organic papers
o1396
Riis and Larsen C13H12O2 doi:10.1107/S1600536805004125 Acta Cryst.(2005). E61, o1396–o1397 Acta Crystallographica Section E
Structure Reports Online
ISSN 1600-5368
2-(1-Naphthyl)propionic acid
Erik Riis* and Sine Larsen
Centre for Crystallographic Studies, Department of Chemistry, Universitetsparken 5, DK-2100 Copenhagen, Denmark
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 122 K
Mean(C–C) = 0.002 A˚
Rfactor = 0.055
wRfactor = 0.161
Data-to-parameter ratio = 14.2
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography
Printed in Great Britain – all rights reserved
2-(1-Naphthyl)propionic acid, C13H12O2, is one of the chiral compounds which exhibit the highest difference (80 K) in melting points between the racemic and enantiomeric crystals. We report here the structure of the racemic compound.
Comment
The structure determination of the title compound, (I), was undertaken as part of an investigation of racemic and enan-tiomeric crystals of chiral compounds, which focuses on the relations between differences in physicochemical properties
(e.g.melting point) and crystal packing. The unit-cell
param-eters of (I) have been reported previously (Husebye, 1961), but no coordinates have been published or are available in the Cambridge Structural Database (Version of November 2004; Allen, 2002). The preparative route for racemic 2-(1-naphthyl)propionic acid is shown in the scheme below. It differs from the previously used procedure (Hechtet al., 1978) by using butyllithium instead of sodium to deprotonate the starting material, 1-naphthylacetic acid.
The carboxylic acid group of 2-(1-naphthyl)propionic acid is hydrogen bonded to the carboxylic acid group of another molecule related by inversion symmetry, forming cyclic carboxylic acid dimers. This packing motif is common in other aromatic monofunctional carboxylic acids (Sørensen & Larsen, 2003). In addition, the carboxy group serves as an acceptor for two C—H hydrogen bonds (Table 1) from two other molecules related by the twofold screw axis and the glide
plane, respectively. These interactions are presumably
important for the arrangement of the naphthyl groups into a herring-bone pattern, in which the naphthyl groups have
C O interactions on one side and C—H on the
opposite side. Since these interactions are between molecules
of opposite chirality, they can not exist in the enantiomeric crystals, which could explain the higher melting point of the racemic acid.
Experimental
Butyllithium in hexane (20 mmol) was added dropwise to a stirred solution of 1-naphthylacetic acid (1.86 g, 10 mmol) in THF under a nitrogen atmosphere and cooled with solid carbon dioxide. The mixture turned red, indicating the formation of the highly conjugated dianion. MeI (10.5 mmol) was then added slowly and the mixture was allowed to reach room temperature. In order to protonate the anion, the solution was then extracted with water and 4MHCl was added followed by evaporation. This yielded an orange oil (1.95 g, 9.75 mmol). The oil was recrystallized fromn-heptane to yield a white powder (1.60 g, 80% yield). Further recrystallization was required to obtain small crystals.
Crystal data
C13H12O2
Mr= 200.24
Monoclinic,P21=c
a= 7.8089 (4) A˚
b= 8.8904 (5) A˚
c= 14.7466 (12) A˚
= 96.143 (7)
V= 1017.89 (11) A˚3
Z= 4
Dx= 1.307 Mg m 3
MoKradiation
Cell parameters from 11792 reflections
= 1.4–37.0
= 0.09 mm1
T= 122.0 (10) K Plate, colorless 0.480.360.09 mm
Data collection
Nonius KappaCCD diffractometer
!and’scans
Absorption correction: none 25823 measured reflections 2456 independent reflections 2071 reflections withI> 2(I)
Rint= 0.046
max= 28.0
h=10!10
k=11!11
l=19!19
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.055
wR(F2) = 0.161
S= 1.05 2456 reflections 173 parameters
Only coordinates of H atoms refined
w= 1/[2(F
o2) + (0.0798P)2
+ 0.705P]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.005 max= 0.54 e A˚3 min=0.25 e A˚3
Table 1
Hydrogen-bonding geometry (A˚ ,).
D—H A D—H H A D A D—H A
O1—H1 O2i
0.91 (3) 1.74 (3) 2.6511 (17) 176 (2) C9—H9 O1ii
1.06 (2) 2.54 (2) 3.4631 (19) 145 (2) C8—H8 O2iii
0.96 (3) 2.64 (2) 3.4331 (18) 140 (2)
Symmetry codes: (i)x;y;z; (ii) 1x;1
2þyþ;12zþ; (iii) 1þx;12yþ;12þzþ.
The positions of H atoms were refined. For H atoms bound to carbon, Uiso(H) = 1.2Ueq(H), while Uiso(H) was refined for the
hydroxyl H atom, H1.
Data collection:KappaCCD Software(Nonius, 1997); cell refine-ment: DIRAX/LSQ (Duisenberg et al., 2003); data reduction:
EvalCCD(Duisenberget al., 2003); program(s) used to solve
struc-ture:SIR97(Altomareet al., 1999); program(s) used to refine struc-ture: SHELXL97(Sheldrick, 1997); molecular graphics:ORTEPII
(Johnson, 1976) andMERCURY(Brunoet al., 2002); software used to prepare material for publication:SHELXL97.
Mr Flemming Hansen is thanked for technical assistance with the data collection. This work was supported by a grant from The Danish Natural Science Research Council.
References
Allen, F. H. (2002).Acta Cryst.B58, 380–388.
Altomare, A., Burla, M. C., Camalli, M., Cascarano, G., Giacovazzo, C., Guagliardi, A., Moliterni, A. G. G., Polidori, G. & Spagna, R. (1999).J. Appl. Cryst.32, 115–119.
Bruno, I. J., Cole, J. C., Edgington, P. R., Kessler, M., Macrae, C. F., McCabe, P., Pearson, J. & Taylor, R. (2002).Acta Cryst.B58, 389–397.
Duisenberg, A. J. M., Kroon-Batenburg, L. M. J. & Schreurs, A. M. M. (2003).
J. Appl. Cryst.36, 220–229.
Hecht, S. S., Loy, M., Mazzarese, R. & Hoffmann, D. (1978).J. Med. Chem.21, 38–44.
Husebye, S. (1961).Acta Chem. Scand.15, 1215–1222.
Johnson, C. K. (1976).ORTEPII. Report ORNL-5138. Oak Ridge National Laboratory, Tennessee, USA.
[image:2.610.314.565.70.232.2]Nonius (1997).KappaCCD Software. Nonius BV, Delft, The Netherlands. Sheldrick, G. M. (1997).SHELXL97. University of Go¨ttingen, Germany. Sørensen, H. O. & Larsen, S. (2003).Acta Cryst.B59, 132–140.
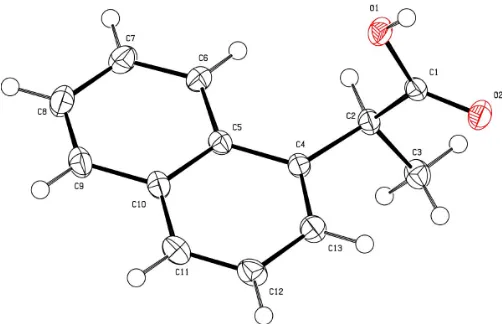
Figure 1
[image:2.610.313.565.285.404.2]AnORTEPII(Johnson, 1976) drawing of the title molecule. Displace-ment ellipsoids are drawn at the 50% probability level and H atoms are shown as spheres with fixed radius.
Figure 2
supporting information
sup-1 Acta Cryst. (2005). E61, o1396–o1397
supporting information
Acta Cryst. (2005). E61, o1396–o1397 [https://doi.org/10.1107/S1600536805004125]
2-(1-Naphthyl)propionic acid
Erik Riis and Sine Larsen
2-(1-naphtyl)propionic acid
Crystal data
C13H12O2
Mr = 200.24 Monoclinic, P21/c Hall symbol: -P 2ybc
a = 7.8089 (4) Å
b = 8.8904 (5) Å
c = 14.7466 (12) Å
β = 96.143 (7)°
V = 1017.89 (11) Å3
Z = 4
F(000) = 424
Dx = 1.307 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 11792 reflections
θ = 1.4–37.0°
µ = 0.09 mm−1
T = 122 K Needle, colorless 0.48 × 0.36 × 0.09 mm
Data collection
KappaCCD diffractometer
Radiation source: fine-focus sealed tube area–detector scans to fill Ewald sphere. 25823 measured reflections
2456 independent reflections
2071 reflections with I > 2σ(I)
Rint = 0.046
θmax = 28.0°, θmin = 2.3°
h = −10→10
k = −11→11
l = −19→19
Refinement
Refinement on F2 Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.055
wR(F2) = 0.161
S = 1.05 2456 reflections 173 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Only H-atom coordinates refined
w = 1/[σ2(F
o2) + (0.0798P)2 + 0.705P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.005 Δρmax = 0.54 e Å−3 Δρmin = −0.25 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O2 −0.12399 (15) 0.14248 (13) 0.02487 (8) 0.0257 (3)
O1 0.08662 (14) 0.03349 (13) 0.11681 (8) 0.0243 (3)
C5 0.21503 (17) 0.34088 (16) 0.27007 (10) 0.0183 (3)
C1 −0.03778 (17) 0.13286 (16) 0.09869 (10) 0.0187 (3)
C4 0.09722 (18) 0.33986 (16) 0.18954 (9) 0.0185 (3)
C10 0.36082 (18) 0.43767 (17) 0.27460 (10) 0.0204 (3)
C13 0.1256 (2) 0.43464 (18) 0.11929 (10) 0.0237 (3)
C6 0.1943 (2) 0.25099 (18) 0.34812 (10) 0.0227 (3)
C2 −0.06012 (18) 0.23639 (17) 0.17781 (10) 0.0198 (3)
C8 0.4576 (2) 0.3505 (2) 0.42651 (11) 0.0281 (4)
C12 0.2686 (2) 0.53317 (18) 0.12373 (11) 0.0259 (3)
C9 0.48078 (19) 0.43895 (19) 0.35368 (11) 0.0258 (3)
C11 0.3845 (2) 0.53331 (19) 0.19959 (11) 0.0255 (3)
C3 −0.22741 (19) 0.32487 (19) 0.16441 (11) 0.0239 (3)
C7 0.3111 (2) 0.2565 (2) 0.42375 (11) 0.0278 (3)
H13 0.037 (3) 0.435 (2) 0.0645 (14) 0.028*
H6 0.096 (3) 0.188 (3) 0.3501 (14) 0.027*
H2 −0.066 (3) 0.172 (2) 0.2287 (14) 0.024*
H8 0.540 (3) 0.353 (2) 0.4801 (15) 0.034*
H12 0.279 (3) 0.600 (3) 0.0735 (15) 0.031*
H9 0.588 (3) 0.511 (3) 0.3521 (15) 0.031*
H11 0.472 (3) 0.596 (3) 0.1996 (15) 0.031*
H3A −0.227 (3) 0.392 (2) 0.1114 (15) 0.029*
H3B −0.237 (3) 0.391 (2) 0.2183 (14) 0.029*
H3C −0.324 (3) 0.255 (2) 0.1582 (14) 0.029*
H7 0.293 (3) 0.192 (3) 0.4768 (15) 0.033*
H1 0.094 (3) −0.026 (3) 0.067 (2) 0.054 (7)*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O2 0.0243 (5) 0.0257 (6) 0.0251 (5) 0.0065 (4) −0.0058 (4) −0.0060 (4)
O1 0.0232 (5) 0.0231 (6) 0.0251 (5) 0.0063 (4) −0.0043 (4) −0.0043 (4)
C5 0.0168 (6) 0.0170 (6) 0.0213 (6) −0.0002 (5) 0.0028 (5) −0.0025 (5)
C1 0.0159 (6) 0.0158 (6) 0.0242 (7) −0.0020 (5) 0.0007 (5) −0.0018 (5)
C4 0.0187 (6) 0.0184 (6) 0.0184 (6) −0.0020 (5) 0.0012 (5) −0.0011 (5)
C10 0.0166 (6) 0.0202 (7) 0.0243 (7) −0.0001 (5) 0.0018 (5) −0.0057 (5)
C13 0.0279 (7) 0.0241 (7) 0.0194 (6) −0.0035 (6) 0.0034 (5) 0.0010 (5)
C6 0.0232 (7) 0.0227 (7) 0.0220 (7) 0.0002 (6) 0.0009 (5) 0.0028 (5)
C2 0.0173 (6) 0.0200 (7) 0.0218 (7) −0.0023 (5) −0.0001 (5) −0.0020 (5)
C8 0.0258 (7) 0.0300 (8) 0.0262 (8) 0.0052 (6) −0.0073 (6) −0.0048 (6)
C12 0.0322 (8) 0.0235 (7) 0.0229 (7) −0.0063 (6) 0.0071 (6) 0.0017 (6)
C9 0.0188 (7) 0.0264 (8) 0.0314 (8) −0.0007 (6) −0.0010 (6) −0.0069 (6)
C11 0.0243 (7) 0.0233 (7) 0.0295 (8) −0.0058 (6) 0.0058 (6) −0.0028 (6)
supporting information
sup-3 Acta Cryst. (2005). E61, o1396–o1397
C7 0.0329 (8) 0.0271 (8) 0.0227 (7) 0.0023 (7) −0.0005 (6) 0.0032 (6)
Geometric parameters (Å, º)
O2—C1 1.2201 (18) C8—C7 1.414 (2)
O1—C1 1.3190 (17) C12—C11 1.362 (2)
C5—C4 1.4223 (19) O1—H1 0.92 (3)
C5—C10 1.4229 (19) C13—H13 1.00 (2)
C5—C6 1.425 (2) C6—H6 0.95 (2)
C1—C2 1.511 (2) C2—H2 0.95 (2)
C4—C13 1.372 (2) C8—H8 0.97 (2)
C4—C2 1.5298 (19) C12—H12 0.96 (2)
C10—C9 1.416 (2) C9—H9 1.06 (2)
C10—C11 1.423 (2) C11—H11 0.88 (2)
C13—C12 1.415 (2) C3—H3A 0.98 (2)
C6—C7 1.364 (2) C3—H3B 1.00 (2)
C2—C3 1.520 (2) C3—H3C 0.98 (2)
C8—C9 1.359 (2) C7—H7 0.99 (2)
C4—C5—C10 119.07 (13) C4—C13—H13 117.4 (12)
C4—C5—C6 123.50 (13) C12—C13—H13 120.2 (12)
C10—C5—C6 117.43 (13) C7—C6—H6 117.8 (12)
O2—C1—O1 123.48 (13) C5—C6—H6 121.0 (12)
O2—C1—C2 123.23 (13) C1—C2—H2 105.3 (12)
O1—C1—C2 113.27 (12) C3—C2—H2 107.6 (12)
C13—C4—C5 118.96 (13) C4—C2—H2 112.1 (12)
C13—C4—C2 118.48 (13) C9—C8—H8 120.1 (14)
C5—C4—C2 122.56 (12) C7—C8—H8 120.2 (13)
C9—C10—C5 119.95 (14) C11—C12—H12 122.1 (13)
C9—C10—C11 120.39 (14) C13—C12—H12 118.5 (13)
C5—C10—C11 119.66 (13) C8—C9—H9 122.7 (12)
C4—C13—C12 122.40 (14) C10—C9—H9 116.3 (12)
C7—C6—C5 121.12 (15) C12—C11—H11 116.6 (14)
C1—C2—C3 112.56 (12) C10—C11—H11 122.9 (14)
C1—C2—C4 107.21 (11) C2—C3—H3A 109.8 (12)
C3—C2—C4 111.86 (12) C2—C3—H3B 109.9 (12)
C9—C8—C7 119.66 (14) H3A—C3—H3B 106.6 (17)
C11—C12—C13 119.37 (14) C2—C3—H3C 109.1 (13)
C8—C9—C10 120.96 (15) H3A—C3—H3C 112.6 (17)
C12—C11—C10 120.51 (14) H3B—C3—H3C 108.9 (17)
C6—C7—C8 120.86 (15) C6—C7—H7 118.9 (13)
C1—O1—H1 109.5 (17) C8—C7—H7 120.2 (13)
C10—C5—C4—C13 −1.4 (2) O1—C1—C2—C4 69.85 (15)
C6—C5—C4—C13 177.72 (14) C13—C4—C2—C1 62.21 (17)
C10—C5—C4—C2 178.32 (13) C5—C4—C2—C1 −117.49 (15)
C6—C5—C4—C2 −2.6 (2) C13—C4—C2—C3 −61.64 (18)
C6—C5—C10—C9 1.7 (2) C4—C13—C12—C11 1.0 (3)
C4—C5—C10—C11 1.1 (2) C7—C8—C9—C10 −0.4 (2)
C6—C5—C10—C11 −178.01 (14) C5—C10—C9—C8 −1.1 (2)
C5—C4—C13—C12 0.3 (2) C11—C10—C9—C8 178.58 (15)
C2—C4—C13—C12 −179.38 (14) C13—C12—C11—C10 −1.3 (2)
C4—C5—C6—C7 −179.87 (15) C9—C10—C11—C12 −179.48 (15)
C10—C5—C6—C7 −0.8 (2) C5—C10—C11—C12 0.2 (2)
O2—C1—C2—C3 14.7 (2) C5—C6—C7—C8 −0.7 (2)
O1—C1—C2—C3 −166.73 (13) C9—C8—C7—C6 1.4 (3)
O2—C1—C2—C4 −108.71 (15)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
O1—H1···O2i 0.91 (3) 1.74 (3) 2.6511 (17) 176 (2)
C9—H9···O1ii 1.06 (2) 2.54 (2) 3.4631 (19) 145 (2)
C8—H8···O2iii 0.96 (3) 2.64 (2) 3.4331 (18) 140 (2)