comment
reviews
reports
deposited research
interactions
information
refereed research
Protein family review
The Smads
Liliana Attisano*
†
and Si Tuen Lee-Hoeflich
†
*Department of Anatomy and Cell Biology and †Department of Medical Biophysics, University of Toronto, 1 King’s College Circle, Toronto, Ontario M5S 1A8, Canada.
Correspondence: Liliana Attisano. E-mail: [email protected]
Summary
The large transforming growth factor-β(TGFβ) superfamily of secreted proteins regulate the growth, development and differentiation of cells in diverse organisms, including nematode worms, flies, mice and humans. Signals are initiated upon binding of TGFβ superfamily members to cell-surface serine/threonine kinase receptors and are then propagated by the intracellular mediators known as Smads. Activation of Smads results in their translocation from the cytoplasm into the nucleus, where they activate or repress transcription together with transcription factors so as to regulate target gene expression. Most Smads consist of two conserved domains, Mad homology (MH) domains 1 and 2, which are separated by a non-conserved linker region. These domains lack enzymatic activity and, instead, Smads mediate their effects through protein-protein and protein-DNA interactions. Targeted disruption of Smad genes in mice has revealed their importance in embryonic development, and a tumor-suppressor role for Smads in human cancers has been described. Smads therefore play an essential role in mediating TGFβ-superfamily signals in development and disease.
Published: 2 August 2001
GenomeBiology2001, 2(8):reviews3010.1–3010.8
The electronic version of this article is the complete one and can be found online at http://genomebiology.com/2001/2/8/reviews/3010 © BioMed Central Ltd (Print ISSN 1465-6906; Online ISSN 1465-6914)
Gene organization and evolutionary history
The Smads are a group of related intracellular proteins criti-cal for transmitting to the nucleus signals from the trans-forming growth factor-β (TGFβ) superfamily at the cell surface (reviewed in [1-10]). Although related to each other, Smads are structurally distinct from other intracellular effector proteins. The prototypic members of the Smad family, Mad and Sma, were first described in Drosophilaand Caenorhabditis elegans, respectively [6,8]. Related proteins in Xenopus, humans, mice and rats were subsequently iden-tified, and all family members are now known as Smads, a contraction of the invertebrate gene names. More recently, related proteins have also been described in zebrafish and the helminth parasite Schistosoma mansoni. Functional studies have demonstrated that Smads, which range from about 400 to 500 amino acids in length, can be grouped into three subfamilies, the receptor-regulated Smads (R-Smads), the common Smads (co-Smads) and the inhibitory Smads (I-Smads), each of which plays a distinct role in the pathway (Figure 1). Representative members for all three subfamilieshave been identified in most animal species, and the highest degree of sequence conservation is observed among the members of each subfamily. For instance, DrosophilaMad and human Smad1, members of the R-Smad subgroup, are 82% identical in amino-acid sequence. Across subfamilies, the highest degree of conservation is observed in the carboxy-terminal Mad homology 2 (MH2) domain, with amino acid sequence identities ranging from 32% to 97% in the human Smads.
been determined in either the mouse or human genome, and in all cases the genes consist of 6-12 exons. Alterna-tively spliced mRNA species for Smads 2, 4, 5, 6 and 8 have also been described [11].
Characteristic structural features
Most Smads have two conserved domains at their amino (MH1) and carboxyl (MH2) termini that are separated by a proline-rich linker region of varying length (Figure 1a-c). In the inhibitory Smads, Smad6 and Smad7 and DAD, the MH1 domains are replaced by divergent amino-termini that share regions of similarity within the inhibitory Smad subgroup. Although the structure of a full-length Smad has not yet been determined, crystallographic analysis of individual Smad domains has provided insights into the important structural features of the MH1 and MH2 domains (Figure 1b,c).
The MH1 domain
Certain R- and co-Smads have DNA-binding activity, binding a core DNA consensus sequence of GNCN. Although this interaction is of relatively low specificity, DNA binding has been shown to be vital for the transcriptional activation of specific target genes. The crystal structure of the MH1 domain of Smad3 bound to an 8 base-pair Smad-binding element (GTCTGTCT) demonstrates that the MH1 domain forms a compact globular fold that uses a highly conserved 11-residue β hairpin to contact DNA in the major groove (Figure 1b [9,12,13]). In Smad2, a 30 amino-acid insertion encoded by exon 3 is thought to displace the β-hairpin loop, providing a structural explanation for Smad2’s lack of DNA-binding activity.
[image:2.609.55.298.81.725.2]Smad-dependent activation of target promoters depends upon translocation of R-Smad-co-Smad complexes from the
Figure 1
Primary structure and relationships of Smads. (a)The conserved Mad homology 1 (MH1) and Mad homology 2 (MH2) domains are separated by a proline-rich non-conserved linker region. (b)Structure of the Smad3 MH1 domain (reproduced with permission from [13]). H2, helix H2; β, βhairpin, which contacts DNA. (c)Structure of the Smad4 MH2 and Smad activation domains (reproduced with permission from [26]). H1-H5, helices; L1-L3, loops. New features not present in the first Smad4 MH2 crystal structure [17] are colored green. The location of bound sulfate ions from the crystallization medium are in blue. (d)Relationship dendrogram for the Smad family, including members from Drosophila(D), C. elegans(C), S. mansoni(S) and human (the remainder). The subgrouping of Smads into the common Smads (co-Smads), receptor-regulated (R-Smads) and inhibitory (I-(R-Smads) is indicated. C. elegans
Smads, which have not been subject to extensive
biochemical characterization, have been excluded from this subgrouping. This dendrogram was generated using the MacVector program.
Smad4β (X) Smad4 Medea (D)
sma4 (C) sma3 (C) sma2 (C) Mad (D) Smad8
Smad5 Smad1
dSmad2 (D) Smad3
Smad2 Smad2 (S)
daf3 (C) daf14 (C) daf8 (C)
DAD (D)
(D) Smad7 Smad6
Common Smads (Co-Smads)
R-Smads (BMP-regulated)
R-Smads (TGFβ/Activin
regulated)
Inhibitory Smads (I-Smads)
MH1 non-conserved MH2
linker
(a)
(b)
(c)
(d)
L4
L1
L2
H3
TOWER
H5
H4
H3
H1
H2
L1
L3
L2
H1
N
H2
β
-hairpin
comment
reviews
reports
deposited research
interactions
information
refereed research
cytoplasm to the nucleus. R-Smads enter the nucleus after receptor-mediated phosphorylation, whereas Smad4 requires association with an R-Smad partner for nuclear accumula-tion. A basic helix (H2) in the MH1 domain, consisting of a typical nuclear localization signal (KKLKK), has been shown to be essential for Smad3 nuclear import (Figure 1b [14,15]). Translocation of Smad3 into the nucleus requires interaction with importin β, and it is interesting to note that the pres-ence of the insertion encoded by exon 3 in the MH1 domain of Smad2 prevents its interaction with importin β[15]. This result and other data [16] suggest that other determinants, such as the MH2 domain, may also be involved in regulating nuclear accumulation of R-Smads. The interaction of Smads with several transcription factors,including Jun, TFE3, Sp1 and Runx, also occurs through the MH1 domain; detailed analysis of the determinants for these types of interactions have not yet been conducted, however.
The MH2 domain
The MH2 domain does not bind DNA and instead is a multi-functional region that mediates differential association with a wide variety of proteins. Many of these interactions serve to provide specificity and selectivity to Smad function. The first crystal structure of a Smad MH2 domain to be solved was that of the co-Smad Smad4 [9,12,17]. This study revealed that the MH2 domain is composed of five αhelices (H1 to H5) and three loops (L1, L2 and L3) that enclose a
β sandwich (Figure 1c [17]). Smads exist as monomers and trimers, and although there is some controversy as to the precise composition, stoichiometry and formation of the oligomers, it is clear that the MH2 domain is critical for mediating interactions in the oligomers. Analysis of the trimeric Smad4 crystal demonstrated that the loop-helix region (L1, L2, L3 and H1) of one subunit makes extensive contacts with the three-helix bundle (H3, H4 and H5) of
another subunit and that many conserved residues in Smads are located within the trimer interface [17].
Propagation of TGFβsignals is mediated by the direct asso-ciation of R-Smads with the TGFβ receptor complex. The R-Smads are then directly phosphorylated by the type I TGFβ receptor kinase on the last two serines of a conserved SSXS motif located at the extreme carboxyl terminus of the MH2 domain. Biochemical analyses have shown that specific Smad-receptor interactions are mediated by the L3 loop in the R-Smads and the L45 loop in the type I receptor. Further insights into this interaction were provided by the structure of the Smad2 MH2 domain crystallized in complex with the Smad-binding domain (SBD) of SARA (Smad anchor for receptor activation), a protein that functions to recruit Smads to the TGFβreceptor [9,12,18]. The overall topology of the R-Smad MH2 domain is similar to that of Smad4, but the R-Smads also have a basic pocket on one surface that lies adjacent to loop 3 [18]. As Smad4 does not interact with receptors, it is thought that this basic pocket may serve as a docking site for the phosphorylated and activated type I receptor. The crystal structure of the Smad2 MH2 domain in complex with the SARA SBD also revealed an unusual arrangement in which the 40-residue SBD is in an extended conformation that forms a proline-rich coil, an αhelix and a
[image:3.609.54.563.119.289.2]β strand that contacts the three helix bundle (H3, H4 and H5) and a βstrand of the Smad2 MH2 domain. It is thought that the interaction of SARA with the βsheet is required for specificity, whereas contact with the three-helix bundle con-tributes to the binding affinity. It is currently not clear whether other proteins that interact with the Smad MH2 domain might also adopt a similar interaction interface; functional studies have indicated that α-helix 2 of the MH2 domain is important for the interaction of Smad2 with the transcription factor FAST (FoxH1) [19].
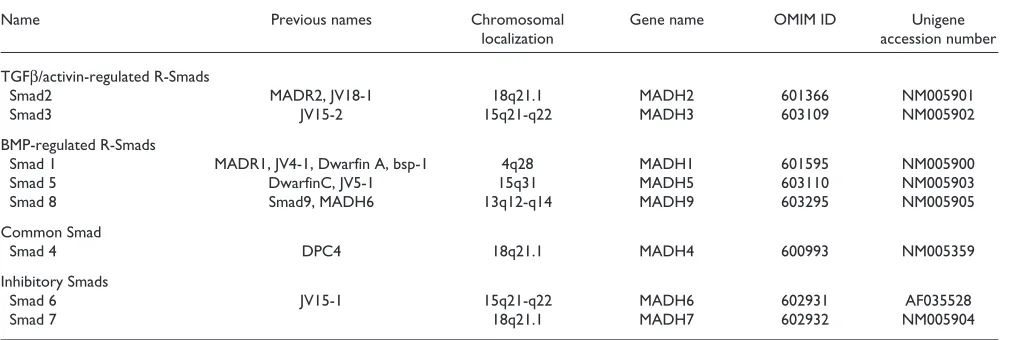
Table 1
Chromosomal localization of human Smads
Name Previous names Chromosomal Gene name OMIM ID Unigene
localization accession number
TGFβ/activin-regulated R-Smads
Smad2 MADR2, JV18-1 18q21.1 MADH2 601366 NM005901
Smad3 JV15-2 15q21-q22 MADH3 603109 NM005902
BMP-regulated R-Smads
Smad 1 MADR1, JV4-1, Dwarfin A, bsp-1 4q28 MADH1 601595 NM005900
Smad 5 DwarfinC, JV5-1 15q31 MADH5 603110 NM005903
Smad 8 Smad9, MADH6 13q12-q14 MADH9 603295 NM005905
Common Smad
Smad 4 DPC4 18q21.1 MADH4 600993 NM005359
Inhibitory Smads
Smad 6 JV15-1 15q21-q22 MADH6 602931 AF035528
Smad 7 18q21.1 MADH7 602932 NM005904
The Smad linker region
The linker region that connects the MH1 and MH2 domains contains a number of important peptide motifs. These include potential sites for phosphorylation by mitogen-activated protein kinases (MAPKs) - this phosphorylation can block R-Smad function [1-5] - and a nuclear export signal located within exon 3 of the co-Smad Smad4 [15,20,21]. R-Smads and I-Smads also contain a conserved proline-tyrosine (PY) motif that mediates interaction with the WW domains in the Smad-interacting proteins Smurf1 and Smurf2. Smurfs are E3 ubiquitin ligases of the C2-WW-HECT domain class that catalyze ubiquitin-mediated degradation of certain Smads and Smad-associated proteins, including the nuclear oncoprotein SnoN and the TGFβ-receptor complex [22-25]. The linker region of Smad4 also contains a Smad acti-vation domain (or SAD) that is required for transcriptional activation. A crystal structure of a fragment of Smad4 that includes the SAD and MH2 domain revealed that the SAD contacts a Smad4-specific sequence in the MH2 domain [9,12,26]. This stabilizes a glutamine-rich α-helical extension termed the TOWER, which, together with the proline-rich SAD, may form a transcriptional activation surface [26].
Localization and function
Developmental expression patternsIn general, all Smads are widely expressed throughout embryonic development and in most adult tissue and cell types. Analysis of mouse embryos has revealed, however, that there is some variation in the pattern, timing and level of expression of the individual Smads [27,28]. For instance, the inhibitory Smads, Smad6 and Smad7, are highly expressed in the developing cardiovascular system, although each also displays distinct expression patterns in non-cardiovascular tissues, including intramembranous bone and testis. The co-Smad Smad4 is ubiquitously expressed throughout embryonic development, with particularly high levels being detected in the epithelial crypts of the gut. Interestingly, the R-Smads display overlapping expression patterns; at least one of the BMP-regulated Smads (Smad1, Smad5 and Smad8) and one TGFβ/activin-regulated Smad (Smad2 or Smad3) is expressed in every tissue [27,28].
Function
Members of the TGFβ superfamily signal by inducing the stable assembly of heteromeric complexes of transmembrane type I and type II serine/threonine-kinase receptors. Within this complex the type II receptor kinase phosphorylates the type I receptor, which subsequently initiates downstream sig-naling to the Smad pathway. Smads then propagate the TGFβ signal from the cell surface into the nucleus [1-5].
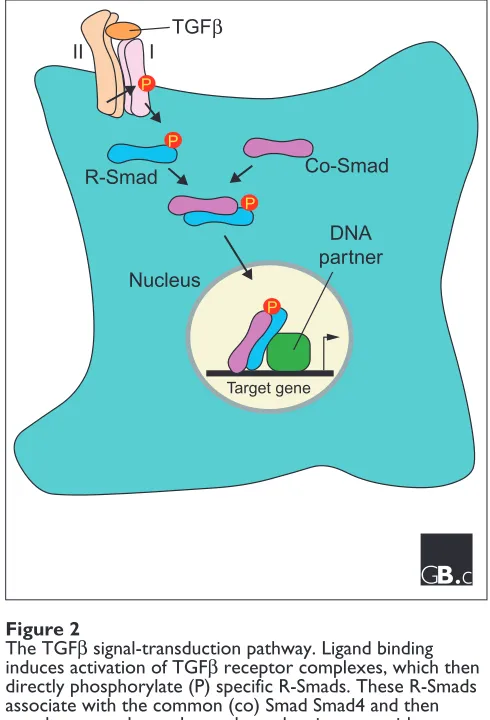
As mentioned above, three functional classes of Smads have been defined, each of which plays a distinct role in the signaling pathway (Figure 2). The activated type I receptors associate with specific R-Smads and phosphorylate them on
motif. The recognition of different R-Smads by the various type I receptor kinases is highly specific. Thus, the TGFβand activin type I receptors, TβRI (ALK5) and ActRIB (ALK4), respectively, activate both Smad2 and Smad3, which are closely related, whereas ALK1 and the BMP type I receptors ALK2, ALK3 and ALK6 all target Smads 1, 5 and 8. Phospho-rylated R-Smads then dissociate from the receptor and form a heteromeric complex with the co-Smad, Smad4. Although in mammals there is only one co-Smad, in Xenopusa second co-Smad, Smad4β, has been identified.
The R-Smad-co-Smad heteromeric complex then translocates to the nucleus to modulate the activity of specific promoters. Although Smads can directly bind DNA with low affinity and low specificity, they rely on interactions with various DNA-binding partners to target specific genes for transcriptional regulation. For instance, the TGFβ/activin-regulated Smads, Smad2 and Smad3, directly associate with DNA-binding partners such as FoxH1 (FAST), AP-1, TFE3, Sp1, Mixer, Runx2, LEF1/TCF and Miz1. Much less is known about the binding partners of BMP-regulated Smads, but identified nuclear partners include OAZ, Runx2 and Hoxc-8/9. Once localized to appropriate target promoters, Smads can then positively or negatively regulate transcriptional activity by recruiting coactivators, such as CBP/p300, or corepressors, including TGIF and Ski/Sno, which bind deacetylases.
Unlike the R-Smads, the I-Smads, Smad6 and Smad7, are potent antagonists of TGFβ signaling pathways. I-Smads, which do not have a carboxy-terminal SSXS motif, function by stably binding to activated receptor complexes, thus blocking access to and phosphorylation of the respective R-Smads by the type I receptor kinase. In addition, Smad7 can concomitantly induce ubiquitin-mediated degradation of active receptor complexes through its ability to recruit Smurfs, members of the C2-WW-HECT domain E3 ligase family [24,25]. Unlike the R- and co-Smads, which translo-cate from the cytoplasm to nucleus upon activation of the signaling pathways, Smad7 resides in the nucleus, and ligand stimulation results in its export into the cytoplasm where it can bind to receptors to manifest its inhibitory effects. In addition to TGFβ-independent signals, expression of I-Smadgenes is stimulated by TGFβand BMPs, thereby providing for negative feedback of the pathway.
Important mutants
Smad3-deficient mice are viable. Several groups have inde-pendently targeted the Smad3gene, and each reports distinct phenotypes, including defects in T-cell or splenocyte respon-siveness, presence of colorectal cancers and the development of a degenerative joint disease resembling osteoarthritis. Mice lacking the inhibitory Smad Smad6 have cardiovascu-lar abnormalities, including hyperplasia of the cardiac valves and outflow tract septation defects, suggesting that Smad7 cannot substitute for Smad6 even though both are highly expressed in the cardiovascular system. The phenotypes of mice lacking Smads 1, 7 and 8 remain to be determined. It is interesting to note that mice heterozygous for Smad2, Smad3 or Smad4 also display varying defects, indicating that Smadgene dosage is important [29].
TGFβ is a potent inhibitor of cellular proliferation of many normal cell types, suggesting that loss of TGFβ responsive-ness may be an important step during tumor progression. Consistent with this, mutations in Smads have been impli-cated in a number of human cancers (reviewed in [2,7,30]. Smad4 was originally identified as a potential tumor-sup-pressor gene in pancreatic carcinoma, and mutations in col-orectal, lung and pancreatic tumors have also been reported. Smad4is also mutated in families with familial juvenile poly-posis, an inherited syndrome associated with an increased risk of gastrointestinal cancer. Consistent with these observa-tions, Smad4heterozygote mice develop intestinal polyposis and invasive carcinomas. Smad2 has also been shown to harbor mutations in colorectal and lung tumors. Thus, Smads that mediate TGFβ signals appear to represent a class of tumor-suppressor genes important in human cancer. To date, there is no evidence that inactivating mutations in the other Smads are associated with cancers or other human diseases.
Frontiers
Since the first genetic description of Smads in 1995 and the initial biochemical characterizations of the proteins in 1996,
our understanding of how Smads function to mediate TGFβ signaling has grown considerably. The availability of complete human and Drosophilagenome sequences has confirmed that the full complement of Smads is now known. In addition, structural, biochemical and cell-biological approaches have culminated in the development of a model that provides a molecular description of how Smads transmit TGFβ super-family signals (Figure 2). With this basic framework in hand, current research efforts are directed towards reaching a more detailed mechanistic understanding of the signaling process. An area of particular interest is how localization of Smads and their association with other proteins is con-trolled. The phosphorylation of Smads by the TGFβreceptor complex is essential for initiating the signaling cascade, and recruitment of R-Smads to the receptor is thought to be facili-tated by SARA, but the subcellular compartment in which these events occur, and even whether there is a SARA-like protein for the BMP-regulated Smads, is not known. Insights into what determines the subcellular localization and nuclear accumulation of Smads will also be invaluable for enhancing our understanding of how their nuclear activities are mani-fested. Recent evidence has shown that Smads associate with E3 ubiquitin-ligases, are themselves ubiquitinated and degraded, and can serve as adapters to mediate ubiquitin-mediated degradation of other proteins [22-25,31]. Thus, it will be important to understand how a cell maintains the deli-cate balance between Smad and ubiquitin-ligase protein levels to ensure appropriate responsiveness to TGFβ -super-family signals. Although Smads are known to function in the nucleus as transcriptional regulators, little is understood of what determines whether Smads positively or negatively reg-ulate transcription. Furthermore, very few DNA-binding partners for the BMP-regulated Smads are known.
Cells receive multiple simultaneous signals, and the interac-tion of the TGFβpathway components with effectors of other signaling pathways has been described. Thus, future efforts will also focus on developing a better understanding of how
comment
reviews
reports
deposited research
interactions
information
[image:5.609.53.553.111.266.2]refereed research
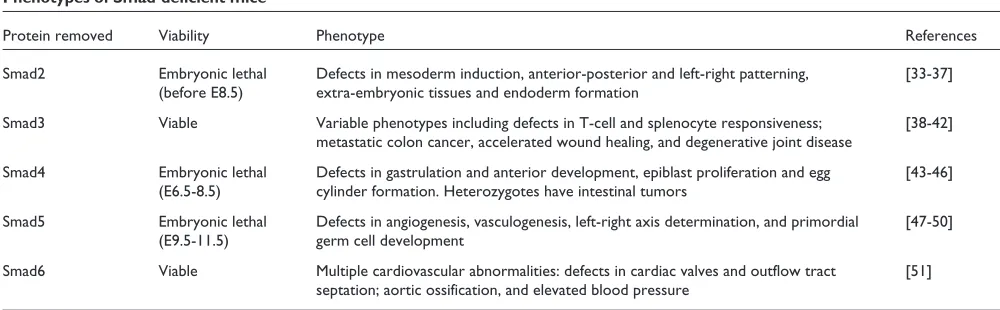
Table 2
Phenotypes of Smad-deficient mice
Protein removed Viability Phenotype References
Smad2 Embryonic lethal Defects in mesoderm induction, anterior-posterior and left-right patterning, [33-37] (before E8.5) extra-embryonic tissues and endoderm formation
Smad3 Viable Variable phenotypes including defects in T-cell and splenocyte responsiveness; [38-42] metastatic colon cancer, accelerated wound healing, and degenerative joint disease
Smad4 Embryonic lethal Defects in gastrulation and anterior development, epiblast proliferation and egg [43-46] (E6.5-8.5) cylinder formation. Heterozygotes have intestinal tumors
Smad5 Embryonic lethal Defects in angiogenesis, vasculogenesis, left-right axis determination, and primordial [47-50] (E9.5-11.5) germ cell development
and whether Smads cross-talk with other signaling path-ways. Current research efforts, including the search for novel Smad-interacting proteins, will undoubtedly shed light on these questions and may reveal new insights that challenge existing paradigms.
TGFβsuperfamily members play critical roles in numerous developmental events from cell-fate determination to organogenesis, and there is great interest in understanding these events. Examination of the effects of gene disruption in mice has revealed important information on the role Smads play in the earliest events. With the exception of Smad3 and Smad6, however, mice deficient in Smads die early in embryonic life; future work directed towards understanding their role in later development will thus require the genera-tion of condigenera-tional alleles. In addigenera-tion, the phenotype of mice lacking Smad1, Smad7 or Smad8 is eagerly awaited. These studies in mice will be bolstered by the analysis of Smad function in several other genetically manipulatable model systems, including C. elegans, Drosophilaand zebrafish.
β
human disorders, including fibrosis, hypertension, osteo-porosis, atherosclerosis and cancer, making this pathway an excellent target for therapeutic intervention [2,7,30]. Fur-thermore, mutations in components of the TGFβsignaling pathway have been associated with a number of hereditary diseases including persistent Müllerian duct syndrome, hereditary hemorrhagic telangiactasia, hereditary chon-drodysplasia, familial primary pulmonary hypertension and hereditary non-polyposis colorectal cancer [2,30]. Of the Smads, only Smad4 has been shown to be associated with a hereditary disease, namely juvenile polyposis syndrome. Thus, it will be important to determine whether other hered-itary syndromes can be attributed to mutations in Smads. In addition, the pathological implications of Smad hemizygos-ity or Smad dysfunction in other diseases, including cancer, is a worthy undertaking, as this may provide a target for the development of novel clinical treatments.
References
1. Attisano L, Wrana JL: Smads as transcriptional co-modulators.
Curr Opin Cell Biol2000, 12:235-243.
This reference and [2-8] are general reviews that cover the molecular details of TGFβ-superfamily signaling pathways and their involvement in disease.
2. Massagué J, Blain SW, Lo RS: TGFββsignaling in growth control, cancer and heritable disorders.Cell2000, 103:295-309. 3. ten Dijke P, Miyazono K, Heldin C-H: Signaling inputs converge
on nuclear effectors in TGF-ββ signaling. Trends Biochem Sci
2000, 25:64-70.
4. Wrana JL: Regulation of Smad activity.Cell2000,100:189-192. 5. Zhang Y, Derynck R: Regulation of Smad signalling by protein
associations and signalling crosstalk. Trends Cell Biol 1999, 9:274-279.
6. Padgett RW: TGFββsignaling pathways and human diseases.
Cancer Metastasis Rev1999, 18:247-259.
7. de Caestecker MP, Piek E, Roberts AB: Role of Transforming Growth Factor-ββsignaling in cancer. J Natl Cancer Inst 2000, 92:1388-1402.
8. Raftery LA, Sutherland DJ: TGF-beta family signal transduction in Drosophila development: from Mad to Smads. Dev Biol
1999, 210:251-268.
9. Attisano and Wrana Lab[http://ana30.med.utoronto.ca] In addition to providing an overview of the work conducted in the Wrana and Attisano laboratories, the site provides information on the TGFβsignaling pathway, including crystal structures of pathway compo-nents and a narrated signaling movie.
10. Wrana JL: Crossing Smads. Science’s STKE 2000 [http://stke. sciencemag.org/cgi/content/abstract/OC_sigtrans;2000/23/re1] An online review of the Smad pathway in Science’s Signal Transduction Knowledge Environment.
11. GeneCards[http://bioinformatics.weizmann.ac.il/cards]
An encyclopedia of human genes, proteins and diseases, maintained by the Weizmann Institute of Science. Search under the Smad gene name to obtain an integrated summary of specific available links to major data sources.
12. Shi Y: Structural insights on Smad function in TGFββ signal-ing.Bioessays2001, 23:223-232.
A review focused on structural and crystallographic analysis of TGFβ pathway components.
13. Shi Y, Wang YF, Jayaraman L, Yang H, Massague J, Pavletich NP: Crystal structure of a Smad MH1 domain bound to DNA: insights on DNA binding in TGF-beta signaling. Cell 1998, 94:585-594.
This study revealed that Smads bind DNA through a β-hairpin loop, a novel DNA-binding motif.
[image:6.609.54.299.88.448.2]14. Xiao Z, Liu X, Henis YI, Lodish HF: A distinct nuclear localiza-tion signal in the N terminus of Smad3 determines its Figure 2
The TGFβsignal-transduction pathway. Ligand binding induces activation of TGFβreceptor complexes, which then directly phosphorylate (P) specific R-Smads. These R-Smads associate with the common (co) Smad Smad4 and then translocate to the nucleus, where they interact with a variety of DNA-binding partners to regulate gene expression. See text for further details.
R-Smad
Co-Smad
TGF
β
Nucleus
DNA
partner
P
P
P
P
Target gene
ligand-induced translocation. Proc Natl Acad Sci USA 2000, 97:7853-7858.
Demonstration that nuclear translocation of R-Smads requires a deter-minant in the MH1 domain.
15. Kurisaki A, Kose Y, Yoneda Y, Heldin C-H, Moustakas A: Trans-forming Growth Factor-ββinduces nuclear import of Smad3 in an importin-ββand Ran-dependent manner. Mol Biol Cell
2001, 12:1079-1091.
This paper suggests that nuclear accumulation of Smad2 and Smad3 might occur in a distinct manner.
16. Xu L, Chen Y-G, Massagué J: The nuclear import function of Smad2 is masked by SARA and unmasked by TGFββ -depen-dent phosphorylation.Nat Cell Biol2000, 2:559-562.
This paper suggests that Smad2 nuclear localization is mediated by the MH2 domain.
17. Shi Y, Hata A, Lo RS, Massagué J, Pavletich NP: A structural basis for mutational inactivation of the tumour suppressor Smad4.Nature1997, 388:87-93.
The first crystal structure of a Smad domain, the Smad4 MH2. 18. Wu G, Chen YG, Ozdamar B, Gyuricza CA, Chong PA, Wrana JL,
Massagué J, Shi Y: Structural basis of Smad2 recognition by the Smad anchor for receptor activation. Science 2000, 287:92-97.
This crystallographic study reveals how Smad2 associates with SARA and provided insights into how Smad2 might interact with the TGFβ -receptor complex.
19. Chen Y-G, Hata A, Lo RS, Wotton D, Shi Y, Pavletich N, Massagué J: Determinants of specificity in TGF-ββ signal transduction.
Genes Dev1998, 12:2144-2152.
This paper demonstrates that four residues in the L45 loop of the type I receptor and two residues in the L3 loop of Smads establishes the specificity of R-Smad-receptor interaction.
20. Pierreux CE, Nicolas FJ, Hill CS: Transforming growth factor ββ -independent shuttling of Smad4 between the cytoplasm and nucleus.Mol Cell Biol2000, 20:9041-9054.
This reference and [21] demonstrate that the cytoplasmic localization of Smad4 is the result of active nuclear export.
21. Watanabe M, Masuyama N, Fukuda M, Nishida E: Regulation of intracellular dynamics of Smad4 by its leucine-rich nuclear export signal.EMBO Rep2000, 1:176-182.
22. Zhu H, Kavsak P, Abdollah S, Wrana JL, Thomsen GH: A SMAD ubiquitin ligase targets the BMP pathway and affects embry-onic pattern formation.Nature1999, 400:687-693.
The first description of the role of C2-WW-HECT domain E3 ligases such as Smurf1 in the ubiquitin-mediated degradation of Smads. 23. Bonni S, Wang H-R, Causing CG, Kavsak P, Stroschein SL, Luo K,
Wrana JL: TGF-ββinduces assembly of a Smad2–Smurf2 ubiq-uitin ligase complex that targets SnoN for degradation.Nat Cell Biol2001, 3:587-595.
This reference and [24,25] demonstrate that Smads can function as adapters for the E3 ligases Smurf1 and Smurf2 to induce degradation of Smad-associated proteins.
24. Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, Wrana JL: Smad7 binds to Smurf2 to form an E3 ubiqui-tin ligase that targets TGFββreceptor for degradation.Mol Cell2000, 6:1365-1375.
25. Ebisawa T, Fukuchi M, Murakami G, Chiba T, Tanaka K, Imamura T, Miyazono K: Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J Biol Chem2001, 276:12477-12480. 26. Qin B, Lam SS, Lin K: Crystal structure of a transcriptionally
active Smad4 fragment.Structure Fold Des1999, 7:1493-1503. The crystal structure of the Smad4 MH2 domain together with the transcriptional activation domain located in the adjacent linker region is described.
27. Luukko K, Ylikorkala A, Mäkelä TP: Developmentally regulated expression of Smad3, Smad4, Smad6 and Smad7 involved in TGF-beta signaling.Mech Dev2001, 101:209-212.
This reference and [28] provide a comprehensive description of the expression patterns of Smads in mouse embryos.
28. Flanders KC, Kim ES, Roberts AB: Immunohistochemical expression of Smads1-6 in the 15-day gestation mouse embryo: signaling by BMPs and TGF-ββs. Dev Dyn 2001, 220:141-154.
29. Weinstein M, Yang X, Deng C-X: Functions of mammalian Smad genes as revealed by targeted gene disruption in mice.Cytokine Growth Factor Rev2000, 11:49-58.
A comprehensive review of the phenotypes of Smad-deficient mice. 30. Blobe GC, Schiemann WP, Lodish HF: Role of Transforming
Growth Factor ββ in human disease. New Eng J Med 2000, 342:1350-1358.
A medically oriented review that focuses on TGFβin human disease. 31. Fukuchi M, Imamura T, Chiba T, Ebisawa T, Kawabata M, Tanaka K,
Miyazono K: Ligand-dependent degradation of Smad3 by a ubiquitin ligase complex of roc1 and associated proteins.
Mol Biol Cell2001, 12:1431-1443.
32. Online Mendelian Inheritance in Man (OMIM) [http://www.ncbi.nlm.nih.gov/omim]
A catalog of human genes and genetic disorders. Search for Smads using the identifiers provided in Table 1 to obtain a summary of the human genes and associated genetic disorders.
33. Waldrip WR, Bikoff EK, Hoodless PA, Wrana JL, Robertson EJ: Smad2 signalling in extraembryonic tissues determines anterior-posterior polarity of the early mouse embryo.Cell
1998, 92:797-808.
This reference and [34-37] describe the early embyronic defects in Smad2-deficient mice.
34. Weinstein M, Yang X, Li C, Xu X, Goday J, Deng C-X: Failure of egg cylinder elongation and mesoderm induction in mouse embryos lacking the tumor suppressor Smad2.Proc Natl Acad Sci USA1998, 95:9378-9383.
35. Nomura M, Li E: Smad2 role in mesoderm formation, left-right patterning and craniofacial development.Nature1998, 393:786-790.
36. Heyer J, Escalante-Alcade D, Lia M, Boettinger E, Edelmann W, Stewart CL, Kucherlapati R: Postgastrulation Smad2-deficient embryos show defects in embryo turning and anterior mor-phogenesis.Proc Natl Acad Sci USA1999, 96:12595-12600. 37. Tremblay KD, Hoodless PA, Bikoff EK, Robertson EJ: Formation of
the definitive endoderm in mouse is a Smad2-dependent process.Development 2000, 127:3079-3090.
38. Ashcroft GS, Yang X, Glick AB, Weinstein M, Letterio JJ, Mizel DE, Anzano M, Greenwell-Wild T, Wahl SM, Deng C, Roberts AB: Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat Cell Biol 1999, 1:260-266.
This reference and [39-42] describe the varying phenotypes observed by different groups in the viable Smad3 null mice.
39. Zhu Y, Richardson JA, Parada LF, Graff JM: Smad3 mutant mice develop metastatic colorectal cancer.Cell1998, 94:703-714. 40. Datto MB, Frederick JP, Pan L, Borton AJ, Zhuang Y, Wang X-F:
Targeted disruption of Smad3 reveals an essential role in Transforming Growth Factor beta-mediated signal trans-duction.Mol Cell Biol1999, 19:2495-2504.
41. Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, Roberts AB, Deng C: Targeted disruption of Smad3 results in impaired mucosal immunity and diminished T cell respon-siveness to TGF beta.EMBO J1999, 18:1280-1291.
42. Yang X, Chen L, Xu X, Li C, Huang C, Deng X-X: TGF-ββ/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage.J Cell Biol
2001, 153:35-46.
43. Sirard C, de la Pompa JL, Elia A, Itie A, Mirtsos C, Cheung A, Hahn S, Wakeham A, Schwartz L, Kern SE, et al.: The tumor suppressor gene Smad4/Dpc4 is required for gastrulation and later for anterior development of the mouse embryo.Genes Dev1997, 12:107-119.
This reference and [44] demonstrate the requirement for the common Smad Smad4 in the earliest stages of mouse development.
44. Yang X, Li C, Xu X, Deng C: The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc Natl Acad Sci USA 1998, 95:3667-3672.
45. Takaku K, Oshima M, Miyoshi H, Matsui M, Seldin MF, Taketo MM: Intestinal tumorigenesis in compound mutant mice of both DPC4 (Smad4) and Apc Genes.Cell1998, 92:645-656. This reference and [46] use mouse models to demonstrate the involve-ment of Smad4 in colorectal cancer.
46. Takaku K, Miyoshi H, Matsunaga A, Oshima M, Taketo MM: Gastric and duodenal polyps in Smad4 (dpc4) knockout mice.Cancer Res1999, 59:6113-6117.
47. Chang H, Huylebroeck D, Verschueren K, Guo Q, Matzuk MM, Zwijsen A: Smad5 knockout mice die at mid-gestation due to
comment reviews reports deposited research interactions information
1999, 126:1631-1642.
This reference and [48-50] demonstrate the importance of Smad5 in angiogenesis, left/right patterning and primordial-germ-cell formation. 48. Yang X, Castilla LH, Xu X, Li C, Gotay J, Weinstein M, Liu PP, Deng
C-X: Angiogenesis defects and mesenchymal apoptosis in mice lacking SMAD5.Development 1999, 126:1571-1580. 49. Chang H, Zwijsen A, Vogel H, Huylebroeck D, Matzuk M: Smad5 is
essential for left-right asymmetry in mice. Dev Biol 2000, 219:71-78.
50. Chang H, Matzuk MM: Smad5 is required for mouse primordial germ cell development.Mech Dev2001, 104:61-67.
51. Galvin KM, Donovan MJ, Lynch CA, Meyer RI, Paul RJ, Lorenz JN, Fairchild-Huntress V, Dixon KL, Dunmore JH, Gimbrone MAJ, et al.: A role for Smad6 in development and homeostasis of the cardiovascular system.Nat Genet2000, 24:171-174.
![Figure 1Primary structure and relationships of Smads. domain (reproduced with permission from [13])](https://thumb-us.123doks.com/thumbv2/123dok_us/8603973.865512/2.609.55.298.81.725/figure-primary-structure-relationships-smads-domain-reproduced-permission.webp)