a , ' u
-Ch a r a c t e r i s a t i o n o f B c e l l e p i t o p e s o n h u m a n CHORIONIC GONADOTROPIN
M arie Catherine Françoise C H A R R E L -D E N N IS
A thesis subm itted for the degree o f Doctor o f Philosophy
Departm ent o f Im m unology and M olecular Pathology
University College London
London
All rights reserved
INFORMATION TO ALL USERS
The quality of this reproduction is dependent upon the quality of the copy submitted.
In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uest.
ProQuest U643814
Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author.
All rights reserved.
This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC.
ProQuest LLC
789 East Eisenhower Parkway P.O. Box 1346
Ab s t r a c t
A B cell epitope is defined as part o f the antigen that is recognised by any given antibody. Despite the identification of common features, such as size or general shape, by the analysis of crystal structures of antigen/antibody complexes, relatively little is understood concerning B cell epitopes compared to T cell epitopes. This project aimed to characterise B cell epitopes on the P-subunit of human chorionic gonadotropin (hCG), because the availability of a crystal structure and a well-defined panel o f monoclonal antibodies made p-hCG an ideal model. The production of hCG is largely restricted to pregnancy and its presence is necessary for the implantation of the fertilised egg. It has thus been employed in clinical trials as a potential contraceptive vaccine. The information gained during this project could increase our general knowledge concerning B cell epitopes, which ultimately could be helpful in the design of a safe birth control vaccine. Four separate approaches toward defining B cell epitopes on p-hCG were investigated: • The feasibility o f identifying antibody-protected amino acids on a linear peptide
epitope was examined using mass spectrometry in deuterium exchange experiments. The results obtained revealed the limitations of this technique, particularly in relation to the proportion of amide hydrogens engaged in hydrogen bonds and the presence of water molecules at the antibody/antigen interface.
• A peptide library was screened with hCG-specific monoclonal antibodies to identify and characterise mimotopes of the relevant epitope. Although peptides mimicking the antigenicity were identified and information concerning the antigenic profile of this epitope were gained, the peptides did not elicit hCG-reactive antibody responses in vivo.
• Site-specific mutagenesis o f P-hCG combined with expression on the surface of mammalian cells was employed to define residues involved in an hCG-specific epitope.
et la sagesse à ne pas s*en soucier. ”
Ac k n o w l e d g e m e n t s
I would first like to thank Professor Ivan M. Roitt, Dr. Peter J. Delves and Dr. Torben Lund for the opportunity to come to London and work in their laboratory. Their knowledge, optimism and support have been invaluable over the past few years. I would also like to acknowledge Professor Mike Steward and Dr. Caroline Stanley for their guidance during the construction and analysis of the peptide libraries. Professor Tom Rademacher and Professor Carol Robinson must also be thanked for their collaboration and expertise in the complex field o f mass spectrometry. Dr. Charles Kelly has been of great help in the analysis of the results obtained using surface plasmon resonance technology.
My appreciation goes to Dr. Jean-Marc Reyrat, his passion for Science and his professionalism have motivated my decision to take this path, not always easy but always interesting.
In addition, I must extend my thanks to the members of the MMBU and the “little lab”, both past and present, who have always been generous with their time, material and expertise. In particular, I would like to thank Dr. Solveig Hannesdottir (#SS1), who has been a precious moral help in the past 4 years and such a good fiiend. A special mention to Solveig, Jonathan, Ruth, Kevin, Marta, Mike, Kate and the others for the numerous K&Q meetings that permitted me to forget that it rains once too many times in this country.
I would like to thank my family for their constant support and encouragement throughout this PhD and the circuitous path that led to it. Merci.
Title...1
Abstract... 2
Acknowledgements...4
List of figures...13
List of tables...15
List of abbreviations...16
I. General Introduction... 19
1.1. Overview of the Immune System...20
1.1.1. Innate immunity...20
1.1.1.1. The receptors of the innate immunity... 20
1.1.1.2. The instructive role of innate immunity... 21
1.1.2. Acquired immunity... 22
1.1.2.1. The cellular players of acquired immunity...22
1.1.2.1 .a. T helper cells... 22
1.1.2.1 b. Cytotoxic T cells...22
1.1.2.1.C.B cells...23
1.1.2.2. The recognition receptors... 23
1.1.2.2.a. TheTC R ... 23
1.1.2.2.b. The BCR... 24
1.1.2.2.C. Evolution of the B cell recognition receptor throughout the primary immune response 26 1.2. The antigen/antibody complex...27
1.2.1. The antibody... 27
1.2.2. The antigen... 28
1.2.2.1. Nature of the factors determining immunogenicity of an antigenic determinant... 28
1.2.2.2. Identification of B cell qpitopes...29
1.2.2.2.a. Fragmaitation of the antigen...29
1.2.2.2.b. Mutants of the antigen... 30
1.2.2.2.C. Determination of the structure of the antigen/antibody complex... 30
1.2.2.3. Intrinsic characteristics of an epitope... 31
1.2.2.3.a. The structural nature of an epitope... 31
1.2.2.3.b. Critical residues in epitopes...33
1.2.3. The antibody/antigen interaction... 34
1.2.3.1. Thmnodynamics of the interaction...34
1.2.4. Predicting B cell epitopes...36
1.3. Human chorionic gonadotropin...38
1.3.1. The essential role of hCG during pregnancy... 38
1.3.2. Expression ofhC G ... 41
1.3.3. Structure of hCG... 41
1.3.3.1. The glycoprotein hcmnone femily...41
1.3.3.2. The primary structure of hCG...42
1.3.3.2.a. The a-subunit...42
1.3.3.2.b. The P-subunit...42
1.3.3.3. Secondary and tertiary structure of hCG...45
1.3.3.4. The glycosylation of hC G ...47
1.3.3.5. Assembly and secretion of the heterodimer...47
1.3.4. Epitopes of hCG... 48
1.3.4.1. T cell epitopes... 48
1.3.4.2. B cell epitopes... 48
1.3.4.2.a. Epitopes located on the a-subunit...49
1.3.4.2.b. Epitopes located on the P-subunit... 50
1.3.4.2.C. Epitopes located on the holo-hormcne...51
1.4. An overpopulated world... 53
1.4.1. The currait situation... 53
1.4.2. One solution: the contraceptive vaccine... 53
1.4.3. Hormone vaccines... 53
1.4.3.1. Vaccines under development... 55
1.4.3.1.a. The GnRH and FSH vaccines...55
1.4.3.1.b. The hCG vaccines ...55
1.4.3.1.e. The hCG vaccines require improvements... 57
1.5. Aim... 58
II. Materials and Methods... 59
n .l. Materials...60
II. 1.1. Standard buffers and solutions...60
n.1.2. Suppliers... 62
n.1.3. Description and source of Escherichia coli strains... 64
II. 1.4. Description and sources of plasmids...64
n. 1.5. Descriptirm and source of the cell line...68
II. 1.6. Antibodies...68
n.1.7. Peptides and proteins... 72
II. 1.8. Prima-s... 72
IL2.1. Bacterial Methods...73
11.2.1.1. Propagation of bacteria...73
II.2.12. Long term storage of bacteria... 73
11.2.1.3. Preparation of competent E. coli cells... 74
11.2.1.4. Transformation of bacteria... 74
11.2.2. DNA methods... 75
11.2.2.1. Preparation of plasmid DNA from K coli cells... 75
II.2.2.1.a. Small-scale plasmid DNA extraction from transformed bacteria... 75
n.2.2.1.b. Large-scale plasmid DNA extraction from transformed bacteria... 75
11.2.2.2. Separation of DNA fragments by agarose gel electrophoresis...77
11.2.2.3. Restriction enzyme digests of plasmid D N A ... 77
n.2.2.4. Removal of the 5' phosphate group from DNA (dephosphorylation)... 78
11.2.2.5. Purification of DNA fragm aits...78
IL2.2.6. DNA ligation reaction:... 78
11.2.2.7. Amplification of DNA by PC R ...79
11.2.2.8. DNA Sequencing... 79
11.2.3. Cell culture... 80
n.2.3.1. Culture conditions...80
IL2.3.2. Routine cell passage...80
11.2.3.3. Freezing and recovery of cell stocks... 80
n.2.3.4. Transient transfection of plasmids... 81
n.2.3.4.a. DEAE transfection method...81
IL2.3.4.b. Calcium phosphate transfection method... 81
11.2.3.4.c. Liposome transfection method... 81
11.2.3.4.d. Electroporation...82
11.2.3.5. Construction of stable cell lines...82
11.2.3.6. Analysis of the transfection... 83
11.2.3.6.a. Microscopy analysis... 83
11.2.3.6.b. Fluorescence-activated cell sorter (FACS)... 83
11.2.4. Protein methods...83
n.2.4.1. Protein quantification... 83
II.2.4.2. SDS-polyacrylamide gel electrophoresis (SDS-PAGE)...84
n.2.4.3. Coomassie staining of SDS-polyacrylamide gel... 84
11.2.4.4. Silver staining of SDS-polyacrylamide gel...84
n.2.4.5. Transfer of proteins to nitrcx^llulose memlrane (Western blotting)... 85
11.2.4.6. Immunodetection of proteins on Western blot... 85
n.2.4.7. Preparaticm of IgG Fab fi-agmait... 85
n.2.4.8. Screening of the combinatorial peptide library... 86
II.2.4.10. Method for SPOT analysis...86
n.2.4.11. En2yme-linked immimoabsorbent assay (ELISA)... 87
II.2.4.12. SMART size exclusion chromatography column... 88
11.2.5. Immunisation... 88
11.2.6. Serum extraction from blood sample... 89
11.2.7. Mass Spectrometry...89
n.2.8. Surfece plasmon resonance technology...90
III. Identification of antibody-protected residues of a linear epitope in deuterium exchange experiments... 91
m i . Introduction... 92
m.1.1. The mass spectrometer... 92
III. 1,1.1. The ion source... 92
III. 1.1.l.a. The desorption techniques...93
m.1.1.l.b. The spray techniques... 94
III. 1.1.2. The mass analyser...96
III. 1.2. Fractional abilities of the mass spectrometer...98
III. 1.3. MS reveals a broad range of information frcm the sequence to the structure of a protein 99 in . 1.3.1. Determination of the mass of a protein... 99
III. 1.3.2. Protein sequencing...100
m . 1.3.3. Protein conformational studies... 101
III. 1.3.3.a. Charge state distribution (CSD)... 101
ni.l.3.3.b. Deuterium/hydrogen exchange studies... 102
m.l.3.3.c. Chemical modifications... 104
III. 1.3.4. Studies of non-covalent protein complexes...105
III. 1.3.5. The antibody/antigen non-covalent complex...106
m.l.3.5.a. Analysis by enzymatic degradation... 106
III. 1.3.5.b. Analysis by chemical modification... 107
III. 1.4. The OT3A2 antibody binds to the hCG C-terminal peptide... 107
m.1.5. Aim of the study...107
m.2. Results...108
111.2.1. Structural study of the peptide 127-145...108
111.2.2. Determination of the mass... 108
111.2.2.1. Determination of the mass using MALDI-FT-MS... 108
ni.2.2.2. Determination of the mass using ESI-FT-MS...109
in.2.3. Determination of the optimal conditims of fi-agmentation... 113
m.2.3.1. Using MS/MS...113
111.2.3.2. Using nozzle-skimmer potential... 113
m.2.4. Identificatim of the Augments created... 117
IIL2.5.1.b.ThepH...123
IIL2.6. Does the Fab fragment of an antibody protect the residues of the epitope from exchange?.. 125
ni.3. Discussion... 132
ni.3.1. Determination of the mass and sequence of the peptide 127-145...132
III.3.2. The peptide 127-145 does not have an inherent structure... 133
in.3.3. Deuterium exchange e?q)eriment... 133
m .3.4. OT3A2 Fab fragment does not protect the residues of the epitope from deuterium exchange ... 135
in.3.4.1. The nature of antigen/antibody interactions...135
ni.3.4.2. Exchange occurs during or after ionisation...136
in.3.5. Possible experimental modifications... 137
IV. Selection and analysis of eight-mer peptides mimicking a conformational epitope ofp-hC G ... 138
IV.l. Introd action... 139
rv.1.1. Synthesis of the peptide libraries... 139
rV. 1.2. Information gained by the use of peptide libraries...141
IV, 1.2.1. Information gained on linear epitopes... 141
rV. 1.2.2. Information gained on conformational epitopes... 143
rV. 1.2.3. Information gained on hCG using peptide libraries...146
IV. 1.3. Aim...146
rv.2. Results...147
rV.2.1. Presentation of the experimental design...147
rV.2.2. Screening of the chemical combinatorial library...147
rV.2.3. Functional analysis on SPOT membrane... 150
rV.2.3.1. Peptide design... 150
rv.2.3.2. Identification of potential mimotopes on the nitrocellulose membrane... 153
rV.2.3.2.a. Detection of the binding by a P-galactosidase-conjugated antibody... 153
rv.2.3.2.b. Detection of the binding by a HRP-conjugated antibody... 157
rV.2.4. Antigenic and immunogenic studies of potential mimotopes... 161
rv.2.4.1. Antigenicity studies...161
IV.2.4.2. Immunogenicity studies... 166
rV.2.4.2.a. Immunogenicity studies on the sera of mice immunised with M A P-M l... 168
rV.2.4.2.b. Immunogenicity studies on the sera of mice immunised with MAP-M2... 169
IV.2.4.2.C. Immunogenicity studies on the sera of mice immunised with MAP-M3... 170
rV.2.4.2.d. Immunogenicity studies on the sera of mice immunised with MAP-M4...171
rv.3. Discussion... 174
rv.3.1. The binding of INN-2 mAh to the peptides of the sub-library is specific...175
rV.3.2. Identification of critical characteristics in the epitope recognised by lNN-2 mAb...175
rv.3.3. Hypothetical mapping of the epitope recognised by lNN-2 mAb on the crystal structure of hCG... 176
rv.3.4. Recognition patterns of the membrane by lNN-32 mAb and lNN-20 mAb... 177
rv.3.5. Mimotopes were not identified but antigenic mimics were distinguished... 178
V. Functional characterisation of a conformational epitope using a mutagenesis-based method...181
V.l. Introduction... 182
V. 1.1. The mapping of conformational epitopes using mutagenesis has been successfully applied to P-hCG...182
V .l.2. The pi and pT epitopes... 183
V .l.2.1. p i and p i' are hCG-specific, conformational and distinct...183
V. 1.2.2. Residues involved in the p i and PT epitopes... 183
V .l.3. Identification of residues possibly involved in the p i epitope...184
V. 1.3.1. Hypothesis based on the crystal structure analysis...184
V. 1.3.2. Hypothesis based on the glycosylation studies... 187
V .l.4. Aim ... 187
V.2. Results...188
V.2.1. Construction of surface expressed mutants and wild type p-hCG... 188
V.2.1.1. Introduction of the mutations into the P-hCG gene... 188
V.2.1.2. Construction of plasmids permitting mammalian cell surface expression of the mutants and wild type p-hCG...195
V.2,1.2.a. Construction of plasmids for transient transfection into mammalian cells...195
V.2.1.2.b. Construction of plasmids allowing stable transfection... 196
V.2.2. Surface expression of the proteins... 199
V.2.2.1. Transient expression of the proteins...199
V.2.2. l.a. Detmnination of the most appropriate method of transfection...199
V.2.2. l.b. Transient expression of wild type P-hCG... 201
V.2.2.2. Stable expression of the proteins... 201
V.2.3. Antigenic analysis of the proteins... 204
V.3. Discussion...206
V.3.1. Expression systems... 206
V.3.2. Characterisation of P-hCG-specific epitopes using a mutagenesis-based method... 207
V.3.2.1. The (31 antigenic region... 207
VI. Antigenic characterisation of the p-hCG mutant R68£... 211
VI.1. Introduction... 212
VI. 1.1. Biosensors permit the characterisation of the antibody/antigen interaction... 212
VI.1.1.1. The principle of surface plaanon resonance...212
VI. 1.1.2. Characta-isation of antigen/antibody interaction using SPR... 213
VI. 1.2. The mutant P-hCG R68E...214
VI.12.1. Previous antigenic diaracterisation... 214
VI. 1.2.2. Production of the mutant P-hCG R68E...215
VI. 1.3. Aim...216
VI.2. Results...217
VI.2.1, Production, detection and characterisatiœi of wild type P-hCG and mutant P-hCG R68E... 217
VI.2.1.1. Construction of the baculovirus expression vectors...217
VI.2.1.2. Identification of optimal conditions of production... 217
VI.2.1.3. Physical characteristics of the p-oteins produced... 219
VI.2.2. Determination of the binding kinetics...221
VI.2.2.1. Experiments controlling for non-specific binding... 222
VI.2.2.2. The bivalaicy of antibody stabilises the Hisg-tagged protein... 225
VI.2.2.3. Production of the Fab fragments...225
VI.2.2.4. Determinatimi of the binding kinetics of the Fab fragments to Bac hCG WT and Bac P-hCG R68E...231
VI.2.2,4.a. Kinetics equations...231
VI.2.2.4.b. Determinaticm of the kinetics constants... 233
VIJ. Discussion...240
VI.3.1. Baculovirus-derived P-hCG is structurally similar to human-derived P-hCG...240
VI.3.2. The binding of the antibodies to Bac p-hCG follows the 1:1 Langmuir model... 241
VI.3.3. The dynamic of the interaction between the antibodies and Bac p-hCG... 241
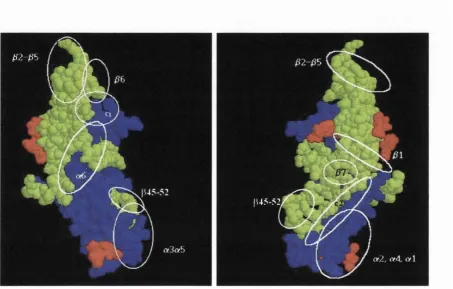
VI.3.4. Characterisation of epitopes of Bac P-hCG R68E... 242
VI.3.4.1. Characta-isation of the binding to the P3 to P5 epitopes... 242
VI.3.4.2. Characterisation of the binding to the P6 and p7 epitopes...243
VI.3.4.3. Characterisation of the binding to the p i epitope...243
VI.3.5. Epitope specificity of the immune response...244
VII. Conclusions... 246
Vn.1. Achievements... 247
VII. 1.1. Epitope mapping of P-hCG... 247
VIL 1.1.2. The P6 and P? epitopes...249
Vn. 1.1.3. A CTP epitope...250
VU. 1.2. Understanding B cell epitopes... 250
Vn.2. Future work... 252
VII.2.1. Epitope mapping of P-hCG...252
VII.2.2. The hCG vaccine...252
Figure 1.1 : Ribbon diagram o f a soluble IgG l...25
Figure 1.2: Gene rearrangements o f the human heavy chain o f a secreted IgM...25
Figure 1.3: Prevalence of amino acid residues on the surface of a protein and in an epitope... 34
Figure 1.4: Pattern o f plasma hormones during pregnancy...40
Figure 1.5: Organisation o f the genes coding for the a - and p-subunit o f hCG... 44
Figure 1.6: Crystal structure representation of the hCG heterodimer... 46
Figure 1.7: Epitopes localisation on hCG...52
Figure 1.8: Hormones involved in the production o f gametes, sex steroids and maintenance o f pregnancy...54
Figure lU.l: Schematic representation o f the thermally assisted electrospray ionisation source with counter-current gas... 95
Figure III.2: Principle o f ion cyclotron resonance... 97
Figure III.3: Principle o f image current detection... 97
Figure UI.4: Sequence of the peptide 127-145 at pH 7.0...108
Figure III. 5: Positive ion MALDI-FT-MS and ESI-FT-MS spectra o f peptide 127-145 in deionised H2O at pH 7.0...110
Figure III.6: Positive ion ESI-FT-MS spectra of peptide 127-145 in deionised H2O at pH 7. 0... I l l Figure m.7: Graph showing the different fragments obtained as a function of the potential applied in the nozzle-skimmer region... 114
Figure HI.8: Positive ion ESI-FT-MS spectra of peptide 127-145 in deionised H2O at pH 7.0 under optimal conditions of fragmentation... 115
Figure III.9: Pattern o f fragmentation o f a peptide... 117
Figure HI. 10: Diagram showing the sequence of the peptide 127-145 and the different points of fragmentation under optimal conditions of fragmentation...119
Figure III.l 1 : Diagram showing the protection from deuterium exchange of the residues forming the epitope...120
Figure IQ. 12: Positive ion ESI-FT-MS spectrum of peptide 127-145 in deionised D2O at pH 7.0,25°C for 1 second...122
Figure 111.13: Positive ion ESI-FT-MS spectrum of peptide 127-145 in deionised H2O for Is at 4°C, pH 1.5... 124
Figure 111.14: Positive ion nanoflow-ESI-TOF-MS spectrum of the Fab fragment/peptide complex in deionised H2O at pH 7.0... 126
Figure HI. 15: Positive ion nanoflow-ESI-TOF-MS spectrum o f the Fab fragment/peptide complex in deionised H2O at pH 7.0 after collision induced dissociation... 128
Figure HI. 16: Positive ion nanoflow-ESI-TOF-MS spectrum of the Fab fragment/peptide complex in deionised D2O at pH 7.0,4°C for 1 second... 129
Figure III. 17: Positive ion nanoflow-ESI-TOF-MS spectrum o f the peptide in deionised D2O at pH 7.0,4°C for 1 second, after collision in the cell...130
Figure III.l 8: Positive ion nanoflow-ESI-TOF-MS spectrum of the peptide 127-145 in deionised D2O at pH 7.0,25°C for 1 minute... 131
Figure 111.19: Ribbon diagram of the crystal structure of the OT3A2 Fab fragment complexed with a peptide representing the residues 132-145 o f p-hCG... 136
Figure IV. 1 : Screening o f the combinatorial library by INN-32 mAb... 149
Figure IV.3: Nitrocellulose membrane probed by an anti-measles virus antibody... 154
Figure IV.4: Nitrocellulose membrane probed by the INN-2 monoclonal antibody...155
Figure IV.5: Nitrocellulose membrane probed by the INN-32 monoclonal antibody.... 155
Figure IV.6: Nitrocellulose membrane stained by antibodies detected by a HRP-conjugated antibody... 159
Figure IV.7: Schematic representation o f a tetrameric multiple antigen peptides... 161
Figure IV.8: Conqjetition assays with INN-2 mAh...163
Figure FV.9: Competition assays with INN-32 mAh... 164
Figure IV. 10: Direct ELISA analysis o f the recognition o f the MAPs by INN-20 mAb. 165 Figure IV.l 1 : Analysis o f the mice sera prior to immunisation...167
Figure IV. 12: Determination of the capacity o f the sera of mice immunised with MAP-M l to recognise hCG and MAP-MAP-MAP-Ml in an ELISA...168
Figure IV. 13: Determination of the capacity o f the sera o f mice immunised with MAP-M2 to recognise hCG and MAP-MAP-M2 in an ELISA...169
Figure IV. 14: Determination o f the capacity of the sera o f mice immunised with MAP-M3 to recognise hCG and MAP-MAP-M3 in an ELISA...170
Figure IV. 15: Determination of the capacity o f the sera o f mice immunised with MAP-M4 to recognise hCG and MAP-MAP-M4 in an ELISA...171
Figure IV. 16: Determination o f the capacity o f the sera o f mice immunised with MAP-MS to recognise hCG and MAP-MAP-MS in an ELISA... 172
Figure IV. 17: The immunogen-binding sera cross-react with other MAPs... 173
Figure IV. 18: Hypothetical mapping o f the p i epitope... 177
Figure V .l: Identification of residues potentially involved in the pi epitope...186
Figure V.2: Construction of the P-hCG genes mutated at positions 13 and 61, and subcloning into the pBluescript.SK+... 190
Figure V.3: A. Elimination of undesired mutations o f the pBluescript.phCG.M13. B Construction o f mammalian expression vectors containing the mutant 13 p-hCG. 193 Figure V.4: Restriction analysis o f the pcDNA3 constructs...197
Figure V.5: DNA sequences o f the p-hCG genes WT, M l 2, Ml 3, Ml 9.3 and M61... 198
Figure V.6: Comparison between eflBciency and toxicity of transfection using DEAE technique with pCDM8.GFP in CHO cells... 200
Figure V.7: Transient transfection of CHO cells with pCDM8.GFP using the liposome technique... 200
Figure V.8: Stable expression of GFP, p-hCG WT, M l2, M13 and M61 in CHO cells. 203 Figure V.9: Binding of the antibodies to the p-hCG wild type and mutants... 205
Figure VI. 1 : Description of a Biosensor...213
Figure VI.2: Western blot analysis o f the production of Bac P-hCG WT and Bac p-hCG R68E proteins as a function of the time post infection... 218
Figure VI.3: Quantification o f Bac P-hCG WT and Bac P-hCG R68E by ELISA. 218 Figure VE.4: Size exclusion chromatography o f Bac p-hCG R68E on Sepharose® 12 PC 3.2/30 and SMART system... 220
Figure VI.5: Description of the surface plasmon resonance experiment... 221
Figure VI.6: Sensorgrams of experiments controlling for non-specific binding... 223
Figure VI.7: Analysis o f the quality o f the papain digestion of the five antibodies... 227
Li s t o f t a b l e s
Table II. 1: Standard buffers and solutions... 61
Table II.2: Description and source of bacterial strains...64
Table II.3: Description and source of plasmids... 67
Table II.4: Description and source of the cell line... 68
Table II.5: Description and source of the antibodies...71
Table II.6: Description and source of the peptides and proteins...72
Table Ü.7: Sequence and name o f the primers... 72
Table III.l : Calculated and experimental monoisotopic m/z ratio for the peptide 127-145 carrying one, two, three, four, five and six charges... 109
Table III.2: Size, charge and sequence of the fi-agments obtained under optimal conditions of fi-agmentation... 118
Table III.3: Number of hydrogens exchanged with deuterium as a fimction of the time of exchange and the temperature... 121
Table III.4: Number o f peptide-associated deuteriums non-exchanged for hydrogens as a function of the pH and number of scans recorded at 4°C...123
Table IV.l: Examples of peptides mimicking various ligands identified using peptide libraries... 140
Table IV.2: Antigenic mimics and mimotopes of conformational B cell epitopes... 144
Table IV.3: Sequence o f the peptides selected in the eight-mer combinatorial library with the INN-2 and INN-32 antibodies... 150
Table IV.4: Sequences o f the analogues and lead sequences synthesised on a nitrocellulose membrane... 152
Table IV.5: Summaiy o f the peptide recognition on the membrane by the INN-2 and INN-20 monoclonal antibodies... 160
Table IV.6: Summary o f the recognition of the dififerent sequences on beads, on the membrane, and as a MAP...166
Table IV.7: Immunisation of BALB/c mice by MAP...166
Table V.l: Description of the DNA fi-agments loaded in agarose gels I, II and m ...190
Table V.2: Mutations discovered with the sequencing o f the pBluescript.phCG.M13.. 192
Li s t o f a b b r e v i a t i o n s
Aa amino acid
Ab antibody
ADCC antibody-dependent cell-mediated cytotoxicity
AP alkaline phosphatase
APC antigen-presenting cell
APS ammonium persufate
ASN antisense
ATCC American type culture collection
ATP adenosine triphosphate
BCR B cell receptor
bp base pair
BSA bovine serum albumin
C Celsius
cAMP cyclic adenosine 3',5-monophosphate CBB carbonate bicarbonate buffer
cDNA complementary DNA
CDR complementary determining region
CHO Chinese hamster ovary
CIAP caff intestinal alkaline phosphatase CID collision-induced dissociation
CMV cytomegalovirus
CRM charged residue model
CSD charge state distribution
CTL cytotoxic T lymphocyte
CTP carboxy terminal part
D deuterium
Da Dalton
ddHzO double-distilled water
ddNTP dideoxynucleoside triphosphate DMEM Dulbecco’s modified Eagle’s medium
DMF di-methyl-formamide
DMSG dimethylsulphoxide
DNA deoxyribonucleic acid
dNTP deoxynucleoside triphosphate
DT diphtheria toxoid
DTT dithiothreitol
ECL™ enhanced chemiluminescence
EDTA ethylenediaminetetra-acetic acid
EOF epidermal growth fector
ELISA enzyme-linked immunoabsorbent assay
ESI electrospray ionisation
eV electron Volt
Fab fi-agment antigen-binding
FACS fluorescence-activated cell sorter
Fc fi'agment crystallisable
FA Freund’s adjuvant
FT-MS Fourier transform mass spectrometry
g gram
GFP green fluorescent protein
GnRH gonadotropin-releasing hormone
GTE glucose Tris-EDTA
HBS HEPES buffered saline
HESS Hank’s balanced salt solution
hCG human chorionic gonadotropin
H hydrogen
HEL hen egg-white lysozyme
HEPES N-2-hydroxyethyl piperazine-N'2-ethansul HPLC high performance liquid chromatography
HRP horseradish peroxidase
HSD hetero-species dimer
lAA isoamyl alcohol
IBAR Institute for Biomedical Aging Research
IBM ion evaporation model
IL interleukin
IFN interferon
IPTG isopropyl P-D-thiogalactopyranoside
IRMA immunoradiometric assay
kbp kilo base pair
1 litre
lb pound
LB Luria Bertani medium
LH luteinising hormone
EPS lipopolysaccharide
m meter
M mol/1
mAb monoclonal antibody
MALDI matrix-assisted laser desorption ionisation
MAP multiple antigen peptide
MCS multiple cloning site
M-CSF macrophage colony-stimulating factor MES 2-[N-morpholino] ethanosulfonic acid MHC major histocompatibility conçlex
MS mass spectrometry
MW molecular weight
m/z mass to charge ratio
N.D. not determined
NGF nerve growth fector
NK natural killer cell
NMR nuclear magnetic resonance
NTA nitrilotriacetic acid
OD optical density
Pa Pascal
PBS phosphate-buffered saline
PCR polymerase chain reaction
PDB protein data bank
PDGF-B platelet-derived growth factor B
PIPES piperazine N-N’-bis [2-ethanol sulfonic acid]
PRR pattem-recognition receptor
rpm revolution per minute
RU resonance unit
SDS sodium dodecyl sulphate
SN sense
SPR sur6ce plasmon resonance
TAB Tris-acetate-EDTA buffer
TBE Tris-boric acid-EDTA buffer
TBS Tris buffered saline
IC R T cell receptor
TdT terminal deoxynucleotidyl transferase
TE tris-EDTA buffer
TEMED N,N,N’,N’-tetramethylethylene-diamine TEA trifluoro acetic acid
TGF-P2 transforming growth factor P2
TNF tumour necrosis factor
TOF time of flight
TSEB tissue-specific element binding protein
TSH thyroid stimulating hormone
TT tetanus toxoid
UCL University College London
uv
ultravioletUK United Kingdom
USA United States o f America
WT wild type
General Introduction
The first section (I.l) of this introduction defines, succinctly, the role of B cells in the immune system. The second section (1.2) focuses on the B cell receptor and the pattern recognised on the antigen, the B cell epitope. The following section of this introduction (1.3) reviews human chorionic gonadotropin (hCG). Finally, the potential role of hCG in the development o f a contraceptive vaccine will be introduced (1.4). With this background the context for the work o f this thesis will be set.
1.1. Ov e r v ie w o f t h e Im m u n e Sy s t e m
The immune system is composed o f various mechanisms that are complementary and modulate each other with the aim o f defending the body against infectioa An overview of the defence mechanisms involved in the protection against pathogens is briefly presented, with an emphasis on the mechanisms o f recognition by the immune system of the pathogen.
To fight against infectious/foreign agents, vertebrates have developed two interrelated levels o f protection, innate immunity and acquired immunity. Innate immunity forms a first line o f defence that may be strengthened, if necessary, by extremely specific cell eflTectors constituting the acquired immunity.
1.1.1. In n a t ei m m u n i t y
The elements of the innate immunity include physical barriers, secretory molecules and cellular components. The role o f the anatomical barriers is to prevent foreign agents firom invading the body. If penetration occurs, the soluble and the cellular effectors constituting innate immunity identify the presence o f a foreign substance and are activated. Their activation results in an immediate maximal response aiming at the eradication of the foreign agent. This response is germ line encoded and thus is not intrinsically affected by contact with the antigen.
1.1.1.1. The receptors of the innate immunity
surfece or soluble receptors, also referred to as pattem-recognition receptors (PRRs), identify the specific composition of surface carbohydrate (Stahl, 1992) and lipid stmctures (Akira, 2001; Ulevitch and Tobias, 1995; Ulevitch and Tobias, 1999) of bacteria and viruses. The pathogen-associated molecular patterns (PAMPs) recognised by PRRs do not evolve throughout life, insuring the maintenance of the inherent ability to distinguish between pathogen and self. Reciprocally, innate immunity is unable to adapt to certain contemporary organisms and is inflexible with regard to the genetic variability o f micro-organisms.
Natural killer (NK) cells are innate immune cells that recognise intracellular pathogens and tumours (reviewed in (Moretta et a l, 2002)). A balance between signals transmitted by activating and inhibitory receptors regulates the fimction o f NK cells. Activating receptors recognise ligands on the surface o f tumours or pathogen-infected cells that are up-regulated during infection and stress (Moretta et a l, 2001; Lanier, 2001). Inhibitory receptors are specific for major histocompatibility complex class I molecules (Karre et a l, 1986; Ravetch and Lanier, 2000).
Ll.1.2. The instructive role of innate immunity
Once the micro-organism is detected, information concerning this organism are transmitted by innate immunity elements to the acquired immunity effectors (Fearon and Locksley, 1996; Medzhitov and Janeway, 1997). Macrophages and dendritic cells play a critical role as an antigen-presenting ceU (APC) to T cells in secondary lymphoid organs (Bell et a l, 1999). The soluble molecules of the complement system focus the attention of follicular dendritic cells and B cells on the foreign element in the lymphoid follicle, resulting in the stimulation of B cells (Fearon and Carroll, 2000; Nielsen et a l, 2000). In addition, a permanent communication between the various elements of the immune system is maintained by way of cytokines (Mackay, 2001). Cellular components such as macrophages and NK cells secrete cytokines that modulate acquired immunity, by regulating the growth and differentiation of T and B lymphocytes (Mackay, 2001).
General Introduction
1.1.2. Ac q u ir e di m m u m t y
Ll.2.1. The cellular players of acquired immunity
The eflfector cells o f the specific arm of the immune system are helper T cells, cytotoxic T cells and B cells.
1.1.2.1. a. T helper cells
CD4'*’ T helper cells initiate and enhance cellular and humoral responses by the production o f specific cytokines and by cell to cell contacts. The specific T cell receptor (TCR) on CD4^ cells recognises linear peptides, fi*om processed antigen, associated with the major histocompatibility complex (MHC) class II molecules on APC, i.e. dendritic cells, macrophages and activated B lymphocytes (Engelhard, 1994a; Karlsson et a l,
1994; Salazar-Fontana and Bierer, 2001). The T cell requires co-stimulation in order to become activated and this second signal results fi’om the binding o f the surface molecules, B7 (CD80/CD86) on the APC and CD28 on the T cell (Jenkins et a l, 1991). Two fimctionally distinct T helper subsets, Thl and Th2, have been characterised in mouse (Mosmann et a l, 1986; Mosmann and Cofifinan, 1989) and human (Romagnani, 1991). Thl cells produce interferon-y, tumour necrosis factor (TNF) and interleukin 2 (IL-2), and provide helper activity primarily for cell-mediated immunity. Th2 helper cells produce IL-4, IL-5 and IL-10, which stimulate the humoral arm of acquired immunity (Del Prete
et a l, 1991; Mosmann and Sad, 1996; Romagnani, 1994; Salgame et a l, 1991).
1.1.2.1.b. Cytotoxic Tcells
Infected cells expose peptides from viral proteins at the cell surfece in association with their MHC class I molecules (Natarajan et a l, 1999). The recognition of the MHC I- peptide complex by the TCR in association with CD8 triggers the CD8^ cytotoxic T lymphocytes (CTL) (Engelhard, 1994a; Engelhard, 1994b; Garcia et a l, 1999; Townsend
et a l, 1986). These activated CTL develop into effectors in the presence o f cytokines produced by T helper cells. CTL can destroy the infected cell by direct cell-to-cell contact or by the release of molecules that possess killing activity (Podack, 1995; Trapani et a l,
intracellular infections (Kaufinann, 1988; Kaufinann, 1993; McMichael and Askonas, 1978; McMichael et a l, 1983; Trapani et a l, 1999).
L 1.2.Le. B cells
B cells utilise their B cell receptor (BCR) to recognise the surface o f foreign agents. As for T cells, B cells require two signals to become fully activated. The first signal is the binding o f the antigen to the BCR. This induces internalisation of the antigen, which leads to processing for subsequent presentation of the antigenic peptides to T helper cells by MHC class H. This cognate recognition of MHC Il-peptide complex by an activated T helper cell allows the B cell to receive the second signal through CD40- CD154 contact (Clark and Lane, 1991; Mefifre et a l, 2000; Parker, 1990). The activated B cells proliferate and difierentiate into antibody-secreting plasma cells and memory B cells. Extracellular infectious agents are the principal targets of antibodies. The binding o f antibodies can neutralise microbial toxins. However, the main action o f antibodies is to opsonise the foreign agent and thereby trigger phagocytosis by macrophages and neutrophils. In addition, binding o f antibodies to pathogens induces the activation o f the classical pathway o f the complement system, which will indirectly enhance antigen presentation and thereby ultimately lead to the destruction of pathogens. Furthermore, antibodies have the potential to induce lysis of infected cells by NK cells, through a mechanism known as antibody-dependent cell-mediated cytotoxicity (ADCC).
I.I.2.2. The recognition receptors
In contrast to the innate immunity germ line encoded receptors that recognise unique features o f an extended range of organisms, the recognition receptors (TCR and BCR) of acquired immunity are generated by somatic gene recombination and are characterised by their capacity to specifically identify individual pathogens. In addition, this veiy flexible system, which copes with the genetic variability of micro-organisms, selects and maintains the most appropriate responses for contemporary infectious agents on an immediate rather than evolutionary time scale.
I.1.2.2.a. The TCR
The TCR is a transmembrane molecule composed of two discrete chains (Haskins
General Introduction
associated with the CD3 complex. The three complementary determining regions (CDR) of each variable domain establish contact with the peptide presented by an appropriate MHC molecule (Garcia et a l, 1999). The TCR chain results of the random association of gene segments, a variable (V), diversity (D), joining (J) and constant (C) segment (Chien
et a l, 1984; Hedrick et a l, 1984b). The arrangement o f gene segments from each gene cluster (V, D, J and C gene cluster), splicing inaccuracies and random interchain association create an extraordinary large repertoire of T cells (Hedrick et a l, 1984a). Individual T cells produce TCRs with a unique specificity. T cells expressing receptors o f appropriate aflBnity for the antigen and that receive co-stimulation are triggered, expand and develop into effector cells. Once the infection is eliminated, a small portion o f the antigen-specific T cells are maintained as memory T cells (reviewed in (Dutton et a l,
1998; Sprent and Surh, 2001)). These memory T cells will react quickly, strongly and specifically to subsequent infection with the same antigen.
I.L2.2.b. The BCR
7i
Figure 1.1: Ribbon diagram of a
soluble IgGI.
Figure based on (Harris e t al., 1998). The antibody molecule is composed of two heavy (light blue and yellow) and two light (green and dark blue) chains that are linked by disulphide bonds. The variable domain of the heavy ( Vh) and
light chain ( Vl) each contain three CDRs
that make contact with the antigen. The membrane bound antibody presents
heavy chains extended by
transmembrane sequence that allows the anchoring of the antibody in the lymphocyte membrane.
G e rm line V 1-50
VDJ joining
D 1-25
V 1-50
1
B cell DNA
Transcription
P r im a r y t r a n s c r ip t
Splicing
m R N A
^ Translation
P ro te in
J 1-6 C
p Ô y3 y 1 a l y 2 y 4 6 a 2
DJ C
p Ô y 3 y l a l y 2 y 4 c a 2 TdT
^ p 0 y 3 y l a l y 2 y 4 s a 2
TdT
VDJ C
p Ô
V D JC p
IgM
Figure 1.2: Gene rearrangements of the human heavy chain of a secreted IgM.
Gaieral Introduction
1.1.2.2. c. Evolution o f the B cell recognition receptor throughout the primary immune response
(Reviewed in (MacLennan et a l, 2000; Neuberger et a l, 2000))
Once an infectious agent is encountered by the adaptive immune system, a primary immune response against its antigens may be triggered and is initially composed o f low affinity IgM antibodies. During the primary immune response, naïve B cells in secondary lymphoid tissue (i.e. lymph nodes, spleen and mucosa-associated lymphoid tissue) encounter, through their BCR, the antigen and move to the T cell area where they elicit T cell help (Bumet, 1958; Jeme, 1955). Activated B cells start dividing exponentially in the follicle and differentiate into centroblasts to form a germinal centre (reviewed in (MacLennan and Gray, 1986)). During this proliferation phase, antibody class switching occurs from IgM to IgG, A and E under the influence o f cytokines derived from T cells, macrophages and other cell types (Stavnezer, 1996). Switching of class results from the rearrangement of the genes coding for the heavy chain constant region. This modification increases the impact of the antibodies on the immune system (McHeyzer-Williams et a l, 2001; Vora et a l, 1998). Simultaneously, B cells are subject to a high rate of somatic mutation affecting the antibody CDRs, thereby creating a repertoire o f clones of higher, equal and lower affinity (Jacob et a l, 1991; McHeyzer- Williams et a l, 2001; Siekevitz et a l, 1987; Vora et a l, 1998; Wedemayer et a l, 1997; Wysocki et a l, 1986). The clones o f highest affinity are maintained by contact with the antigen presented as immune complex by follicular dendritic cells, and by the association through CD40 with T helper cells (Berek et a l, 1991; Liu et a l, 1989; Liu et a l, 1991). The selected B cells divide into plasma cells that secrete antibodies and into memory B cells that will be ready to quickly produce antibodies of high affinity if the antigen is encountered again.
1
.
2
.
T h e a n t i g e n / a n t i b o d y c o m p l e xAs a result of the complexity, specificity and diversity of the antigen/antibody interaction, the processes underlying the recognition o f an antigen by an antibody have proved difficult to understand. However, the mechanisms of binding are slowly being unraveDed by the analysis o f a few antigen/antibody complexes and common features of immune complexes are beginning to emerge. The advent o f monoclonal antibody technology and X-ray crystallographic studies, and a panel o f biochemistry and thermodynamic studies, have made possible a better understanding of the interactions occurring between antigen and antibody.
1.2.1. Th e a n t ib o d y
General Introduction
side chains, that accommodate the side chains of the antigen residues (Davies and Cohen, 1996; Jones and Thornton, 1996; Jones and Thornton, 1997b). Crystal structures of haptens, peptides and proteins complexed with antibody molecules revealed a distinction between the various types of antigen-binding site on the antibody, respectively concave, ridged and planar (MacCallum et a/., 1996; Webster et aL, 1994; Wilson and Fremont, 1993). A characteristic of the antigen-binding site on the antibody is the high prevalence o f aromatic residues, in particular tyrosine and tryptophan, surrounded by polar residues, in particular serine and asparagine (Amit et a l, 1986; Davies and Cohen, 1996; Jackson, 1999; Kabat et a l, 1977; Lea and Stuart, 1995; Mian et a l, 1991; Padlan, 1990; Padlan et a l, 1989).
1.2.2. Th e a n t ig e n
I.2.2.I. Nature of the factors determining immunogenicity of an antigenic determinant
Antigenicity describes the ability o f a peptide or protein to be recognised by an antibody, whereas immunogenicity is the ability to induce an immune response. It has been demonstrated that the entire surface of a protein can potentially be antigenic; however, following immunisation, the production o f an antibody to each segment o f the protein is not observed (Benjamin et a l, 1984; Geysen et a l, 1987b; Green et a l, 1982; Lemer, 1982). In defined species, it appears that some epitopes are more reactive than others (Geysen et a l, 1987b). Two divergent explanations have been proposed to explain this fact (Berzofsky, 1985). Atassi and collaborators suggested that the capacity o f a protein segment to be immunogenic is due to the intrinsic nature of the segment, composition and shape for instance (Atassi, 1979; Twining et a l, 1981; Twining et a l,
consequently most studies related to antigenic determinants evaluate the intrinsic factors o f an epitope.
Epitopes can contain lipids, carbohydrates, nucleic acids, amino acid residues or simple chemical groups acting as hapten. Epitopes of a proteinaceous nature are emphasised in this presentation. A combination of techniques has permitted the identification of numerous epitopes on antigens, and some intrinsic characteristics can be deduced fi’om the analysis o f these epitopes.
I.2.2.2. Identification of B cell epitopes
The three main approaches that have been developed to determine the localisation, conformation and composition o f B cell epitopes, are: (1) antigenic and immunogenic analysis o f segments o f the antigen, (2) antigenic and immunogenic analysis of mutants of the antigen and (3) determination o f the structure o f the antigen/antibody complex. The fimctional approaches (methods (1) and (2)) define the relative contribution of residues to complex formation and stabilisation, whereas the structural approach (method (3)) defines the spatial arrangements of residues in the antibody/antigen complex. Lysozyme c, fi*om hen egg-white (HEL) in particular, is an antigen that has been used as a prototype protein for investigating antigenic determinants. Examples of epitope mapping strategies applied on lysozyme have been selected.
1.2.2.2. a. Fragmentation o f the antigen
The fi*agmentation of an antigen into small segments can be accomplished by chemical, enzymatic, biochemical or genetic means.
General Introduction
solution in the presence of the antibody. Any peptides missing from the profile were suggested to be involved with the antibody (Kiselar and Downard, 1999b).
The chemical synthesis of peptides or the subcloning of gene fragments into an appropriate vector, such as phage, permit the production o f defined overlapping fragments representing the entire primary sequence or selected regions of the antigen. These methods have the advantages of not requiring any purified original antigen and of ensuring a reproducible high production of fragments. Atassi and collaborators isolated immunologically reactive fragments of HEL and synthesised overlapping peptides o f the selected regions (Atassi, 1978; Atassi and Lee, 1978). The antigenic analysis by anti-HEL antibodies permitted the identification o f the position and size o f antigenic determinants on the crystal structure o f HEL (Kundrot and Richards, 1987). In addition, the capacity of the peptides to induce an immune response recognising HEL was investigated (Amon et a l, 1971).
L2.2.2.b. Mutants o f the antigen
The study of mutants, natural or chemically and molecularly engineered, allows the assessment o f the contribution of particular amino acids in the antigenicity of the protein (Benjamin, 1991; Greenspan and Di Cera, 1999; Minor et aL, 1985). Homologous proteins from different species are natural mutants presenting potentially different antigenicity. An extensive serological analysis has been undertaken by Smith-Gill and collaborators through a study of cross-reactivity with different avian lysozymes (Smith- Gill et aL, 1982). This analysis identified residues determining the specificity of individual antibodies. Alternatively, a comparison o f the sites o f chemical modifications o f the complexed antigen and the sites using the free HEL was used to establish the “foot- printing” of the antibody (Fiedler et aL, 1998; Li et aL, 2001). Site-directed mutations in antigenic sites o f chicken egg white lysozyme were introduced by genetic manipulation; the mutants were analysed in order to evaluate the fimctionality of individual residues (Malcolm et aL, 1989).
I.2.2.2.C. Determination o f the structure o f the antigen/antibody complex
the residues involved in the antibody/antigen interaction. Crystallographic investigations o f antibody/antigen complexes permitted the assessment of the predictions made from functional epitope mapping by direct visualisation of the contacting residues (Padlan,
1990; Sheriff 1993).
In addition, a second structural method, nuclear magnetic resonance (NMR), has been applied to HEL, principally to study folding/unfolding states (Chung et aL, 1997; Eyles et aL, 1994; Maleknia et aL, 1999; Miranker et aL, 1993; Radford et aL, 1992a). However, two studies have shown that residues involved in the binding with the antibody can be identified by comparing the deuterium/hydrogen exchange rate in the presence of the antibody and the exchange rates in the free antigen (Benjamin et aL, 1992; Cheetham
etaL, 1991).
I.2 .2 .3 . In trin sic c h a ra cteristics o f an e p ito p e
1.2.2.3. a. The structural nature o f an epitope
• An epitope is located on the surface o f the native antigen
In 1969, few techniques were available to study epitopes. However, antigenicity and immunogenicity studies on synthetic peptides suggested that a B cell epitope required a minimum of four amino acid residues that must be easily accessible without fragmentation of the protein (Sela, 1969). In addition, it was observed that modification o f the overall conformation of the protein, after dénaturation for instance, or modification o f the primary structure, e.g. change o f stereospecificity, removes the epitope (Sela, 1969). The recognition of an epitope by an antibody depends, therefore, at least on structural elements located on the surface o f the native antigen.
• An epitope can be continuous or discontinuous
General Introduction
often associated with N- and C-terminal sequences (Benjamin et aL, 1984). The size of the area on a globular protein covered by the antibody was estimated, by the analysis of co-con^lex crystal structures, to vary between 650 to 900Â (Davies and Cohen, 1996; Jones and Thornton, 1996; Laver et aL, 1990; Sheriff et aL, 1987; Webster et aL, 1994). On the convoluted surface of a protein, patches o f continuous residues with a similar size are limited, explaining the preponderance of discontinuous epitopes (Barlow et aL, 1986).
• Continuous and discontinuous epitopes have common characteristics: accessibility and protrusion
Studies using fragments of the antigen, particularly synthetic overlapping peptide analogues of the primary sequence, identified numerous epitopes. Most of the epitopes identified were continuous epitopes, although a fi^ction of these peptides map part of discontinuous epitopes. Two to eight residues, six residues in average, are critical for the binding o f the antibody to the peptide (Appel et aL, 1990). Most o f these critical residues are localised on the surface of the antigen; consequently hydrophilicity is a feature of residues forming B cell epitopes (Appel et aL, 1990; Hopp and Woods, 1981). Moreover, these studies showed that the easily accessible protruding parts of the protein surfece, identified by high protrusion index and solvent accessibility, are often the site of recognition (Novotny et aL, 1986a; Novotny et aL, 1986b; Thornton et aL, 1986). A large majority o f these linear epitopes are located on segment of protein having a surfece of convex shape, confirming the determinant role of structural elements in recognition (Craig et aL, 1998; Furie et aL, 1975; Geysen et aL, 1987b; Sachs et aL, 1972). Later on. X-ray crystallographic studies have highlighted a feature o f antigenic peptides, namely the presence o f (J-tums in the antibody/antigen complex (Dyson and Wright, 1995; Garcia
et aL, 1992; Ghiara et aL, 1994; Rini et aL, 1993; Shoham, 1993; Stanfield et aL, 1990; Tormo et aL, 1994). It had been suggested that the binding might induce p-tum conformation in peptides prone to this conformation (Wfilson and Stanfield, 1994). Therefore it is not surprising that a high mobility associated with low packing density are also characteristics o f the epitopes identified using peptide mapping (Geysen et aL,
Stanfield et a l, 1990). Consequently, loops and N- and C-termini, which are fi*equently surface-exposed, protruding and relatively mobile, are often antigenic sites identified by peptide mapping.
New insights into the nature of discontinuous epitopes have been gained through X-ray crystallographic studies of antigen/antibody complexes. Antigen/antibody analysis o f co-crystals confirmed that the residues forming a discontinuous epitope are also usually polar, protruding and accessible (Jones and Thornton, 1997a). In addition, this technique revealed that two to seven segments can interact with the antibody in a conformational epitope (Jones and Thornton, 1996; Sheriff, 1993; Sheriff et a l, 1987).
I.2.2.3.b. Critical residues in epitopes
G eneral Introduction
14
12
10
n.--- *
0
A C D E F G H I K L M N P Q R S T V W Y
Figure 1.3: Prevalence of amino acid residues on the surface of a protein and in an
epitope.
Dr. Andrew Martin (Department of Biochemistry, UCL, UK) determined the prevalence of the 20 amino acids (represented by the one letter symbol on the X-axis) on the surface of protein (blue circles), and he compared this prevalence with their representation in epitopes (pink squares). The Y-axis is an arbitrary unit relative to the number of times each residue has been identified on the surface or in an epitope.
1.2.3. Th e a n t i b o d y/a n t i g e n i n t e r a c t i o n
I.2.3.I. Thermodynamics of the interaction
Three factors permit the antigen and the antibody to overcom e the hydration
repulsion field that exists around biopolymers in aqueous solutions: (1) the protruding
position o f the contact residues; (2) the opposite electric charges o f paratopes and
epitopes; (3) the presence o f hydrophobic spots in paratopes (Sundberg et al, 2000; van
Oss, 1998). Once epitopes and paratopes are in close proximity, they have to overcome
large entropie barriers before they can form a tight association. The loss o f the entropy o f
free rotation and translation o f both molecules, as w ell as the loss o f conformational
entropy o f mobile segments and o f side chains upon binding, is partially counterbalanced
by the entropie contribution arising from the release o f bound water molecules (Kelley
and O'Connell, 1993). In addition, the complementarity of the forces involved in the binding o f the antibody to the antigen provides important enthalpic contributions (Tello et a l, 1994). The antibody/antigen intermolecular forces are non-covalent interactions - mainly hydrogen bonds (H-bonds), van der Waals and electrostatic interactions, such as salt bridges - and they involved, in a conformational epitope, 15 to 20 residues on each partner of the complex (Davies and Cohen, 1996; Jones and Thornton, 1996; Laver et a l,
1990; Sheriff et al., 1987; van Oss, 1998). The antigen/antibody complexes interact predominantly through side-chain-side-chain or side-chain-main-chain mechanism (Jackson, 1999). Energetically, 5 to 6 residues seem to contribute most o f the binding energy (Amit et a l, 1986; Kelley and O'Connell, 1993; Novotny, 1991; Novotny et al.,
1989; Sheriff et al., 1987). This widely held view that antigen/antibody association is invariably mediated by only a few strong non-covalent interactions has been questioned upon the identification of epitopes in which the fi*ee energy of binding arises fi*om many productive interactions distributed over the entire interfece (Dall'Acqua et al., 1996; Dall'Acqua et al., 1998; Goldman et al., 1997). The H-bonds are formed between polar residues on both molecules and sometimes with molecules of water located in the cavities within the interface and on the periphery (Bhat et al., 1994; Braden et al., 1995; Davies and Cohen, 1996; Faelber et al., 2001). In addition to its contribution to charge complementarity, water molecules may increase the packing density of the antibody/antigen interface, which already presents a high degree of shape complementarity (Faelber et al., 2001; Jones and Thornton, 1997a; Tulip et al., 1992). However, no obvious correlation has been established between the nature of the binding forces involved at the interfece and the affinity of the interaction (Webster et al., 1994).
L 2 .3 .2 . C o n fo r m a tio n a l ch a n g e s u p o n b in d in g
General Introduction
disposition of the variable region of the heavy and light chains have been observed as well (Arevalo et a l, 1993a; Arevalo et a l, 1993b; Herron et a l, 1991; Rini et a l, 1992; Stanfield et a l, 1993; Tormo et a l, 1994; Wilson and Stanfield, 1993). A large body of data supports the “induced fit” conformation changes (Arevalo et a l, 1993a; Arevalo et a l, 1993b; Bhat et a l, 1990; Rini et a l, 1992; Schulze-Gahmen et a l, 1993; Stanfield et a l, 1990; Stanfield et a l, 1993; Tormo et a l, 1994). Antibodies are believed to modify the conformation o f their paratope to fit perfectly the shape of the epitope. Alteration in the antigen conformation upon antibody binding has also been observed and allows the participation of previously buried residues in antigen/antibody binding (GetzofiF et a l,
1987). This could complicate greatly the identification of the contact residues in epitopes. Overall, the conformational changes observed in antigens are less than seen in antibodies, and involve mainly the side-chains o f contacting residues (Colman et a l, 1987; Davies and Cohen, 1996; Sheriff et a l, 1987).
1.2.4. Pr e d ic t in g B c e l le p i t o p e s
A number of empirical approaches have been employed to predict B cell epitopes fi*om the primary amino sequence and, when available, from the tertiary structure of the protein (Parker et a l, 1986; Van Regenmortel and Daney de Marcillac, 1988). These approaches are based on the B cell epitope features presented above, such as hydrophilicity, solvent accessibility, protrusion of side chains, mobility and secondary structures. However, these prediction methods have encountered very limited success and seem to identify mainly linear epitopes (Van Regenmortel, 1989; Van Regenmortel and Pellequer, 1994). The failure o f these prediction methods to define inherent conformational epitopes of a protein could have two major causes:
(1) A large majority o f the B cell epitopes analysed to determine the antigenic determinant characteristics have been identified using techniques based on the
(2) Prediction methods attempt to define epitopes only on a structural basis. Crystallographic studies have the advantage o f visualising the epitope in its totality and of
maintaining the antigen in its original conformation. However, X-ray crystallography provides only spatial information of the interaction at a particular time point, without defining the contribution o f each residue to the binding. In addition, the data obtained by fimctional analysis, such as definition of critical residues, have been taken into account only in the perspective o f their structural position on the three-dimensional structure.
Consequently, the addition of fimctional information to the structural definition of an operational epitope is a sine-qua-non condition for the improvement o f the prediction o f conformational B cell epitopes.
General Introduction
L 3. Hu m a n CHORIONIC GONADOTROPIN
1.3.1. Th e e s s e n t ia lr o l e o f hC G d u r in gp r e g n a n c y
Once the follicle reaches maturation in the ovary, it releases the ovum into the feUopian tubes where it can be fertilised. As soon as the ovum has been fertilised, it starts to divide. The zygote reaches the uterus approximately three days after fertilisation when it becomes the blastocyst. The blastocyst consists of two types o f cell, a central cluster of cells called the inner cell mass surrounded by a wall o f trophectoderm cells. In the uterus, the blastocyst continues to grow and develop for an additional six days followed by the invasion of the adjacent uterine epithelium by the trophectoderm cells o f the blastocyst. The outer layer of trophoblast cells loses their cell membranes to form a syncitium (the syncitiotrophoblast or placental tissues), while the inner trophoblast cells form the cytotrophoblast. As the blastocyst implants deeply into the endometrium, the uterine epithelium fuses over it. The trophoblast gradually develops a vascular system that, in association with the developing maternal vascular system, forms the placenta. The growing inner cell mass develops into the foetus. In many ways, the developing foetus and placenta function together as a fetoplacental unit.
One of the earliest functions o f the implanting trophoblast is the synthesis of human chorionic gonadotropin (hCG) (Fox and Kharkongor, 1970; Major et a l, 1967; Pierce and Parsons, 1981). The expression of p-hCG mRNA can be detected as early as the eight cells stage (Bonduelle et a l, 1988), and secretion of hCG by the blastocyte is observed on day 7 (Fishel et a l, 1984; Hay and Lopata, 1988). Production o f hCG is continued by the synciotrophoblast, and its concentration in the maternal circulation reaches a peak about 50-60 days after the last menstrual bleed (Fig. 1.4) (Hobson, 1971; Mishell et a l, 1973; Mishell et a l, 1974). The concentration in the plasma then falls quite sharply to reach a new level that remains relatively constant until the end o f pregnancy, apart from a second small increase observed during weeks 28-36 o f pregnancy.
progesterone-dominated uterus in the absence of oestrogen remains hostile to implantation (Liu et a l, 1995). hCG replaces, by binding the same receptor on ovarian theca, granulosa, luteal and interstitial cells, the luteinising hormone (LH) that by day 24 of the menstrual cycle is only being secreted in small, basal quantities. Binding o f hCG or LH to the receptor increases adenyl cyclase activity mediated by intracellular membrane associated G proteins, resulting in an increase in cyclic adenosine 3', 5-monophosphate (cAMP) (Gudermann et a l, 1992). Ultimately, these events lead to the production o f steroids. Some secondary mediators, such as inositol phosphate or diacylglycerol resulting from the activation of the phospholipase C pathway, might also be involved in mediating the action o f the hormones (Cooke, 1990; Gudermann et a l, 1992). In addition, hCG could stimulate the foetal production o f dehydroepiandrosterone, which can be converted to oestrogen by the placenta (Seron-Ferre et a l, 1978). In the male foetus, hCG stimulates the interstitial cells of Leydig to begin to secrete testosterone, upon binding to CG/LH receptors located on the testicular Leydig cells (Ahluwalia et a l, 1974).
The mechanism regulating the synthesis and release o f hCG in the first few weeks of pregnancy is unknown. However, it has been hypothesised that the biosynthesis of hCG by the placenta can be modulated by numerous substances including:
- epidermal growth factor (EGF) (Morrish et a l, 1987),
- gonadotropin-releasing hormone (GnRH) (Bamea and Kaplan, 1989; Kay and Jameson, 1992; Lin et a l, 1995; Merz et a l, 1991 ; Siler-Khodr et a l, 1986), - y-aminobutyric acid (Licht et a l, 1992),
- P-adrenergic agonists/a-adrenergic agonists (Oike et a l, 1990) - interleukin-1 (IL-1) (Masuhiro et a l, 1991),
- interleukin-6 (IL-6) (Nishino et a l, 1990),
G eneral Introduction
I
100
50 150 200 250 300
Last m enstrual bleed D ays Parturition
Figure 1.4: Pattern of plasma hormones during pregnancy.
Figure based on (Johnson and Everitt, 1999). The human syncitiotrophoblast synthesises hCG (red) within the first two weeks of pregnancy. An important peak of production is observed around the fiftieth day after the last menstrual bleed. The production of oestrogen (black) and of progesterone (green) by the corpus luteum is stimulated by the production of hCG. The production of hCG then falls sharply and is maintained at a constant level, with a small increase towards the end of the pregnancy, until parturition.