgmL-1) were utilized for calculating parameters like Capacity factor (k), Theoretical plates, Resolution, Tailing factor and % RSD. Theoretical plats and Tailing factor parameter were found 6269(ICH≥2000) and 1.058(ICH≤2), respectively. The RSD value of retention time of five injected sample was observed as 0.054% and for area of chromatogram as 0.033%. The stated results for different parameters met the requirements of ICH guidelines.
Stability of DTDH solution: The stability study of DTDH was carried out by preparing solutions of the standard (30 µg mL-1) and sample (30 µgmL-1) and injecting each of those solutions separately at zero min, after four hours and after eight hours of preparation of the solutions. Obtained data are presented below in the Table-1.
If RSD of a drug solution is ≥2.0% then it is considered that the solution is stable. In the present case for the standard as well as for the sample solution, the RSD value was very low (0.51% to 0.55%) and it indicates that the prepare solution with the API and sample f DTDH were stable enough.
Validation study
1. Specificity: For the specificity study,
identification of the active was studied first, comparing the raw material (mobile phase + daclatasvir) with a standard of reference (mobile phase+ daclatasvir reference standard). Another study was carried out to check the absence of interference by the excipients (Placebo) which were present dosage form of DTDH.
It was observed that the injection of placebo did not show any peak under the optimized conditions of HPLC in the present study and no other peak was found with the standard DTDH. It indicates that DTDH standard was free from any impurity. Moreover, peak for the sample coincided with the RT of the standard DTDH. Recovery of DTDH was
with RSD not more than 2% (Table-4) in present
of placebo. It was therefore, concluded that the development method is selective in relation to the excipients of the final preparation.
2. Linearity: The linearity of an analytical method is an assessment of its capability of achieving results that are directly proportional to the concentration of the analyte in the solution. It was established by preparing five different sample solutions of DTDH covering a concentration of 15-45 45μgmL-1 (50% - 150%) and injecting them individually. Peak areas were plotted against actual concentration. The plot was linear from the first sight and the correlation coefficient was observed as (r2 = 0.9997), which was within the limit (r2 =0.995) of ICH guide line. All these results are in support of the linearity and hence it can be said that the RP-HPLC responses are directly proportional to the concentration of the DTDH.
3. Precision Study: In the study of the
instrumental system precision, where RSD of area of standard DTDH was 1.20% and retention time was 0.55%. Six determinations were carried out in a single day (1st day) with six different samples, and average percent of DTDH was found as 99.63% with RSD 0.59% (Table-2). In the second day the assay results of DTDH was found as 99.37% and RSD as 0.99% (data not shown). Since the precision study results were within the ICH limits of ±2.0%, it can be said the proposed method was precise.
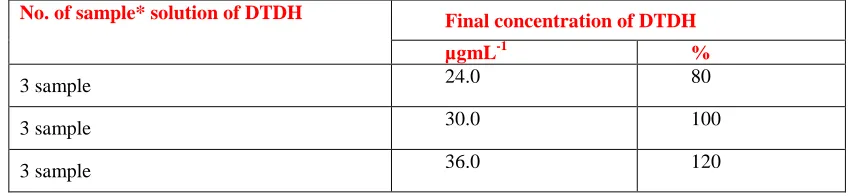
4. Accuracy (recovery): The concentration of
sample prepared for accuracy were 80%, 100% and 120% (24.0, 30.0 and 36.0 µg mL-1). The results obtained from the recovery of nine samples of three different concentrations viz 80%, 100% and 120% (Table-3) indicated that the average recovery was 99.28%, with RSD as 0.80% (Table-4) and thus complied the ICH guideline for % RSD ±2%.
Therefore, it can be concluded that the recovery study of the API in the matrix for the developed method for its (API) assessment from final product was correct, and therefore, the proposed analytical method was sufficiently accurate.
5. Robustness: The method was checked for
Mondal, et al. Int J Pharm 2017; 7(4):
7-1
3
ISSN 2249-1848
limit ≤2%, therefore the proposed RP-HPLCmethod was robust and validated.
CONCLUSION
A simple and quick, new analytical method has been developed to be applied in routine to
determine DTDH and in its tablet dosage form. And the proposed method (RP-HPLC) to determine DTDH in tablet dosage form was found to be linear, precise, accurate, selective and robust.
Table -1: Peak area of the standard (30 µg mL-1) and the sample solutions (30 µg mL-1) of DTDH at different time.
Time Peak area of standard solution
RSD (%)
Peak area of sample solution
RSD (%)
At zero min 2104985
0.55
2209844
0.51
After 4 hours 2118739 2198434
After 8 hours 2095623 2187473
Table-2: Results of precision of DTDH.
Test
Standard weight in mg
Standard Peak area
Sample weight in mg
Sample Peak Area
Assay result in %
Average Assay result in %
1st Test
Sample 1
30.4 2044545
501.5 2582372 99.4
100.0
Sample 2 513.4 2675324 100.6
2nd Test
Sample 1 514.4 2654334 99.6
99.3
Sample 2 514.6 2639876 99.0
3rd Test
Sample 1 506.3 2601349 99.2
99.6
Sample 2 520.3 2695498 100.0
Average Assay results of all test in % 99.63
% of RSD 0.59
Table-3: Samples and their corresponding concentrations
No. of sample* solution of DTDH Final concentration of DTDH
µgmL-1 %
3 sample 24.0 80
3 sample 30.0 100
3 sample 36.0 120
Table-4: Recovery of DTDH API from the simulated samples of DTDH & Excipient.
Sample Conc.
Sample No.
Weight of Standard in mg
Average area of Standard
Weight of Sample
(mg) Area of Sample
Recovery (%) Placebo DTDH
24µg/m l (80 %)
1
30.5 2012103
390.1 96.1 2103284 100.64
2 391.2 96.3 2063245 98.52
3 391.3 96.0 2087463 99.99
30µg/m l (100 %)
1 393.0 120.0 2578208 98.80
2 395.2 121.0 2589876 98.42
3 391.1 120.4 2605705 99.52
36µg/m l (120 %)
1 392.3 144.0 3102726 99.08
2 392.3 144.1 3134584 100.03
3 392.3 144.3 3091283 98.51
Average 99.28
% RSD -0.80
Table-5: Effect of various parameters checked in robustness.
Parameter Condit ion
Absorban ce
Resul ts (%)
Mean value (%)
RSD
ICH Limit (%)
Flow rate
0.8 3382980 99.8
99.65 0.61
NMT 2
1.0 2878683 99.6
1.2 2384988 99.5
pH of buffer
4.8 2728115 98.5
99.23 1.32
5.0 2781828 101.8
5.2 2664734 98.8
MP composition (Buffer : ACN)
56:44 2715653 98.6
99.72 0.89
50:50 2736913 100.3
44:56 2689594 100.3
Temperature
27 2799486 99.7
99.47 0.98
30 2699414 99.6
ISSN
2249-Mondal, et al. Int J Pharm 2017; 7(4):
7-13
1848
Fig. 1: Structural formula of Daclatasvir Di-hydrochloride.
Fig. 2: Chromatogram of Daclatasvir Di-hydrochloride (RT = 6.138 min)
Fig. 3: Plot of concentration vs peak area of DTDH
REFERENCES
2. Guedj J, Dahari H, Rong L, Sansone ND, Nettles RE, Cotler SJ, Layden TJ, Uprichard SL, Perelson AS. Proc Natl Acad Sci U S A, 2013; 110(10): 3991-6.
3. Zhang X. Acta Pharm Sin B, 2016; 6(1):26-31.
4. Gentile I, Borgia F, Coppola N, Buonomo AR, Castaldo G, Borgia G. Curr Med Che, 2014; 21(12): 1391-404.
5. Guideline IH. Validation of analytical procedures: text and methodology. Q2 (R1). 2005; 1. 6. Sarrio RV, Silvestre LJ. Bioserch 1996.
7. Jiang H, Kandoussi H, Zeng J, Wang J, Demers R, Eley T, He B, Burrell R, Easter J, Kadiyala P, Pursley J.J Pharm Biomed Anal, 2015; 107: 409-18.
8. Ariaudo A, Favata F, De Nicolò A, Simiele M, Paglietti L, Boglione L, Cardellino CS, Carcieri C, Di Perri G, D’Avolio A . J Pharm Biomed Anal, 2016; 125: 369-75.